Southern Blotting for DNA Methylation Analysis: A Classic Tool's Modern Role in Epigenetic Research and Drug Development
This article provides a comprehensive guide for researchers and drug development professionals on the application of Southern blotting for DNA methylation analysis.
Southern Blotting for DNA Methylation Analysis: A Classic Tool's Modern Role in Epigenetic Research and Drug Development
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on the application of Southern blotting for DNA methylation analysis. We cover the foundational principles of DNA methylation and Southern blot hybridization, detailing a complete methodological workflow from genomic DNA digestion with methylation-sensitive enzymes to probe design and detection. The guide addresses common troubleshooting scenarios and optimization strategies for sensitivity and specificity. Finally, we critically evaluate Southern blotting's role in the modern epigenetic toolkit, comparing it to next-generation sequencing and PCR-based methods, and discuss its enduring value in validation and clinical assay contexts.
The Bedrock of Methylation Analysis: Core Concepts and Historical Context of Southern Blotting
DNA methylation, a cornerstone of epigenetic regulation, involves the addition of a methyl group to the cytosine base, predominantly at CpG dinucleotides. Within the broader thesis of analyzing DNA methylation patterns via Southern blotting—a foundational yet robust technique for assessing specific genomic loci—this application note explores the critical concepts of CpG islands, their role in gene silencing, and the profound implications for human disease. Southern blotting provides a direct, hybridization-based method to visualize methylation-sensitive restriction enzyme digestion patterns, offering a tangible link between epigenetic modification, gene expression status, and phenotypic outcome.
Core Concepts and Quantitative Data
CpG Islands: Genomic Distribution and Characteristics
CpG islands (CGIs) are genomic regions with high frequency of CpG sites. They are typically associated with gene promoters.
Table 1: Standard Characteristics of CpG Islands
| Parameter | Typical Value | Definition/Notes |
|---|---|---|
| Length | >200 bp | Minimum span to be classified as a CGI. |
| GC Content | >50% | Proportion of Guanine and Cytosine nucleotides. |
| Observed/Expected CpG Ratio | >0.6 | Ratio of observed CpG frequency to the frequency expected from GC content. |
| % of Gene Promoters Associated with CGI | ~70% | Varies by gene class (housekeeping vs. tissue-specific). |
| Methylation State in Normal Somatic Cells | Mostly Unmethylated | Hypermethylation is associated with long-term silencing. |
Gene Silencing and Disease Implications
Aberrant DNA methylation, particularly the hypermethylation of promoter-associated CGIs, is a hallmark of transcriptional silencing and is implicated in numerous diseases, most notably cancer.
Table 2: Association of CGI Hypermethylation with Select Diseases
| Disease/Condition | Frequently Silenced Gene(s) via CGI Methylation | Functional Consequence |
|---|---|---|
| Colorectal Cancer | MLH1 (DNA repair), CDKN2A/p16 (cell cycle) | Genomic instability, unchecked proliferation. |
| Leukemia (AML) | C/EBPα (differentiation) | Blocked cellular differentiation. |
| Neurological (Rett Syndrome) | MECP2 mutations (methyl-CpG binding protein) | Disrupted reading of methylation signals, severe neurodevelopment issues. |
| Atherosclerosis | ESR1 (Estrogen Receptor α) | Altered vascular response, inflammation. |
Experimental Protocols
Protocol 1: Southern Blot Analysis of DNA Methylation at a Specific Loci
Principle: Genomic DNA is digested with methylation-sensitive and methylation-insensitive isoschizomer restriction enzymes (e.g., HpaII and MspI). Differential digestion patterns, visualized by Southern blotting with a locus-specific probe, reveal methylation status.
Detailed Methodology:
- DNA Isolation & Quantification: Isolate high-molecular-weight genomic DNA from target tissue/cells. Quantify via spectrophotometry (e.g., Nanodrop). Ensure A260/A280 ratio ~1.8.
- Restriction Enzyme Digestion: Set up parallel 20 µg DNA digestions overnight:
- Reaction A (Methylation-Sensitive): HpaII (cuts unmethylated CCGG, blocked by methylation at internal C).
- Reaction B (Control): MspI (cuts all CCGG regardless of methylation state).
- Reaction C (Size Control): A rare-cutter (e.g., EcoRI) to generate a large fragment encompassing the region of interest.
- Use manufacturer-recommended buffers and conditions.
- Gel Electrophoresis: Load digested DNA on a 0.8-1% agarose gel. Include a DNA molecular weight ladder. Run at ~1-2 V/cm until optimal separation is achieved.
- Southern Blotting:
- Denature & Neutralize: Depurinate gel briefly in 0.25M HCl. Denature in 0.4M NaOH/0.6M NaCl. Neutralize in 1M Tris-HCl (pH 7.4)/1.5M NaCl.
- Capillary Transfer: Transfer DNA overnight via upward capillary method onto a positively charged nylon membrane using 20X SSC as transfer buffer.
- Immobilization: UV-crosslink DNA to the membrane.
- Hybridization & Detection:
- Pre-hybridize: Incubate membrane in hybridization buffer (e.g., Church buffer: 0.5M NaHPO₄, 7% SDS, 1mM EDTA, pH 7.2) at 65°C for 1-2 hours.
- Probe Preparation: Label a locus-specific, PCR-generated probe (200-500 bp) with [α-³²P] dCTP using a random priming kit. Purify using a spin column.
- Hybridize: Add denatured probe to fresh buffer and incubate with membrane overnight at 65°C.
- Washing: Perform stringent washes (final wash: 0.1X SSC, 0.1% SDS at 65°C).
- Visualization & Analysis: Expose membrane to a phosphorimager screen or X-ray film. Analyze banding patterns.
- Interpretation: Identical patterns in HpaII and MspI digests indicate unmethylated loci. Presence of larger, uncut fragments in HpaII vs. MspI indicates methylation at those restriction sites.
The Scientist's Toolkit: Key Research Reagents & Materials
| Item | Function in Protocol |
|---|---|
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII) | Key tool for initial discrimination; fails to cut when its recognition site's cytosine is methylated. |
| Isoschizomer Control Enzymes (e.g., MspI) | Cuts same sequence regardless of methylation; essential control for DNA integrity and digestion efficiency. |
| Positively Charged Nylon Membrane | Robust solid support for immobilized, denatured DNA for subsequent hybridization. |
| [α-³²P] dCTP (Radioactive) or Digoxigenin-dUTP (Non-radioactive) | Label for generating high-specific-activity probes for sensitive detection. |
| Locus-Specific Oligonucleotides/PCR Probes | Ensures targeted analysis of specific CpG islands or genomic regions of interest. |
| Phosphorimager System or X-ray Film | For high-resolution, quantitative detection of hybridized signal from the Southern blot. |
Protocol 2: Combined Restriction Enzyme & Bisulfite Sequencing for Validation
Principle: This complementary protocol uses sodium bisulfite to convert unmethylated cytosines to uracil (read as thymine in sequencing), while methylated cytosines remain unchanged. Subsequent PCR and sequencing of the CGI of interest provides single-base-pair resolution of methylation status.
Detailed Methodology:
- Bisulfite Conversion: Treat 500 ng - 1 µg genomic DNA with sodium bisulfite using a commercial kit (e.g., EZ DNA Methylation Kit). This deaminates unmethylated C to U.
- PCR Amplification: Design primers specific for the bisulfite-converted, top-strand sequence of your target CGI. Use a polymerase robust for amplifying GC-rich, converted DNA.
- Cloning & Sequencing: Ligate PCR product into a TA-cloning vector. Transform competent bacteria. Pick 10-20 individual colonies for plasmid purification and Sanger sequencing.
- Analysis: Use software (e.g., Quantification Tool for Methylation Analysis) to compare sequence reads to the original genomic sequence. Calculate % methylation at each CpG site.
Visualizations
Diagram 1: CGI Methylation Leads to Gene Silencing
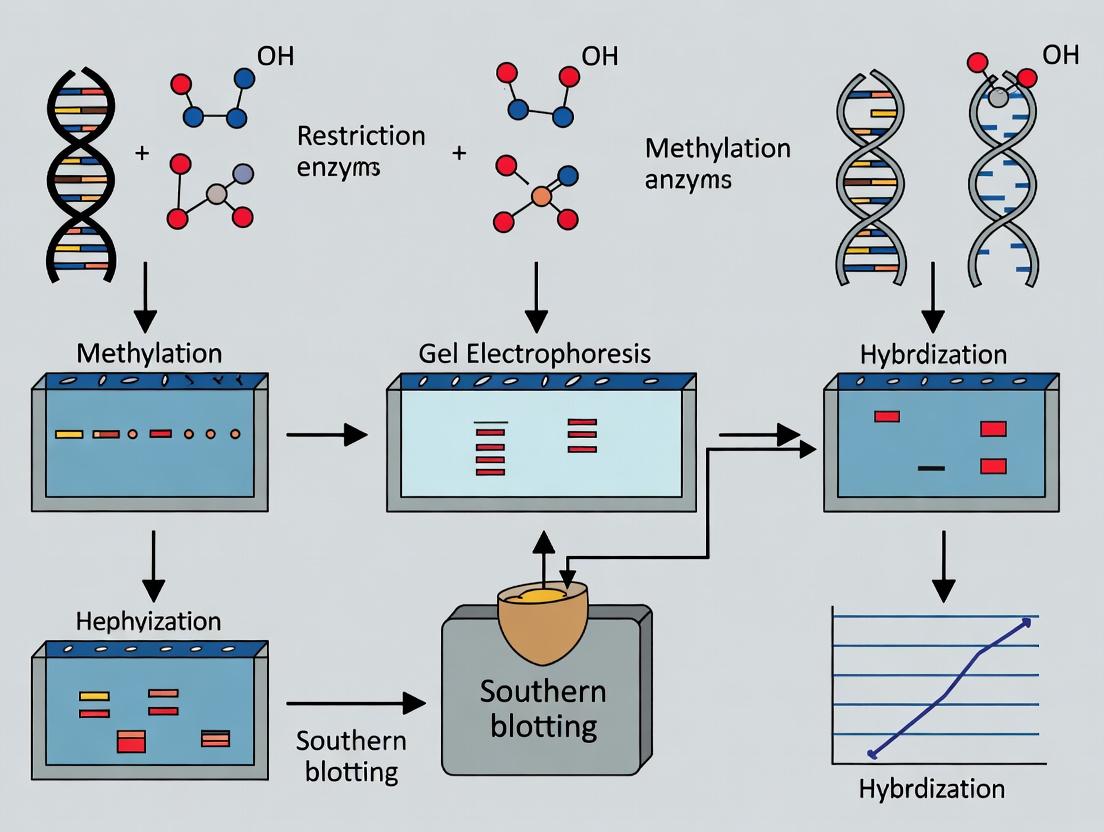
Diagram 2: Southern Blot Methylation Analysis Workflow
Within the context of DNA methylation analysis, Southern blotting remains a foundational technique for assessing specific genomic DNA sequences and their methylation status. While newer methods exist, Southern blotting provides direct, quantitative, and sequence-specific information, often serving as a gold standard for validating results from high-throughput but indirect assays. Its application is critical in epigenetic research, genotyping, transgenic organism analysis, and in drug development for diseases linked to aberrant methylation, such as cancer and neurological disorders.
Principles and Applications in Methylation Analysis
The power of Southern blotting for methylation studies hinges on the use of methylation-sensitive restriction enzymes (MSREs). These enzymes cleave DNA only at unmethylated recognition sites, allowing researchers to infer methylation status based on fragment size patterns after hybridization.
Key Restriction Enzymes for Methylation Analysis
Table 1: Common Methylation-Sensitive Restriction Enzymes
| Enzyme | Recognition Site | Cuts When Site Is... | Typical Application in Southern Blotting |
|---|---|---|---|
| HpaII | CCGG | Unmethylated (on internal C) | Detects methylation at CpG islands. Often used with its methylation-insensitive isoschizomer MspI for comparison. |
| SmaI | CCCGGG | Unmethylated | Analysis of methylation in GC-rich regions. |
| BstUI | CGCG | Unmethylated | Useful for examining methylation in non-CpG contexts (e.g., CHH methylation in plants). |
| EcoRII | CCWGG | Unmethylated | Broader recognition for methylation screening. |
| NotI | GCGGCCGC | Unmethylated | Analysis of large, GC-rich regions and genomic imprinting. |
Table 2: Fragment Pattern Interpretation
| Digestion Scenario | Expected Southern Blot Result | Methylation Inference |
|---|---|---|
| MspI & HpaII both produce small fragments | Multiple bands < 1kb | Target CCGG sites are unmethylated. |
| MspI produces small fragments; HpaII produces one large fragment | Single band > 5kb | Target CCGG sites are fully methylated. |
| MspI produces small fragments; HpaII produces a mix of large and small fragments | Multiple bands of varying sizes | Partial or heterogeneous methylation across the cell population. |
Detailed Protocols
Protocol 1: Genomic DNA Digestion for Methylation Analysis
Objective: To cleave genomic DNA with methylation-sensitive and -insensitive enzymes to generate distinct fragment patterns.
Materials:
- High-molecular-weight genomic DNA (>50 kb).
- Methylation-sensitive restriction enzyme (e.g., HpaII) and its isoschizomer (e.g., MspI).
- Appropriate 10x reaction buffer.
- Nuclease-free water.
- Thermocycler or water bath.
Procedure:
- Set up two parallel digestion reactions for each DNA sample:
- Reaction A (Methylation-Insensitive Control): 5 µg genomic DNA, 10 U MspI, 1x buffer, in 50 µL total volume.
- Reaction B (Methylation-Sensitive Test): 5 µg genomic DNA, 10 U HpaII, 1x buffer, in 50 µL total volume.
- Mix gently and centrifuge briefly.
- Incubate at 37°C for 16-24 hours (overnight) to ensure complete digestion.
- Inactivate enzymes by heating to 65°C for 20 minutes or as per enzyme specifications.
- Proceed to electrophoresis or store at -20°C.
Protocol 2: Gel Electrophoresis and Capillary Transfer
Objective: To separate DNA fragments by size and transfer them to a solid membrane.
Materials:
- Agarose (molecular biology grade).
- 1x TAE or TBE electrophoresis buffer.
- Gel casting tray, comb, and electrophoresis tank.
- DNA molecular weight marker (e.g., lambda HindIII digest).
- Depurination solution: 0.25 M HCl.
- Denaturation solution: 0.5 M NaOH, 1.5 M NaCl.
- Neutralization solution: 0.5 M Tris-HCl, 1.5 M NaCl, pH 7.5.
- Transfer buffer: 20x SSC (3 M NaCl, 0.3 M sodium citrate, pH 7.0).
- Nylon membrane (positively charged).
| Reagent | Function |
|---|---|
| HpaII/MspI Enzymes | Isoschizomer pair for comparative methylation analysis at CCGG sites. |
| Positively Charged Nylon Membrane | Binds negatively charged, denatured DNA fragments covalently via UV crosslinking. |
| [32P]-dCTP or Digoxigenin (DIG)-dUTP | Label for probe synthesis; provides high sensitivity for detection. |
| Denaturation Solution (NaOH/NaCl) | Converts double-stranded DNA to single strands for efficient hybridization. |
| 20x SSC Buffer | High-salt transfer buffer promotes DNA binding to the membrane during capillary action. |
| Formamide | Hybridization buffer component; lowers the melting temperature, allowing specific hybridization at lower temps. |
| Salmon Sperm DNA | Blocking agent to reduce non-specific binding of the probe to the membrane. |
- Whatman 3MM paper, paper towels, glass plate, weight.
- UV transilluminator for crosslinking.
Procedure:
- Cast a 0.8-1.0% agarose gel in 1x TAE buffer.
- Load digested DNA samples and a DNA ladder.
- Run gel at 1-2 V/cm until bromophenol blue dye has migrated adequately (12-16 hours for optimal separation of large fragments).
- Depurination (Optional for >5 kb fragments): Soak gel in 0.25 M HCl for 15 min with gentle agitation to fragment large DNA, improving transfer efficiency.
- Denaturation: Soak gel in denaturation solution for 2 x 15 min.
- Neutralization: Soak gel in neutralization solution for 2 x 15 min.
- Assemble capillary transfer stack (from bottom to top): wick (3MM paper soaked in 20x SSC), gel, membrane, stack of dry 3MM paper, paper towels, weight. Transfer for 12-24 hours.
- Disassemble, rinse membrane in 2x SSC, and UV-crosslink DNA to the membrane.
Protocol 3: Probe Labeling and Hybridization
Objective: To generate a sequence-specific, labeled probe and detect target fragments on the membrane.
Materials:
- DNA template for probe (plasmid, PCR product).
- Random hexamer primers.
- Klenow fragment of DNA polymerase I.
- Labeled nucleotide: [α-32P]dCTP or DIG-dUTP.
- Sephadex G-50 column for purification (if using radioisotope).
- Hybridization bottles and oven, or sealed bags and water bath.
- Pre-hybridization/Hybridization buffer (e.g., QuickHyb or Church buffer).
- Wash solution 1: 2x SSC, 0.1% SDS.
- Wash solution 2: 0.1x SSC, 0.1% SDS.
- Detection system: X-ray film/phosphorimager (radioactive) or chemiluminescence imager (DIG).
Procedure:
- Probe Labeling: Use a random primed labeling kit. Mix 25 ng DNA template, random hexamers, dNTPs (including labeled dCTP/dUTP), and Klenow enzyme. Incubate at 37°C for 30 min. Purify labeled probe from unincorporated nucleotides.
- Pre-hybridization: Place membrane in hybridization tube/bag. Add pre-warmed hybridization buffer (5-10 mL). Incubate at 65°C for 30-60 min with rotation/agitation.
- Hybridization: Denature the purified probe by boiling for 5 min, then chill on ice. Add to fresh, pre-warmed hybridization buffer. Pour off pre-hybridization buffer, add probe/buffer mix. Hybridize at 65°C overnight.
- Washing: Perform sequential washes:
- Wash 1: 2x SSC/0.1% SDS at room temperature for 15 min.
- Wash 2: 2x SSC/0.1% SDS at 65°C for 15 min.
- Wash 3: 0.1x SSC/0.1% SDS at 65°C for 15-30 min (stringency wash).
- Detection: For radioactive probes, expose membrane to X-ray film or phosphorimager screen. For DIG probes, block membrane, incubate with anti-DIG-AP conjugate, wash, incubate with chemiluminescent substrate, and image.
Visualizations
Diagram 1 Title: Southern Blot Workflow for Methylation Analysis
Diagram 2 Title: Methylation-Sensitive Restriction Enzyme Logic
Application Notes
Within the context of a thesis focused on DNA methylation analysis via Southern blotting, methylation-sensitive and methylation-dependent restriction enzymes (MSREs/MDREs) are foundational tools for assessing epigenetic status at specific genomic loci. These enzymes enable the mapping of CpG methylation patterns, crucial for research in gene silencing, genomic imprinting, carcinogenesis, and pharmaceutical development of epigenetic therapies.
HpaII and MspI are the canonical isoschizomer pair. Both recognize the sequence CCGG. HpaII is methylation-sensitive; it cannot cut if the internal cytosine is methylated (C^mCGG). MspI cuts regardless of this internal cytosine methylation but is inhibited by methylation of the outer cytosine. This differential activity allows researchers to discriminate between methylation states.
NotI (recognition site: GCGGCCGC) is often used as a methylation-sensitive enzyme for probing CpG islands, especially in genomic Southern blotting, as its site is frequently found in unmethylated, transcriptionally permissive regions.
Current research trends, confirmed via recent sources, emphasize their use in combination with Southern blotting for validating genome-wide methylation data from techniques like bisulfite sequencing or arrays, providing a gold standard for locus-specific methylation quantification.
Table 1: Key Characteristics of Featured Restriction Enzymes
| Enzyme | Recognition Sequence | Methylation Sensitivity | Primary Application in Methylation Analysis |
|---|---|---|---|
| HpaII | 5'-C↓CGG-3' | Inhibited by 5-mC at internal C (C^mCGG) | Maps methylation at CCGG sites. Uncut band = methylated. |
| MspI | 5'-C↓CGG-3' | Cuts C^mCGG; Inhibited by 5-mC at outer C (^mCCGG) | Control for presence of CCGG site; identifies hemi-methylation contexts. |
| NotI | 5'-GC↓GGCCGC-3' | Inhibited by CpG methylation within its site | Assays methylation status of CpG-rich promoter regions. |
Table 2: Expected Southern Blot Fragment Outcomes Based on Methylation State
| Genomic DNA State at CCGG Site | HpaII Digest | MspI Digest | Interpretation from Southern Blot |
|---|---|---|---|
| Unmethylated | Cut | Cut | Shorter fragment(s) detected |
| Fully Methylated (Internal C) | Uncut | Cut | Longer fragment detected (HpaII) |
| Hemi-methylated (One strand) | Uncut | Cut | Longer fragment detected (HpaII) |
| Outer C Methylated (^mCCGG) | Cut | Uncut | Longer fragment detected (MspI) |
Experimental Protocols
Protocol 1: Southern Blot Analysis of DNA Methylation Using HpaII/MspI
Objective: To determine the methylation status of specific gene loci containing CCGG sites.
I. Genomic DNA Digestion
- Prepare DNA: Isolate high-molecular-weight genomic DNA (≥50 µg/mL) from target cells/tissue using a phenol-chloroform method.
- Set Up Digests: For each sample, set up three parallel 50 µL digestion reactions:
- Reaction A (HpaII): 5 µg DNA, 20 units HpaII, 1X recommended buffer.
- Reaction B (MspI): 5 µg DNA, 20 units MspI, 1X recommended buffer.
- Reaction C (Control/Uncut): 5 µg DNA, no enzyme, 1X buffer.
- Incubate: Digest at 37°C for 16-24 hours (overnight) to ensure complete digestion.
- Precipitate DNA: Add 1/10 volume 3M sodium acetate (pH 5.2) and 2.5 volumes 100% ethanol. Pellet DNA, wash with 70% ethanol, and resuspend in 20 µL TE buffer.
II. Gel Electrophoresis & Southern Transfer
- Load Gel: Load entire digested samples onto a 0.8% agarose gel. Include a DNA molecular weight ladder.
- Electrophorese: Run at 25-30V overnight for optimal separation of large fragments.
- Depurinate & Denature: Soak gel in 0.25M HCl for 15 min (optional, for >10 kb DNA), then in denaturation solution (1.5M NaCl, 0.5M NaOH) for 30 min.
- Neutralize & Transfer: Soak gel in neutralization buffer (1.5M NaCl, 0.5M Tris-HCl, pH 7.4) for 30 min. Transfer DNA to a positively charged nylon membrane via capillary or vacuum blotting for 12-24 hours. UV-crosslink DNA to membrane.
III. Probe Labeling & Hybridization
- Label Probe: Prepare a locus-specific probe (200-500 bp) using a random primed DNA labeling kit with [α-³²P]dCTP or a non-radioactive digoxigenin (DIG) system.
- Pre-hybridize: Place membrane in hybridization tube with 10 mL Church buffer (1% BSA, 1mM EDTA, 0.5M phosphate buffer pH 7.2, 7% SDS). Pre-hybridize at 65°C for 1 hour.
- Hybridize: Add denatured probe directly to the buffer. Hybridize at 65°C for 16-24 hours.
- Wash: Perform stringent washes: 2X SSC/0.1% SDS at room temperature (5 min), then 0.2X SSC/0.1% SDS at 65°C (2 x 15 min).
IV. Detection
- For radioactive probes, expose membrane to a phosphorimager screen or X-ray film at -80°C.
- For DIG-labeled probes, perform immunodetection with anti-DIG-AP conjugate and chemiluminescent substrate, followed by exposure to X-ray film.
Diagram: Workflow for Methylation Analysis by Southern Blotting
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for MSRE Southern Blotting
| Item | Function & Rationale |
|---|---|
| High-Quality Genomic DNA | Intact, high molecular weight DNA is critical for restriction analysis and clear Southern blot bands. Isolated via phenol-chloroform or column-based kits. |
| Methylation-Sensitive Enzymes (HpaII, NotI) | Primary tools for detecting cytosine methylation at their specific recognition sequences. |
| Control Enzyme (MspI) | Provides the cleavage pattern for all CCGG sites regardless of internal cytosine methylation, confirming site presence and serving as a digestion control. |
| Methylation-Insensitive Rare-Cutter (e.g., EcoRI) | Often used in double-digests to generate a manageable fragment size range encompassing the region of interest. |
| Positively Charged Nylon Membrane | For efficient binding and retention of negatively charged, denatured DNA after Southern transfer. |
| Locus-Specific DNA Probe | Radioactively or non-radioactively labeled DNA fragment complementary to the target sequence, enabling specific detection. |
| Church Hybridization Buffer | Phosphate-based buffer allowing high-specificity hybridization at elevated temperatures with low background. |
| Stringent Wash Buffers (SSC/SDS) | Remove non-specifically bound probe post-hybridization, crucial for signal specificity. |
| Phosphorimager System or X-Ray Film | For detection and visualization of hybridized signals from the membrane. |
Introduction The Southern blot, developed by Edwin M. Southern in 1975, is foundational to molecular biology. Beyond its revolutionary role in DNA mapping and fingerprinting, it was the first practical method to provide direct, physical evidence of epigenetic modifications, specifically DNA methylation. This application note details the use of Southern blotting for DNA methylation analysis, a cornerstone technique that established the principle that heritable changes in gene function occur without alteration of the DNA sequence itself.
Thesis Context This protocol is framed within the thesis that Southern blotting provided the first genome-specific, locus-resolution evidence for DNA methylation patterns, directly linking methylation status to gene silencing (e.g., X-chromosome inactivation, genomic imprinting). While newer techniques offer higher throughput, Southern analysis remains a gold standard for validating methylation status at specific loci due to its direct, hybridization-based detection and lack of bisulfite conversion artifacts.
Application Notes: Analyzing Methylation-Sensitive Restriction Patterns
Core Principle: Methylation-sensitive and methylation-dependent restriction enzymes (MSREs/MDREs) are used to digest genomic DNA. Differences in fragment patterns on a Southern blot reveal the methylation status at specific CpG sites within the probed locus.
Key Data from Seminal Studies (Summarized)
Table 1: Landmark Epigenetic Discoveries Enabled by Southern Blotting
| Biological Process | Gene/Locus | Key Methylation-Sensitive Enzyme(s) | Observed Southern Blot Result | Epigenetic Conclusion |
|---|---|---|---|---|
| X-chromosome Inactivation | PGK1, HPRT | HpaII (sensitive), MspI (insensitive) | Different fragment patterns from active vs. inactive X | Inactive X is hypermethylated at promoter CpG islands |
| Genomic Imprinting | Igf2/H19 ICR | HpaII, SacII | Allele-specific digestion patterns | Differential methylation established parent-of-origin expression |
| Cancer & Tumor Suppressors | RB1, BRCA1 | HpaII, EagI | Aberrant fragment sizes/loss in tumors | De novo promoter hypermethylation silences tumor suppressor genes |
Protocol: Southern Blot Analysis of CpG Methylation at a Specific Locus
I. DNA Digestion with Methylation-Sensitive Enzymes
- Prepare DNA Samples: Isolate high-molecular-weight genomic DNA (≥50 µg) from target tissues/cells using phenol-chloroform extraction.
- Set Up Restriction Digests: For each sample, set up two parallel digestions:
- Reaction A (Methylation-Sensitive): 10 µg DNA, 20-50 U HpaII (cuts CCGG only if internal C is unmethylated), appropriate buffer.
- Reaction B (Methylation-Insensitive Control): 10 µg DNA, 20-50 U MspI (cuts CCGG regardless of methylation), appropriate buffer.
- Include a third digestion with a rare-cutter (e.g., EcoRI) for overall genomic mapping if needed.
- Incubate: Digest at 37°C for 12-16 hours to ensure complete digestion.
- Purify & Quantify: Purify digested DNA and measure concentration.
II. Gel Electrophoresis & Blotting
- Load Gel: Load equal masses (2-5 µg) of digested DNA from Reactions A and B adjacent on a large 0.8-1.0% agarose gel. Include a molecular weight ladder.
- Run Gel: Electrophorese at low voltage (1-2 V/cm) for 12-16 hours for optimal separation of large fragments.
- Depurinate & Denature: Soak gel in 0.25 M HCl (15 min), then in denaturation solution (0.5 M NaOH, 1.5 M NaCl; 2 x 15 min).
- Neutralize: Soak gel in neutralization buffer (0.5 M Tris-HCl pH 7.5, 1.5 M NaCl; 2 x 15 min).
- Capillary Transfer: Set up a capillary transfer (Southern, 1975) using a neutral nylon membrane (positively charged) and 20X SSC buffer, transferring for 18-24 hours.
- Crosslink: UV crosslink DNA to the membrane.
III. Hybridization & Detection
- Probe Preparation: Generate a labeled probe (radiolabeled ³²P-dCTP or digoxigenin) complementary to the genomic region of interest, avoiding CCGG sites within the probe sequence itself.
- Pre-hybridize: Incubate membrane in hybridization buffer (e.g., QuickHyb solution) at 65°C for 20 min.
- Hybridize: Add denatured probe to fresh buffer. Hybridize at 65°C for 2-4 hours.
- Washes: Perform stringent washes: 2X SSC/0.1% SDS at room temperature, then 0.1X SSC/0.1% SDS at 65°C for 30 min each.
- Detection: For radiolabeled probes, expose to a phosphorimager screen or X-ray film. For digoxigenin, use chemiluminescent detection with anti-DIG-AP and CSPD substrate.
IV. Data Interpretation Compare fragment sizes between HpaII and MspI digests.
- Same pattern: Target CCGG sites are unmethylated.
- Larger/simpler HpaII pattern: Target CCGG sites are methylated, preventing HpaII cleavage.
Southern Blot Methylation Analysis Workflow
Methylation Status Dictates Southern Blot Pattern
The Scientist's Toolkit: Essential Reagents for Southern-Based Methylation Analysis
Table 2: Key Research Reagent Solutions
| Reagent/Material | Function & Critical Notes |
|---|---|
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII, SacII) | Core tool. Cuts only at unmethylated recognition sequences to reveal methylation status. |
| Methylation-Insensitive Isoschizomers (e.g., MspI for HpaII) | Essential control. Cuts regardless of methylation, confirming sequence presence and digestion efficiency. |
| Positively Charged Nylon Membrane | Binds negatively charged, denatured DNA fragments after transfer. Critical for probe hybridization. |
| High-Specific-Activity ³²P-dCTP or Digoxigenin (DIG)-dUTP | Label for locus-specific probe. ³²P offers high sensitivity; DIG is safer and stable. |
| Stringent Wash Buffers (e.g., 0.1X SSC/0.1% SDS) | Removes non-specifically bound probe, ensuring signal specificity to the target locus. |
| Phosphorimager Screen & Scanner or X-Ray Film | Detection system for radiolabeled probes. Phosphorimager offers quantitative, wider dynamic range. |
| Chemiluminescent AP Substrate (e.g., CSPD for DIG probes) | Non-radioactive detection. Requires anti-DIG-alkaline phosphatase conjugate antibody. |
Application Notes
Within the broader thesis investigating DNA methylation landscapes via Southern blotting, three principal applications demonstrate the technique's enduring value in resolving locus-specific epigenetic states. Southern blotting, through its combination of restriction enzyme digestion and methylation-sensitive probes, provides a robust, quantitative snapshot of allele-specific methylation that is less susceptible to PCR bias.
1. Genomic Imprinting Analysis Genomic imprinting involves parent-of-origin-specific monoallelic gene expression governed by differential methylation at imprinting control regions (ICRs). Southern blotting is critical for diagnosing imprinting disorders (e.g., Prader-Willi/Angelman syndromes, PWS/AS) and for validating epigenetic models in development. Analysis typically targets differentially methylated regions (DMRs) like SNRPN (PWS/AS locus) or H19/IGF2 DMR (Beckwith-Wiedemann syndrome). Digestion with a methylation-sensitive enzyme (e.g., HpaII) alongside its methylation-insensitive isoschizomer (MspI) reveals parental allele-specific patterns.
2. X-Chromosome Inactivation (XCI) Skewing XCI equalizes gene dosage in females by silencing one X chromosome, forming the inactive X (Xi). The extent of skewing, where one X is inactivated in >75% of cells, is clinically relevant in X-linked disorders and autoimmunity. Southern blotting assays methylation at loci like the human ANDrogen Receptor (AR) or FMRI genes, using tri-nucleotide repeat polymorphisms to distinguish alleles. The ratio of digested (active X) to undigested (inactive X) alleles quantifies skewing.
3. Repeat Element Methylation Global hypomethylation of repetitive elements (LINE-1, Alu, satellite repeats) is a hallmark of cancer genomes and genomic instability. Southern blotting provides a reproducible measure of bulk repeat methylation. A consensus sequence probe for LINE-1, combined with a methylation-sensitive enzyme that cuts frequently within the repeat, yields a smear on a gel; increased digestion (hypomethylation) shifts the smear to lower molecular weights.
Quantitative Data Summary
Table 1: Key Loci and Enzymes for Methylation Analysis via Southern Blotting
| Application | Target Locus | Key Restriction Enzymes | Typical Sample Input | Expected Outcome Measure |
|---|---|---|---|---|
| Imprinting | SNRPN DMR (PWS/AS) | HpaII (sensitive) / MspI (insensitive) | 5-10 µg genomic DNA | Parental allele-specific banding pattern; loss of methylated allele in PWS. |
| XCI Skewing | Human AR (CAG repeat) | HpaII + HindIII | 5 µg genomic DNA | Skewing ratio: (% digested allele A / % digested allele B). Skewing >75:25 is significant. |
| Repeat Elements | LINE-1 (consensus sequence) | HpaII or NotI | 5-10 µg genomic DNA | % Methylation = (Intensity of high MW smear / Total intensity) x 100. Cancer samples show 10-30% reduction. |
Table 2: Advantages of Southern Blotting for These Applications
| Feature | Imprinting/XCI | Repeat Elements | Advantage over NGS-based Methods |
|---|---|---|---|
| Allele Specificity | Directly visualizes parental alleles. | Measures bulk, not single-copy, status. | Avoids PCR amplification bias in bisulfite conversion. |
| Quantification | Semi-quantitative band intensity. | Quantitative via phosphorimager analysis. | Provides a physical map of methylation sites. |
| Probe Specificity | High for unique sequences. | High for repeat consensus. | Can distinguish highly homologous sequences. |
Detailed Experimental Protocols
Protocol 1: Imprinting Analysis at the SNRPN Locus Objective: To determine methylation status at the SNRPN CpG island DMR.
- DNA Digestion: Set up two parallel digestions for each sample.
- Reaction A: 5 µg DNA + HpaII (10 U/µg DNA) + appropriate buffer.
- Reaction B (Control): 5 µg DNA + MspI (10 U/µg DNA) + buffer.
- Incubate at 37°C for 16 hours. Heat-inactivate enzymes.
- Gel Electrophoresis: Load digested DNA on a 0.8% agarose gel. Run at 35V for 16 hours alongside a molecular weight marker. Depurinate, denature, and neutralize the gel.
- Southern Transfer: Perform capillary transfer to a positively charged nylon membrane using 20x SSC buffer for 16-24 hours.
- Probe Labeling & Hybridization: Label a PCR-amplified SNRPN-specific probe (e.g., exon α) with [α-³²P] dCTP using random priming. Hybridize to membrane at 65°C in Church buffer for 16 hours.
- Washing & Detection: Wash stringently (e.g., 0.1x SSC, 0.1% SDS at 65°C). Expose to a phosphor storage screen for 1-5 days. Analyze band patterns: a methylated (HpaII-resistant) band ~4.2 kb and an unmethylated (HpaII-digested) band ~0.9 kb.
Protocol 2: XCI Skewing Analysis Using the AR Locus Objective: To calculate the ratio of active X chromosomes from two alleles.
- Double Digestion: Digest 5 µg DNA with HindIII (to fragment DNA) and HpaII (methylation-sensitive) simultaneously. Include a HindIII-only control for each sample to assess total allele input.
- Gel & Blot: Separate on a 0.8% agarose gel. Transfer as in Protocol 1.
- Hybridization: Hybridize with a radiolabeled (CAG)n repeat probe. The HindIII fragment length is polymorphic, differentiating alleles.
- Quantification: Using a phosphorimager, quantify band intensities.
- For each allele (A & B): Intensity in HpaII+HindIII digest (active, unmethylated X).
- For each allele: Intensity in HindIII-only digest (total allele).
- Calculate: % Active X for Allele A = (Intensity A in HpaII digest / Intensity A in Hind digest) x 100.
- Skewing Ratio = % Active Allele A : % Active Allele B.
Protocol 3: LINE-1 Global Methylation Analysis Objective: To assess bulk LINE-1 CpG methylation.
- Digestion: Digest 10 µg DNA with HpaII (or NotI for a CpG-rich site). Include an uncut control and a digestion with a methylation-insensitive frequent cutter (e.g., MseI) as a DNA quality control.
- Gel & Blot: Run on a 1.2% agarose gel to resolve small fragments. Transfer.
- Hybridization: Hybridize with a ³²P-labeled LINE-1 consensus sequence probe (e.g., from the 5' UTR).
- Analysis: The probe hybridizes to a heterogeneous population of fragments. A hypomethylated sample shows a strong, low molecular weight smear (<1.0 kb). A methylated sample shows a higher molecular weight smear. Quantify total signal in defined size ranges (e.g., >4 kb vs. <1 kb) to calculate a methylation index.
Visualizations
Title: Imprinting Analysis Southern Blot Workflow
Title: X-Chromosome Inactivation Skewing Assay
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Southern Blot Methylation Analysis
| Reagent/Material | Function/Description | Key Consideration |
|---|---|---|
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII, NotI) | Cuts only at unmethylated CpG sites within its recognition sequence. | Paired with insensitive isoschizomer (MspI) for control. Stability and star activity must be monitored. |
| Positively Charged Nylon Membrane | Binds negatively charged, denatured DNA fragments post-transfer. | Critical for probe retention during high-stringency washes. |
| [α-³²P] dCTP or Chemiluminescent Labeling Kit | Provides high-sensitivity detection of hybridized probe. | Radiolabeling offers superior quantitation; chemiluminescence is safer. |
| Specific Hybridization Probes | PCR-amplified or cloned DNA fragments complementary to target locus (e.g., SNRPN, AR, LINE-1 consensus). | Must be verified for specificity and lack of repetitive elements (except for repeat analysis). |
| Phosphor Storage Screen & Imager | Detects and quantifies radiolabel or chemiluminescent signal from the blot. | Essential for quantitative comparison of band intensities. |
| High-Purity Genomic DNA | Starting material. Must be largely intact and free of contaminants. | Degraded DNA leads to smearing. Phenol-chloroform extraction is often used. |
| Stringency Wash Buffers (e.g., SSC/SDS) | Removes non-specifically bound probe after hybridization. | Concentration and temperature determine specificity. |
Mastering the Technique: A Step-by-Step Protocol for Methylation-Specific Southern Blotting
Within the broader thesis on DNA methylation analysis using Southern blotting, the initial step of obtaining high-quality, high-molecular-weight genomic DNA is paramount. The integrity and purity of the isolated DNA directly influence the success of subsequent restriction enzyme digestion, gel electrophoresis, and hybridization. This protocol details optimized methods for genomic DNA isolation, quantification, and quality assessment tailored for methylation-specific Southern blot applications.
Key Research Reagent Solutions
The following table lists essential materials and their functions for successful DNA isolation and quantification.
| Reagent/Material | Function in Protocol |
|---|---|
| Lysis Buffer (w/ Proteinase K & SDS) | Disrupts cellular and nuclear membranes, inactivates nucleases, and digests proteins. |
| RNase A | Degrades RNA to prevent interference with downstream quantification and analysis. |
| Phenol:Chloroform:Isoamyl Alcohol | Organic extraction removes proteins, lipids, and other cellular debris from the DNA solution. |
| Isopropanol/Ethanol | Precipitates high-molecular-weight DNA from the aqueous phase. |
| TE Buffer (pH 8.0) | Stabilizes isolated DNA; EDTA chelates Mg2+ to inhibit DNase activity. |
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII, NotI) | Key tools for methylation analysis; their cutting is blocked by CpG methylation. |
| Fluorometric DNA Binding Dye (e.g., Qubit dsDNA HS Assay) | Provides highly specific quantitation of double-stranded DNA, unaffected by RNA. |
| Nanodrop Spectrophotometer | Provides rapid A260/A280 and A260/A230 ratios for assessing DNA purity. |
| Pulsed-Field Gel Electrophoresis (PFGE) Grade Agarose | Allows resolution of very large DNA fragments post-restriction digestion. |
Detailed Protocol: Genomic DNA Isolation (Organic Extraction)
Principle: This method uses gentle lysis to preserve DNA length, followed by organic purification to remove contaminants that can inhibit restriction enzymes.
Materials & Setup
- Lysis Buffer: 10 mM Tris-Cl (pH 8.0), 100 mM EDTA (pH 8.0), 0.5% SDS.
- Proteinase K (20 mg/mL stock).
- RNase A (10 mg/mL, heat-inactivated).
- Phenol:Chloroform:Isoamyl Alcohol (25:24:1).
- Chloroform.
- Isopropanol and 70% Ethanol (ice-cold).
- TE Buffer: 10 mM Tris-Cl, 1 mM EDTA, pH 8.0.
Step-by-Step Procedure
- Cell Lysis: Suspend 1-5 x 10^6 cells or 25 mg of tissue in 500 µL of Lysis Buffer. Add 5 µL of RNase A and 25 µL of Proteinase K. Mix by inversion.
- Incubation: Incubate at 56°C for 3 hours (or overnight for tissues) with gentle agitation.
- Organic Extraction: Cool sample to room temp. Add an equal volume of Phenol:Chloroform:Isoamyl Alcohol. Mix gently by inversion for 10 minutes. Centrifuge at 12,000 x g for 10 minutes at 4°C.
- Aqueous Phase Recovery: Transfer the upper aqueous phase to a new tube. Add an equal volume of chloroform. Mix gently and centrifuge as in step 3.
- DNA Precipitation: Transfer the aqueous phase again. Add 0.7 volumes of room-temperature isopropanol. Mix gently until the DNA thread is visible. Pellet DNA by centrifugation at 12,000 x g for 10 minutes at 4°C.
- Wash: Wash the pellet with 1 mL of ice-cold 70% ethanol. Centrifuge at 12,000 x g for 5 minutes. Carefully decant ethanol.
- Resuspension: Air-dry the pellet for 10-15 minutes. Dissolve DNA in 50-100 µL of TE Buffer by gentle pipetting. Incubate at 4°C overnight for complete dissolution.
DNA Quantification and Quality Assessment
Accurate quantification is critical for normalizing subsequent restriction digests.
Spectrophotometric Analysis (Purity Check)
Use a 1-2 µL aliquot.
| Parameter | Ideal Value | Indication of Problem |
|---|---|---|
| A260/A280 Ratio | 1.8 - 2.0 | Ratio <1.8 suggests protein/phenol contamination. |
| A260/A230 Ratio | 2.0 - 2.2 | Ratio <2.0 suggests guanidine, phenol, or carbohydrate carryover. |
| Concentration (via A260) | N/A | Can be overestimated due to RNA or contaminants. |
Fluorometric Quantification (Accurate Concentration)
Utilizes dsDNA-specific dyes (e.g., Qubit, PicoGreen).
- Prepare standards and working dye solution as per manufacturer.
- Add 1-10 µL of DNA sample to 190-199 µL of working dye. Mix thoroughly.
- Incubate for 2-5 minutes protected from light.
- Read fluorescence. Calculate concentration from the standard curve.
Integrity Assessment by Gel Electrophoresis
Cast a 0.8% agarose gel in 1x TAE.
- Mix 100-200 ng of DNA with loading dye.
- Load alongside a high-molecular-weight DNA ladder (e.g., λ HindIII).
- Run at 5 V/cm for 45-60 minutes.
- Visualize under UV. Intact genomic DNA should appear as a single, tight, high-molecular-weight band with minimal smearing toward the lower sizes.
Data Presentation: Quantification Method Comparison
The following table summarizes key metrics for the primary DNA quantification methods relevant to Southern blotting.
| Method | Principle | Sample Volume | Concentration Range | Speed | Key Advantage | Key Disadvantage for Southern Blot |
|---|---|---|---|---|---|---|
| NanoDrop UV-Vis | Absorbance at 260 nm | 1-2 µL | 2 ng/µL - 15,000 ng/µL | < 1 min | Rapid purity check (A260/280) | Overestimates if RNA/contaminants present; poor sensitivity. |
| Qubit Fluorometry | Fluorescence of dsDNA-binding dye | 1-20 µL | 0.2 ng/µL - 1000 ng/µL (HS Assay) | ~2-3 min | Highly specific to dsDNA; accurate for low conc. | Does not assess purity or integrity. |
| Agarose Gel | Ethidium bromide intercalation | Varies (≥ 20 ng) | Qualitative | 60-90 min | Assesses integrity and size. | Not quantitative; low sensitivity. |
Title: Genomic DNA Isolation and QC Workflow
Title: DNA Quantification and QC Methods
Critical Considerations for Methylation Analysis
- Minimize Shearing: Avoid vortexing, pipetting vigorously, or using narrow-bore tips after lysis to preserve high molecular weight.
- Purity is Crucial: Residual salts, organics, or ethanol can inhibit subsequent methylation-sensitive restriction enzymes, leading to false-positive methylation signals.
- Quantification Normalization: Use fluorometric values for normalizing restriction digest amounts. Equal mass of DNA is critical for comparative Southern blotting.
- Control DNA: Always include a known unmethylated (e.g., peripheral blood) and methylated control DNA in parallel isolations to validate the entire downstream Southern process.
Within the context of a thesis on DNA methylation analysis via Southern blotting, the selection between single and double restriction enzyme digests is a critical strategic decision. This step determines the resolution and specificity with which methylated alleles can be distinguished from their unmethylated counterparts. Single digests, often using methylation-sensitive enzymes (e.g., HpaII), are employed to assess methylation status at specific loci by comparing fragment patterns to a control digest with its methylation-insensitive isoschizomer (e.g., MspI). Double digests, combining a methylation-sensitive enzyme with a frequent-cutter or a second rare-cutter, are used to generate defined, locus-specific fragments suitable for probing, thereby reducing background and improving interpretability in complex genomic DNA.
Quantitative Comparison: Single vs. Double Digest
Table 1: Strategic Comparison of Digest Types for Methylation Analysis
| Parameter | Single Digest | Double Digest |
|---|---|---|
| Primary Purpose | Global methylation screening; comparison of isoschizomer patterns. | Fine mapping; generation of specific, defined fragments for probing. |
| Typical Enzymes Used | HpaII (sensitive), MspI (insensitive), EcoRI, HindIII. | HpaII + EcoRI; NotI + EagI; BstUI + PstI. |
| DNA Amount Required | 5-10 µg per reaction. | 10-20 µg (due to sequential or simultaneous digestion). |
| Incubation Time | 3-16 hours (overnight common). | 3-16 hours per enzyme; can be simultaneous if buffers are compatible. |
| Key Advantage | Simplicity; direct comparison reveals methylation as presence/absence of cut. | Higher specificity; reduces smear, yields precise fragment for probe hybridization. |
| Key Disadvantage | Can produce large or ambiguous fragments; higher background. | Requires buffer compatibility; more complex optimization. |
| Optimal for Southern | Yes, for initial assessment. | Yes, preferred for precise, publication-quality blots. |
Table 2: Common Methylation-Sensitive Restriction Enzymes (MSREs)
| Enzyme | Recognition Site | Methylation Sensitivity | Common Isoschizomer |
|---|---|---|---|
| HpaII | CCGG | Sensitive to hemi- or full methylation at internal C. | MspI (insensitive) |
| SmaI | CCCGGG | Sensitive to methylation at any C. | XmaI (insensitive) |
| BstUI | CGCG | Sensitive to methylation at either C. | None |
| NotI | GCGGCCGC | Sensitive to methylation. | EagI (often similar sensitivity) |
Detailed Experimental Protocols
Protocol 1: Standard Single Digest with a Methylation-Sensitive Enzyme
Objective: To digest genomic DNA for initial methylation screening. Materials: Genomic DNA (5-10 µg), methylation-sensitive restriction enzyme (e.g., HpaII), appropriate 10x reaction buffer, nuclease-free water. Procedure:
- In a sterile microcentrifuge tube, assemble the following on ice:
- Genomic DNA: 5 µg (in ≤ 20 µL volume)
- 10x Reaction Buffer: 5 µL
- Methylation-Sensitive Restriction Enzyme (e.g., HpaII): 20-30 units
- Nuclease-free water to a final volume of 50 µL.
- Mix gently by pipetting. Centrifuge briefly.
- Incubate at the enzyme's optimal temperature (37°C for HpaII) for a minimum of 6 hours, preferably overnight (12-16 hours).
- Inactivate the enzyme by heating at 65°C for 20 minutes or as per manufacturer's instructions.
- Proceed to gel electrophoresis for Southern blotting.
Protocol 2: Sequential Double Digest for Locus-Specific Analysis
Objective: To perform two-enzyme digestion to generate a precise fragment for Southern probing. Materials: Genomic DNA (10-20 µg), two restriction enzymes, compatible 10x reaction buffer or two separate buffers, nuclease-free water. Procedure:
- Check Buffer Compatibility: Consult the manufacturer's chart for a common buffer allowing >50% activity for both enzymes. If none exists, perform sequential digests.
- First Digest:
- Assemble reaction with first enzyme (e.g., EcoRI, a rare-cutter) in its optimal buffer. Use 1 µg DNA per 5-10 units of enzyme.
- Incubate at optimal temperature for 4-6 hours.
- Purify DNA using a standard PCR purification kit or ethanol precipitation. Elute in low-EDTA TE buffer or nuclease-free water.
- Second Digest:
- Use the purified DNA from step 2 as substrate.
- Assemble a new reaction with the second enzyme (e.g., HpaII) in its optimal buffer.
- Incubate at optimal temperature overnight.
- Inactivate the enzyme(s) and purify the DNA if necessary before electrophoresis.
Visualization: Workflow and Decision Pathway
Diagram Title: Decision Workflow for Restriction Digest in Methylation Analysis
Diagram Title: Molecular Principle of Methylation-Sensitive Restriction Digest
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Restriction Digest in Methylation Studies
| Reagent / Solution | Function & Importance in Methylation Analysis |
|---|---|
| High-Molecular-Weight Genomic DNA | Starting material. Integrity is crucial for Southern blotting; sheared DNA produces poor digestion patterns. |
| Methylation-Sensitive Restriction Enzymes (MSREs) | Core reagents (e.g., HpaII, BstUI). Their cleavage is blocked by CpG methylation, enabling differential analysis. |
| Methylation-Insensitive Isoschizomers | Critical controls (e.g., MspI for HpaII). Cut regardless of methylation, confirming the presence of the restriction site. |
| 10x Restriction Enzyme Buffers | Provide optimal ionic strength and pH for enzyme activity. Compatibility is key for double digests. |
| BSA (Bovine Serum Albumin) | Often included in buffers or added separately to stabilize enzymes during long incubations. |
| DNA Purification Kits (Post-Digest) | For cleaning DNA between sequential digests or before gel loading, removing enzymes, salts, and buffers. |
| Molecular Grade Water | Nuclease-free water to prevent degradation of DNA and enzyme during reaction setup. |
This section details the critical transition from restriction digestion to membrane immobilization within a thesis focused on DNA methylation analysis via Southern blotting. Precise execution of these steps is paramount for the accurate transfer and subsequent hybridization of genomic DNA, enabling the assessment of methylation-dependent restriction fragment length polymorphisms (RFLPs).
Agarose Gel Electrophoresis for Genomic DNA
Following restriction enzyme digestion (e.g., with methylation-sensitive enzymes like HpaII or NotI), size separation is achieved through agarose gel electrophoresis.
Protocol:
- Prepare a large (20 cm x 20 cm) 0.8% - 1.0% agarose gel in 1X TAE buffer. For genomic DNA, lower percentage gels improve resolution of large fragments.
- Mix digested DNA samples (10-20 µg per lane) with 6X loading dye. Include a high-molecular-weight DNA ladder (e.g., Lambda HindIII digest).
- Load samples and run the gel at a low voltage (1-2 V/cm) in 1X TAE buffer for 14-16 hours (overnight) to ensure optimal separation of fragments ranging from 1 kb to over 20 kb.
- Stain the gel with ethidium bromide (0.5 µg/mL) or a safer alternative like SYBR Safe for 30-45 minutes with gentle agitation. Destain in deionized water if necessary and visualize under UV light to assess digestion and separation quality. Document the image.
Quantitative Data Summary: Table 1: Agarose Gel Electrophoresis Parameters for Genomic DNA Southern Blotting
| Parameter | Optimal Condition | Purpose/Rationale |
|---|---|---|
| Gel Percentage | 0.8% - 1.0% | Resolves large DNA fragments (1-50+ kb). |
| DNA Load per Lane | 10 - 20 µg | Ensures sufficient signal for detection of low-copy sequences. |
| Voltage Gradient | 1 - 2 V/cm | Prevents smearing and "bouncing" of high-molecular-weight DNA. |
| Run Time | 14 - 18 hours | Ensures complete separation over long distances. |
| Buffer System | 1X TAE | Standard for genomic DNA separation; better resolution for large fragments than TBE. |
Gel Denaturation and Neutralization
Prior to blotting, DNA must be denatured into single strands to facilitate binding to the membrane.
Protocol:
- Following visualization, gently shake the gel in an excess of Denaturation Solution (0.5 M NaOH, 1.5 M NaCl) for 30-45 minutes. This process breaks hydrogen bonds.
- Rinse the gel briefly with deionized water.
- Transfer the gel to an excess of Neutralization Solution (0.5 M Tris-HCl pH 7.5, 1.5 M NaCl) and shake for 30-45 minutes. This brings the pH to a level compatible with subsequent transfer buffers and membrane binding.
Capillary Blotting (Southern Transfer) Setup
The classic upward capillary method reliably transfers DNA from the gel to a solid support.
Protocol:
- While the gel is neutralizing, assemble the transfer stack on a central platform over a large reservoir (e.g., a dish) containing 20X SSC transfer buffer.
- Create a wick from 2-3 sheets of thick filter paper (Whatman 3MM) cut wider and longer than the gel. Wet the wick in 20X SSC and drape it over the platform, ensuring contact with the buffer reservoir on both ends.
- Place the neutralized gel on the wick, avoiding air bubbles.
- Surround the gel with plastic wrap to prevent short-circuiting of the buffer flow.
- Pre-wet the nylon membrane (positively charged for DNA) in deionized water, then equilibrate in 20X SSC for 5 minutes. Place the membrane precisely on top of the gel. Do not move once contact is made.
- Place 2-3 sheets of SSC-wetted filter paper on the membrane, followed by a stack of dry absorbent paper (paper towels or blotting pads) 5-10 cm high.
- Place a glass plate and a weight (~500 g) on top. Allow capillary transfer to proceed for 16-24 hours.
- After transfer, disassemble the stack. Mark the sample lanes and the side of the membrane that was in contact with the gel. Cross-link the DNA to the membrane using UV irradiation (optimal energy ~120 mJ/cm²) or bake at 80°C for 30-60 minutes under vacuum.
Visualization: Capillary Blotting Assembly Workflow
Diagram Title: Capillary Blotting Stack Assembly
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Solutions for Gel Processing and Southern Transfer
| Reagent Solution | Composition | Primary Function |
|---|---|---|
| TAE Buffer (50X Stock) | 2 M Tris base, 1 M Acetic acid, 50 mM EDTA pH 8.0 | Gel running buffer; chelates divalent cations to inhibit nucleases. |
| Denaturation Solution | 0.5 M NaOH, 1.5 M NaCl | Denatures double-stranded DNA into single strands for membrane binding. |
| Neutralization Solution | 0.5 M Tris-HCl (pH 7.5), 1.5 M NaCl | Neutralizes gel pH after denaturation, preparing DNA for transfer in neutral buffer. |
| 20X SSC Transfer Buffer | 3 M NaCl, 0.3 M Sodium Citrate (pH 7.0) | High-salt transfer buffer; promotes efficient binding of DNA to nylon membrane. |
| Positively Charged Nylon Membrane | Nylon matrix with quaternary ammonium groups | Solid support that binds DNA via electrostatic interactions; essential for probe hybridization. |
| High-MW DNA Ladder | Lambda DNA digested with HindIII | Provides size references (kb) for interpreting Southern blot results. |
Within the comprehensive framework of a thesis on DNA methylation analysis using Southern blotting, Step 4 represents a critical juncture determining experimental success. The specificity and sensitivity of methylation-dependent restriction fragment detection are wholly contingent upon meticulous probe design and robust labeling. This protocol details modern strategies to generate high-fidelity probes that differentiate methylated from unmethylated alleles, minimize cross-hybridization, and enable precise quantification, directly supporting downstream applications in epigenetics research and drug development targeting epigenetic modifiers.
Principles of High-Specificity Probe Design for Methylation Analysis
Probes for methylation-specific Southern blotting must satisfy dual criteria: sequence specificity for the target locus and epigenetic specificity to interpret methylation status in the context of restriction digests (e.g., HpaII vs. MspI). Key design parameters are summarized in Table 1.
Table 1: Quantitative Parameters for Optimal Methylation Analysis Probe Design
| Parameter | Optimal Range | Rationale | Impact on Specificity |
|---|---|---|---|
| Probe Length | 200-500 bp | Balances hybridization kinetics (longer) with reduced non-specific binding (shorter). | >300 bp improves signal; <600 bp reduces background. |
| GC Content | 40-60% | Ensures stable hybridization (Tm ~70-85°C). Avoids high GC regions prone to secondary structure. | Outside range lowers Tm, increasing mismatch hybridization risk. |
| Sequence Complexity | Low Repeat Content (<5%) | Minimizes binding to repetitive genomic elements. | High repeat content causes excessive background smear. |
| Tm (Calculated) | 70-85°C | Must be ~5-10°C above final wash stringency temperature. | Dictates wash stringency; critical for allele discrimination. |
| Self-Complementarity | Free Energy > -5 kcal/mol | Prevents intra-probe hybridization, ensuring target availability. | Negative values indicate hairpins, reducing effective probe concentration. |
| Target Region | Flanks CCGG site(s) | Does NOT contain the HpaII/MspI site itself. Binds to stable fragment internal sequence. | Enables detection of all fragments generated by methylation-sensitive digestion. |
Protocol 2.1: In Silico Probe Design and Validation Workflow
- Sequence Retrieval: Using UCSC Genome Browser or ENSEMBL, extract 2-3 kb of genomic sequence surrounding your locus of interest, including all potential HpaII/MspI sites.
- Restriction Site Mapping: Use software like NEBcutter to map all CCGG sites. Identify the fragment sizes expected for methylated and unmethylated alleles post-HpaII digest.
- Candidate Probe Selection: Select a 300-500 bp sequence from within the largest predicted constant fragment (not containing a CCGG). Verify low repeat content using RepeatMasker.
- Thermodynamic Calculation: Calculate Tm using the nearest-neighbor method (e.g., OligoCalc). Adjust length to achieve Tm >75°C.
- Specificity Check: Perform a BLAST search against the relevant genome to ensure uniqueness. Expect a single perfect match.
- Secondary Structure Prediction: Use mFold or UNAFold. Reject probes with predicted stable secondary structures (ΔG < -5 kcal/mol).
Probe Labeling Strategies & Protocols
Non-radioactive labeling, primarily via digoxigenin (DIG), is standard due to safety, stability, and compatibility with chemiluminescent detection. Random primed labeling is preferred for Southern blot probes.
Protocol 3.1: DIG-High Prime DNA Labeling (Roche) Materials: Purified, linearized probe template (25-50 ng), DIG-High Prime (Component: random hexamers, Klenow enzyme, dNTPs including DIG-dUTP), LiCl, EDTA, Ethanol. Procedure:
- Denature 50 ng of purified PCR product or plasmid fragment (in 16 µL H₂O) by boiling for 10 min, then snap-cool on ice.
- Add 4 µL of DIG-High Prime, mix gently, and centrifuge briefly.
- Incubate at 37°C for 1-20 hours (optimal: 3-4 hours).
- Stop reaction by adding 2 µL 0.2M EDTA (pH 8.0) and heating to 65°C for 10 min.
- Precipitate probe: Add 2.5 µL 4M LiCl and 75 µL pre-chilled 100% ethanol. Mix and incubate at -80°C for 30 min.
- Centrifuge at 13,000 rpm for 15 min at 4°C. Wash pellet with 50 µL cold 70% ethanol. Air-dry.
- Resuspend pellet in 50 µL TE buffer or hybridization solution. Store at -20°C.
Table 2: Comparison of Common Labeling Methods
| Method | Typical Yield (DIG-dUTP incorporation) | Optimal Probe Size | Incubation Time | Best For |
|---|---|---|---|---|
| Random Priming | 1 DIG per 25-30 nt | 200-1000 bp | 1-20 hr | Southern blots, long probes, high sensitivity. |
| PCR Labeling | 1 DIG per 30-40 nt | 100-3000 bp | 2-3 hr | Probes from limited template, specific fragment amplification. |
| Nick Translation | 1 DIG per 20-25 nt | >500 bp | 1.5-2 hr | Very long probes (e.g., BAC DNA). |
Hybridization and Stringency Washes for Specificity
High-specificity detection is achieved in the hybridization and wash steps. The key is to use a precisely calculated hybridization temperature (Thyb) and sequential stringency washes.
- Calculation of Thyb: Thyb = Tm(probe) - (20 to 25°C). For a probe with Tm = 78°C, Thyb = 58°C.
- Importance of Formamide: Inclusion of 50% formamide in hybridization buffer allows effective hybridization at this lower, more specific temperature by destabilizing DNA duplexes.
Protocol 4.1: High-Stringency Hybridization and Washes
- Pre-hybridization: Pre-wet membrane in 2x SSC. Incubate in pre-heated DIG Easy Hyb solution at Thyb for 30-60 min in a roller bottle.
- Hybridization: Denature labeled probe (5-25 ng/mL) by boiling for 5 min, chilling on ice. Add to fresh Thyb-preheated DIG Easy Hyb. Incubate membrane with probe solution at Thyb for 16 hours.
- Post-Hybridization Washes:
- Wash 1: 2x SSC, 0.1% SDS at room temperature (2 x 5 min).
- Wash 2: Stringency Wash: 0.5x SSC, 0.1% SDS at Thyb + 5-10°C (2 x 15 min). (This step is critical for removing partially matched probes).
- Proceed to immunological chemiluminescent detection per manufacturer's protocol.
Visualization: Probe Design and Detection Workflow
Probe Design and Detection Workflow
Probe Binds Independent of Methylation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Probe Generation and Detection
| Reagent / Kit | Manufacturer Example | Primary Function in Protocol |
|---|---|---|
| DIG-High Prime | Roche/Sigma-Aldrich | Integrated system for random-primed incorporation of DIG-dUTP into DNA probe. |
| DIG Easy Hyb | Roche/Sigma-Aldrich | Optimized hybridization solution containing formamide and blocking agents, used for both pre-hybridization and hybridization steps. |
| Anti-Digoxigenin-AP, Fab fragments | Roche/Sigma-Aldrich | Alkaline phosphatase-conjugated antibody for specific binding to DIG-labeled probes on the membrane. |
| CDP-Star / CSPD | Roche/Thermo Fisher | Chemiluminescent alkaline phosphatase substrate; emits light upon dephosphorylation for film or digital imaging. |
| Nylon Membrane, Positively Charged | Roche, Amersham, Pall | Membrane for DNA immobilization by capillary transfer; positive charge ensures covalent binding of alkali-blotted DNA. |
| DIG DNA Labeling and Detection Kit | Roche | Comprehensive kit containing all key components (DIG-High Prime, Easy Hyb, Antibody, Substrate, Buffers) for a complete workflow. |
| PCR DIG Probe Synthesis Kit | Roche | For direct incorporation of DIG-dUTP during PCR amplification of the probe template. |
Application Notes
Within the thesis on DNA methylation analysis using Southern blotting, this step is critical for the specific detection of restriction fragments indicative of methylation status. Hybridization employs a labeled probe complementary to the target sequence flanking the restriction site(s) of interest. Following hybridization, stringent washes remove non-specifically bound probe, ensuring that detected signal originates from perfectly matched sequences—a necessity when distinguishing between methylated (uncleaved) and unmethylated (cleaved) DNA fragments. The choice of detection method (radioactive vs. chemiluminescent) profoundly influences sensitivity, exposure time, safety protocols, and waste disposal.
- Radioactive Detection (³²P or ³³P): Traditionally offers the highest sensitivity, capable of detecting low-abundance targets, which is advantageous for samples with limited DNA or partial methylation. It provides a direct, quantitative linear relationship between signal intensity and probe concentration. However, it requires stringent safety measures, generates hazardous waste, and has a short probe shelf-life due to isotope decay.
- Chemiluminescent Detection: A non-radioactive alternative using enzyme-conjugated probes (e.g., horseradish peroxidase - HRP) that catalyze a light-emitting reaction. It offers improved safety, longer probe stability (months to years), and is compatible with standard lab equipment. Modern enhanced substrates have narrowed the sensitivity gap with radioactivity for most applications. It is the preferred method in clinical and diagnostic settings.
Experimental Protocols
Protocol 5.1: Hybridization and Stringency Washes for Southern Blots
Objective: To hybridize a labeled probe to immobilized DNA on a membrane and perform washes to achieve specific binding.
Materials: Pre-hybridization/Hybridization buffer (e.g., Church & Gilbert buffer: 1% BSA, 1 mM EDTA, 0.5 M NaHPO₄ pH 7.2, 7% SDS), labeled DNA probe, wash buffer I (2X SSC, 0.1% SDS), wash buffer II (0.5X SSC, 0.1% SDS), wash buffer III (0.1X SSC, 0.1% SDS), hybridization oven or water bath, nylon membrane with transferred DNA.
Procedure:
- Pre-hybridization: Place the dried, UV-crosslinked membrane in a hybridization tube. Add an appropriate volume of pre-warmed pre-hybridization buffer (0.1 mL/cm² of membrane). Incubate with rotation at the hybridization temperature (typically 65°C for DNA probes) for 1-4 hours to block non-specific binding sites.
- Probe Preparation: Denature the labeled probe (double-stranded DNA) by heating to 95°C for 5 minutes, then immediately chill on ice.
- Hybridization: Add the denatured probe directly to the hybridization buffer in the tube. Incubate with rotation at the appropriate temperature (65°C for high stringency) for 12-16 hours (overnight).
- Stringency Washes:
- Low Stringency: Discard hybridization solution. Add a large volume of Wash Buffer I at room temperature. Incubate with rotation for 5 minutes. Repeat once. This removes unbound probe.
- High Stringency: Wash the membrane twice with Wash Buffer II pre-warmed to 65°C for 15 minutes each. For maximum stringency, one final wash with Wash Buffer III at 65°C for 15 minutes may be performed to remove probe bound to sequences with low homology.
- Proceed to Detection: Remove the membrane from the tube and proceed immediately to the appropriate signal detection protocol.
Protocol 5.2: Signal Detection via Autoradiography (³²P)
Objective: To visualize radioactive signal from a hybridized membrane.
Materials: Washed membrane, phosphor screen or X-ray film, film cassette, -80°C freezer (for film) or phosphorimager scanner.
Procedure:
- After the final wash, briefly blot the membrane on filter paper to remove excess liquid. Do not let the membrane dry completely if re-probing is intended.
- Wrap the damp membrane in clear plastic wrap.
- In a darkroom, place the wrapped membrane in a film cassette.
- Place a sheet of X-ray film on top of the membrane. For quantitative analysis, place a phosphor screen in contact with the membrane.
- Seal the cassette and expose at -80°C (for film) or at room temperature (for phosphor screen) for several hours to several days, depending on signal strength.
- Develop the film using an automatic processor or manually. For phosphor screens, scan using a phosphorimager.
Protocol 5.3: Signal Detection via Chemiluminescence (HRP-Conjugated Probe)
Objective: To visualize chemiluminescent signal from a hybridized membrane.
Materials: Washed membrane, blocking buffer (5% non-fat dry milk in TBST), detection reagent (e.g., Luminol/H₂O₂ substrate), substrate buffer, HRP-conjugated streptavidin (for biotinylated probes) or anti-digoxigenin antibody (for DIG-labeled probes), wash buffer (TBST: Tris-buffered saline with 0.1% Tween-20), imaging system (CCD camera or chemiluminescence imager).
Procedure:
- Blocking: After the final stringency wash, rinse the membrane briefly in substrate buffer. Incubate the membrane in blocking buffer for 60 minutes at room temperature with gentle agitation.
- Conjugate Binding: Dilute the HRP-conjugate (e.g., Streptavidin-HRP at 1:20,000) in fresh blocking buffer. Incubate the membrane in this solution for 30-60 minutes at room temperature with agitation.
- Washing: Wash the membrane 3-4 times for 5-10 minutes each with a large volume of TBST to remove unbound conjugate.
- Equilibration: Briefly rinse the membrane in substrate buffer for 5 minutes.
- Substrate Incubation: Mix the luminol and peroxide components of the chemiluminescent substrate as per manufacturer's instructions. Place the membrane face-up on a sheet of plastic. Pipette the substrate mixture evenly over the membrane, ensuring complete coverage. Incubate for 5 minutes.
- Imaging: Drain excess substrate, wrap the membrane in plastic wrap, and place it in an imaging cassette. Acquire the image using a CCD camera system, typically with exposure times ranging from 10 seconds to 30 minutes.
Data Presentation
Table 1: Quantitative Comparison of Radioactive vs. Chemiluminescent Detection
| Parameter | Radioactive Detection (³²P) | Chemiluminescent Detection (HRP) |
|---|---|---|
| Typical Sensitivity | 0.1 - 1 pg of target DNA | 1 - 10 pg of target DNA (with enhanced substrates) |
| Linear Dynamic Range | ~3-4 orders of magnitude | ~3 orders of magnitude |
| Typical Exposure Time | 1 hour - 7 days | 10 seconds - 30 minutes |
| Probe Stability | Short (half-life of isotope: ³²P=14.3 days; ³³P=25.4 days) | Long (months to years at -20°C) |
| Safety & Regulation | High (radiation safety protocols, licensed disposal) | Low (standard chemical safety) |
| Quantitative Analysis | Direct (signal proportional to radioactivity) | Indirect (signal depends on enzyme kinetics) |
| Primary Cost Driver | Radioisotope purchase, waste disposal | Enzyme conjugate, substrate kits |
| Best For | Ultimate sensitivity, quantitation, low-abundance targets | Routine analysis, high-throughput labs, clinical settings |
Visualization
Diagram Title: Signal Detection Workflow for Southern Blot Analysis
Diagram Title: Chemiluminescent Signal Generation Pathway
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Hybridization & Detection
| Item | Function in Protocol | Key Considerations for Methylation Analysis |
|---|---|---|
| Specific DNA Probe | Binds complementarily to target sequence adjacent to restriction site(s) interrogated for methylation. | Must be designed for sequences outside the restriction sites used (e.g., NotI, EagI, HpaII) to detect both cut and uncut fragments. |
| Hybridization Buffer (Church & Gilbert) | Provides ions for hybridization, blocking agents (BSA) to reduce background, and SDS to prevent non-specific binding. | High SDS concentration (7%) allows for high-temperature hybridization, increasing stringency and specificity critical for methylation studies. |
| Stringency Wash Buffers (SSC/SDS) | Removes imperfectly matched or nonspecifically bound probe. Lower SSC concentration increases stringency. | High-stringency washes (e.g., 0.1X SSC) are essential to distinguish between perfectly matched target sequences and partial homology. |
| Radioactive Nucleotides (α-³²P-dCTP) | Incorporated into probe via labeling; decays to emit beta particles for detection. | Provides high sensitivity needed for detecting rare alleles or partially methylated DNA in a heterogeneous sample. |
| Non-Radioactive Labeling Kit (DIG or Biotin) | Incorporates haptens into probe for subsequent enzyme-conjugate binding. | Offers safer, stable probes ideal for long-term studies requiring repeated analysis of multiple patient samples over time. |
| HRP-Streptavidin Conjugate | Binds with high affinity to biotinylated probes; HRP enzyme catalyzes chemiluminescent reaction. | Concentration and incubation time must be optimized to minimize background while maintaining signal for low-copy number methylated bands. |
| Enhanced Chemiluminescent (ECL) Substrate | Luminol-based solution oxidized by HRP in the presence of H₂O₂, producing sustained light emission. | Modern "enhanced" substrates provide signal amplification, approaching radioactive sensitivity for most Southern blot applications. |
| Phosphor Screen & Imager | Captures and digitizes radioactive emission; offers a wider linear dynamic range than X-ray film. | Critical for quantitative analysis comparing band intensities between samples (e.g., methylation percentage). |
Within the broader thesis on DNA methylation analysis using Southern blotting, accurate interpretation of autoradiograph banding patterns is the critical final step. This protocol details the methodology for reading these patterns and converting them into quantitative methylation status data, essential for research in epigenetics, oncology, and therapeutic development.
Key Principles of Band Interpretation
A standard Southern blot assay for methylation uses methylation-sensitive restriction enzymes (e.g., HpaII) alongside their methylation-insensitive isoschizomers (e.g., MspI). The presence or absence of restriction sites due to CpG methylation generates distinct fragment sizes detectable with a locus-specific probe.
Core Interpretation Logic:
- Unmethylated DNA: Cut by HpaII, producing shorter fragments.
- Methylated DNA: Resistant to HpaII, producing longer (uncut) fragments or fragments cut only at distal unmethylated sites.
- Complete Digestion Control: MspI digests all DNA regardless of methylation, indicating total DNA loaded and probe efficacy.
- Partial Methylation: Appears as a mixture of both cut and uncut bands, indicating a heterogeneous cell population or allele-specific methylation.
Quantitative Data Analysis Protocol
Materials and Software
- High-resolution scanned autoradiograph or phosphorimage.
- Image analysis software (e.g., ImageJ, Image Studio Lite, or proprietary scanner software).
- Spreadsheet software (e.g., Microsoft Excel, Google Sheets).
Step-by-Step Quantification Method
- Image Calibration: Set the image scale to pixel units. Ensure non-saturating signal intensity.
- Lane Profile Analysis: Define lanes and draw rectangular regions of interest (ROIs) around each distinct band.
- Background Subtraction: Measure background intensity from an adjacent area with no bands and subtract.
- Integrated Density Measurement: For each band ROI, record the Integrated Density Value (IDV) or Volume Intensity.
- Data Normalization:
- Normalize the IDV of each HpaII band to the total signal in the corresponding MspI complete digest lane to account for lane-to-lane loading differences.
- Alternatively, normalize to an internal control band if present.
- Methylation Percentage Calculation:
- For a simple two-band system (cut vs. uncut):
% Methylation = (Intensity of Uncut Band / (Intensity of Cut Band + Intensity of Uncut Band)) * 100 - For complex multi-band patterns, the proportion of signal in higher molecular weight (less digested) bands relative to total signal is calculated.
- For a simple two-band system (cut vs. uncut):
Data Presentation Table: Example Quantification Output
Table 1: Quantitative Methylation Analysis of the MGMT Promoter in Glioma Cell Lines
| Cell Line / Sample | MspI Total Signal (IDV) | HpaII Cut Band (IDV) | HpaII Uncut Band (IDV) | Normalized Uncut Fraction | Methylation Status (%) |
|---|---|---|---|---|---|
| U87-MG (Control) | 15,250 | 12,100 | 450 | 0.036 | 3.6% (Unmethylated) |
| T98G | 14,980 | 2,150 | 10,050 | 0.824 | 82.4% (Hypermethylated) |
| Patient Derived Xenograft A | 16,750 | 6,340 | 7,880 | 0.554 | 55.4% (Partially Methylated) |
| Normal Brain Tissue | 15,500 | 14,200 | 155 | 0.011 | 1.1% (Unmethylated) |
IDV: Integrated Density Value. Normalized Uncut Fraction = [Uncut IDV / (Cut IDV + Uncut IDV)], adjusted by *MspI loading factor.*
Detailed Experimental Protocol: Southern Blot for Methylation Analysis
Genomic DNA Digestion
- Reaction Setup: In separate tubes, digest 5-10 µg of genomic DNA with:
- Tube 1: Methylation-sensitive enzyme (e.g., HpaII, 20 U/µg DNA).
- Tube 2: Methylation-insensitive control (e.g., MspI, 20 U/µg DNA).
- Tube 3: No enzyme (undigested control).
- Incubate at 37°C for 16 hours (overnight). Purify DNA by ethanol precipitation.
Gel Electrophoresis and Blotting
- Load digested DNA onto a 0.8-1.2% agarose gel. Include a molecular weight ladder.
- Run gel at low voltage (1-2 V/cm) until optimal separation is achieved.
- Depurinate, denature, and neutralize the gel in sequence.
- Transfer DNA onto a positively charged nylon membrane via capillary or vacuum transfer.
Probe Labeling and Hybridization
- Probe Preparation: Label a locus-specific PCR fragment or plasmid (200-500 bp) with [α-³²P]dCTP using a random prime labeling kit.
- Pre-hybridize membrane in Church buffer (1% BSA, 1 mM EDTA, 0.5 M NaHPO₄ pH 7.2, 7% SDS) at 65°C for 1 hour.
- Add denatured probe and hybridize at 65°C for 16-20 hours.
Washing and Detection
- Wash membrane sequentially with low-stringency (2X SSC, 0.1% SDS) and high-stringency (0.2X SSC, 0.1% SDS at 65°C) buffers.
- Expose membrane to a phosphor storage screen for 24-72 hours.
- Scan the screen using a phosphorimager for quantitative analysis.
Visualizations
Title: Southern Blot Methylation Analysis Workflow
Title: Interpreting Bands: Enzyme Action & Methylation State
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Southern Blot Methylation Analysis
| Item | Function & Rationale |
|---|---|
| Methylation-Sensitive Restriction Enzyme (e.g., HpaII, NotI) | Cleaves only unmethylated recognition sequences, enabling discrimination of methylation status. |
| Isoschizomer Control Enzyme (e.g., MspI for HpaII) | Cuts the same sequence regardless of methylation, serving as a digestion and loading control. |
| Positively Charged Nylon Membrane | Binds negatively charged DNA permanently after alkaline transfer, essential for probe hybridization. |
| [α-³²P]dCTP or Chemiluminescent Label | Radioactive or non-radioactive label for generating high-sensitivity, locus-specific hybridization probes. |
| Phosphor Storage Screen & Imager | For quantitative, high-resolution detection of radioactive signals from the blot. |
| High-Quality Agarose | For optimal separation of large DNA fragments (1-20 kb) typical in Southern blot analysis. |
| Church Hybridization Buffer | Low-background, efficient hybridization buffer for use with radioactive probes. |
| Molecular Weight Ladder (DNA Marker) | Essential for accurately determining the size of detected restriction fragments. |
| DNA Polymerase & Random Primers Kit | For efficient labeling of probe DNA fragments via random primed synthesis. |
Solving Common Pitfalls: Troubleshooting and Enhancing Southern Blot Sensitivity & Reproducibility
Application Notes & Protocols Context: DNA Methylation Analysis via Southern Blotting
Within a thesis investigating epigenetic alterations in oncogenesis via Southern blotting, achieving optimal signal-to-noise ratio is paramount. Poor signal or excessive background compromises data integrity, particularly when detecting subtle methylation changes at low-abundance genomic loci. This protocol details systematic optimization of probe-specific activity and hybridization stringency to resolve these issues, enabling precise mapping of methylation-sensitive restriction patterns.
Table 1: Optimization Variables and Their Impact on Signal & Background
| Parameter | Low Signal Cause | High Background Cause | Optimal Range (Current Best Practice) |
|---|---|---|---|
| Probe Specific Activity (dpm/μg) | <1 x 10⁹ | >1 x 10¹⁰ | 2-5 x 10⁹ dpm/μg |
| Hybridization Temperature | Too high (>Tm-5°C) | Too low ( | Tm -10°C to Tm -12°C |
| Formamide Concentration | >55% (v/v) | <45% (v/v) | 50% (v/v) |
| Salon Concentration (SSC) | 6x SSC (too high) | <2x SSC (too low) | 2.5x - 4x SSC |
| Blocking Agent (Denhardt’s/SSDNA) | Insufficient concentration | Excessive, viscous solution | 5x Denhardt’s, 100μg/mL sheared salmon sperm DNA |
| Post-Hybridization Wash Stringency | Too stringent (e.g., 0.1x SSC, 65°C) | Too permissive (e.g., 2x SSC, RT) | Primary: 2x SSC, 0.1% SDS, 42°C. Secondary: 0.5x SSC, 0.1% SDS, 55°C |
| Autoradiography Exposure Time | Too short | Too long | 24-72 hours at -80°C with intensifying screen |
Table 2: Probe Labeling Methods Comparison (Radioactive)
| Method | Typical Specific Activity | Recommended for Methylation Analysis | Key Advantage | Protocol Duration |
|---|---|---|---|---|
| Random Priming | 1-5 x 10⁹ dpm/μg | Preferred (high complexity genomic DNA) | High efficiency, reproducible | 1-2 hours |
| Nick Translation | 5 x 10⁸ - 1 x 10⁹ dpm/μg | Suitable (larger DNA fragments) | Incorporates label along fragment | 1.5 hours |
| PCR-based Labeling | 1-3 x 10⁹ dpm/μg | Locus-specific probes | High purity, no template removal | 2-3 hours |
Detailed Experimental Protocols
Protocol 3.1: High Specific Activity Probe Synthesis by Random Priming
Objective: Generate a (^{32}\text{P})-dCTP-labeled probe with specific activity of 2-5 x 10⁹ dpm/μg. Materials: 25-50 ng linearized template DNA, Random Hexamer Primers, Klenow Fragment (exo-), [α-(^{32}\text{P})]dCTP (3000 Ci/mmol), dNTP mix (dATP, dGTP, dTTP at 0.5 mM each), Sephadex G-50 spin columns.
- Denature 25ng of purified DNA probe template (25-800 bp) in 10μL by heating to 95°C for 5 min, snap-chill on ice.
- On ice, prepare reaction mix: 10μL denatured DNA, 2μL 10X Random Primers Buffer, 1μL dNTP mix (lacking dCTP), 5μL [α-(^{32}\text{P})]dCTP (50 μCi), 1μL Klenow Fragment (5 U/μL). Adjust total volume to 20μL with nuclease-free water.
- Incubate at 37°C for 60 minutes.
- Stop reaction by adding 2μL 0.5M EDTA (pH 8.0).
- Purify labeled probe using a pre-equilibrated Sephadex G-50 spin column per manufacturer instructions to remove unincorporated nucleotides. Elute in 50μL TE buffer (pH 8.0).
- Quantify specific activity by scintillation counting (1μL aliquot). Use immediately or store at -20°C for up to 72 hours.
Protocol 3.2: Optimized Hybridization for Methylation-Specific Southern Blots
Objective: Hybridize probe to genomic DNA transferred to nylon membrane with maximal specificity and minimal background. Pre-hybridization:
- Place membrane (with cross-linked DNA) in a hybridization tube. Add 10-20 mL of pre-warmed Church & Gilbert Hybridization Buffer (0.5M NaPO₄ pH 7.2, 7% SDS, 1% BSA, 1mM EDTA). For lower background, 50% formamide can be added, requiring adjustment of temperature (see below).
- Pre-hybridize in a rotary hybridization oven for a minimum of 2 hours at the determined hybridization temperature. Hybridization Temperature Calculation (for formamide buffers): [ T{hyb} = 81.5°C - 0.65(\% \text{GC}) + 16.6 \log{10}[Na^+] - 0.6(\% \text{formamide}) - (600/l) ] Where (l) = probe length in bp. For a 500bp probe with 50% GC in Church buffer (~0.5M Na⁺), using 0% formamide: (T{hyb} \approx 68°C). Using 50% formamide: (T{hyb} \approx 42°C). Hybridization:
- Denature purified probe (from Protocol 3.1) by heating to 95°C for 5 min, snap-chill on ice.
- Add denatured probe directly to the hybridization tube containing fresh, pre-warmed Church & Gilbert buffer (5-10 mL). Use 1-2 x 10⁶ dpm/mL hybridization buffer.
- Hybridize for 16-20 hours (overnight) at the calculated temperature with continuous rotation. Post-Hybridization Washes (Critical for Background Reduction):
- Primary Wash: Discard radioactive hybridization buffer. Wash membrane with 200mL of Wash Buffer I (2X SSC, 0.1% SDS) at room temperature for 15 min with agitation.
- Secondary Wash: Wash with 200mL of Wash Buffer II (0.5X SSC, 0.1% SDS) pre-heated to 55°C. Incubate at 55°C with agitation for 15 min. Monitor background with a Geiger counter; repeat if necessary.
- Blot membrane on Whatman paper to remove excess buffer, wrap in plastic wrap, and expose to phosphorimager screen or X-ray film at -80°C.
Visualization: Workflows & Logical Decision Trees
Diagram 1: Troubleshooting Signal & Background in Southern Blotting
Diagram 2: Optimized Southern Blotting Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Probe & Hybridization Optimization
| Reagent/Category | Specific Product/Example | Function in Optimization | Critical Parameter |
|---|---|---|---|
| Radiolabeled Nucleotide | [α-(^{32}\text{P})]dCTP, 3000 Ci/mmol | Primary label for high-specific activity probes | Freshness (<2 weeks from calibration date) is crucial for high specific activity. |
| Labeling Enzyme | Klenow Fragment (exo-) | Incorporates labeled dNTPs during random priming. | High concentration (5-10 U/μL) for efficient synthesis. |
| Hybridization Buffer | Church & Gilbert Phosphate Buffer (0.5M NaPO₄, 7% SDS) | Provides consistent stringency, lower background than SSC-based buffers. | pH must be 7.2; SDS concentration critical for blocking. |
| Blocking Agents | Sheared Salmon Sperm DNA (ssDNA), Denhardt's Solution (BSA, Ficoll, PVP) | Binds non-specific sites on membrane to prevent probe adherence. | ssDNA must be properly denatured (boiled & snap-chilled) before use. |
| Membrane | Positively Charged Nylon (e.g., Hybond-N+) | High nucleic acid binding capacity and durability through stringent washes. | Consistent lot-to-lot quality is essential for reproducible background. |
| Wash Solutions | SSC (20X Stock), SDS (10% Stock) | Precisely control stringency in post-hybridization washes to remove mismatched probe. | Accurate molarity and temperature are non-negotiable for specificity. |
| Purification Column | Sephadex G-50 Fine Spin Columns | Removes unincorporated nucleotides post-labeling, reducing background. | Must be pre-equilibrated and not overloaded for optimal flow-through. |
Within the framework of DNA methylation analysis via Southern blotting, the integrity of restriction enzyme digestion is paramount. Incomplete or inconsistent digestion, often stemming from suboptimal enzyme activity or impurities in the reaction, leads to artifactual bands, erroneous methylation quantification, and irreproducible data. This application note provides protocols to validate enzyme performance and reaction purity, ensuring reliable interpretation of methylation patterns essential for epigenetic research and drug development targeting epigenetic modifiers.
Protocol 1: Titration of Restriction Enzyme Units for Complete Genomic DNA Digestion
Objective: To empirically determine the optimal units of restriction enzyme required for complete digestion of a target genomic DNA sample, controlling for potential inhibitors.
Materials:
- Genomic DNA (e.g., from HeLa cells, 500 ng/µL).
- Restriction Enzyme (e.g., HpaII, methylation-sensitive).
- Corresponding recommended 10X Reaction Buffer.
- Nuclease-free water.
- Control unmethylated and methylated DNA (e.g., from CpG methyltransferase-treated cells).
- Thermostable water bath or PCR cycler.
- Agarose gel electrophoresis system.
Procedure:
- Prepare a series of 50 µL reactions containing 2 µg of genomic DNA, 1X reaction buffer, and varying amounts of enzyme (e.g., 0, 5, 10, 20, 40 units).
- Incubate at the enzyme's optimal temperature (37°C for HpaII) for 16 hours (overnight) to challenge enzyme longevity.
- Heat-inactivate the enzyme as per manufacturer's instructions.
- Resolve the digested DNA alongside a molecular weight ladder on a 0.8% agarose gel.
- Visualize using ethidium bromide or SYBR Safe staining. Complete digestion is indicated by a clear, smear-free pattern of distinct bands specific to the enzyme used.
Data Interpretation: The minimal unit yielding a digestion pattern identical to that of higher concentrations is the optimal amount. Persistent high-molecular-weight smearing indicates incomplete digestion, necessitating more enzyme, longer incubation, or purity assessment.
Table 1: Example Titration Data for HpaII Digestion of HeLa Genomic DNA (2 µg, 16 hr incubation)
| Enzyme Units (U) | Digestion Completeness (Visual Gel Score) | Notable Artifacts |
|---|---|---|
| 0 | None (Undigested high MW smear) | High MW smear only |
| 5 | Partial (Smear + faint expected bands) | Incomplete pattern |
| 10 | Mostly Complete | Minimal smear |
| 20 | Complete (Clear band pattern) | None |
| 40 | Complete (Clear band pattern) | None |
Protocol 2: Assessing Reaction Purity via Spiked Control DNA Digestion
Objective: To diagnose reaction inhibition caused by impurities in the DNA sample (e.g., salts, phenol, ethanol, protein) versus issues with the enzyme itself.
Materials:
- Test genomic DNA sample.
- Purified control DNA (e.g., unmethylated λ DNA).
- Restriction enzyme with known activity on control DNA (e.g., EcoRI for λ DNA).
- Recommended 10X Reaction Buffer.
- Nuclease-free water.
Procedure:
- Set up two parallel 25 µL digestions:
- Reaction A (Control): 200 ng λ DNA + 1X buffer + 10 U enzyme.
- Reaction B (Spiked Test): 1 µg test genomic DNA + 200 ng λ DNA + 1X buffer + 10 U enzyme.
- Incubate at optimal temperature for 2 hours.
- Analyze digestion completeness by agarose gel electrophoresis.
Data Interpretation: If digestion of the λ DNA is complete in Reaction A but incomplete in Reaction B, inhibitors are present in the genomic DNA preparation. If both are incomplete, the enzyme or buffer may be compromised.
Table 2: Diagnostic Outcomes for Spiked Control Assay
| Result (Gel Analysis) | Diagnosis |
|---|---|
| λ DNA digested in both A and B | No significant inhibitors in test DNA. Enzyme is active. |
| λ DNA digested in A only, not in B | Inhibitors present in test genomic DNA sample. Requires re-purification. |
| λ DNA not completely digested in either A or B | Problem with enzyme, buffer, or reaction conditions (e.g., temperature). |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Valid Digestion in Methylation Analysis
| Item | Function/Benefit |
|---|---|
| High-Fidelity Restriction Enzymes | Ensures lot-to-lot consistency, minimal star activity, and robust performance in long incubations required for genomic DNA. |
| DNA Cleanup & Concentration Kits (Spin-Column) | Removes salts, organics, and enzymes post-digestion or pre-digestion to eliminate inhibitors and ensure reaction purity. |
| Methylation-Sensitive Control DNAs (e.g., in vitro methylated & unmethylated) | Provides unambiguous positive/negative controls for digestion efficiency and enzyme specificity. |
| Thermostable Bath or PCR Cycler | Provides precise, stable incubation temperature to prevent enzyme denaturation and ensure consistent activity. |
| Spectrophotometer (NanoDrop) with A260/A230 | Assesses DNA purity; a low A260/A230 ratio (<2.0) indicates contaminating organics (phenol, guanidine) that inhibit enzymes. |
| Gel Loading Dye with Tracking Dyes | Allows visual monitoring of electrophoresis progress without UV visualization, confirming buffer conductivity. |
Visualization: Diagnostic Workflow for Incomplete Digestion
Diagram Title: Troubleshooting Flowchart for Failed Restriction Digest
Visualization: Key Factors in Restriction Digestion Efficiency
Diagram Title: Interplay of Factors in Digest Efficiency
1. Introduction Within the framework of a thesis on DNA methylation analysis via Southern blotting, the clarity and interpretability of the final autoradiograph are paramount. Faint or smeared bands represent a critical failure point, obscuring methylation patterns and compromising data validity. This application note addresses the two primary culprits: degraded/poor-quality genomic DNA and suboptimal electrophoresis conditions. We present targeted protocols and parameter optimizations to resolve these issues, ensuring high-integrity data for research and drug development applications.
2. Troubleshooting DNA Integrity for Methylation Analysis High-molecular-weight (HMW), intact genomic DNA is non-negotiable for Southern blotting, especially when restriction enzymes like HpaII and MspI (sensitive to methylation status) are used. Degraded DNA leads to a smear, while protein or salt contamination can inhibit restriction digestion, causing faint or absent bands.
Table 1: Quantitative Impact of DNA Quality on Southern Blot Results
| DNA Quality Parameter | Optimal Value/Range | Effect if Suboptimal | Observed Artifact |
|---|---|---|---|
| A260/A280 Ratio | 1.8 - 2.0 | Phenol or protein contamination | Faint bands, high background |
| A260/A230 Ratio | >2.0 | Salt or solvent carryover | Inhibition of restriction digestion |
| Fragment Size (Pulse-field gel) | >50 kbp | Mechanical or nuclease degradation | Smear, no discrete bands |
| DNA Concentration (for digestion) | 0.5 - 1.0 µg/µL | Too low: faint bands; Too high: incomplete digestion | Faint or smeared bands |
Protocol 2.1: Phenol-Chloroform-Isoamyl Alcohol (PCI) Extraction for HMW DNA Objective: To obtain protein-free, HMW genomic DNA from mammalian tissue for methylation analysis. Reagents: Lysis Buffer (10 mM Tris-HCl pH 8.0, 0.1 M EDTA pH 8.0, 0.5% SDS, 20 µg/mL RNase A), PCI (25:24:1), Chloroform, 70% Ethanol, TE Buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Procedure:
- Homogenize 25 mg tissue in 500 µL Lysis Buffer. Incubate at 56°C for 2 hours.
- Cool to RT. Add 500 µL PCI. Mix gently by inversion for 10 minutes.
- Centrifuge at 12,000 x g, 15 min, 4°C. Transfer aqueous (top) phase to a new tube.
- Add 500 µL chloroform. Mix gently. Centrifuge at 12,000 x g, 10 min, 4°C.
- Transfer aqueous phase. Add 0.5 volumes of 7.5 M ammonium acetate and 2 volumes of 100% ethanol. Precipitate at -20°C for 1 hour.
- Pellet DNA (12,000 x g, 30 min, 4°C). Wash with 70% ethanol. Air-dry.
- Resuspend in 50 µL TE buffer overnight at 4°C. Quantify via spectrophotometer.
Protocol 2.2: DNA Integrity Verification by Pulse-Field Gel Electrophoresis (PFGE) Objective: To visually confirm DNA is of sufficient size (>50 kbp) for Southern analysis. Procedure:
- Cast a 1% certified PFGE agarose gel in 0.5X TBE.
- Mix 200 ng DNA with loading dye. Load alongside a HMW ladder (e.g., 50-1000 kbp).
- Run on PFGE system with appropriate pulse times (e.g., 1-20 sec switch time) for 16 hours at 6 V/cm, 14°C.
- Stain with ethidium bromide (0.5 µg/mL) for 30 min, destain in water for 30 min, image. A single, tight, high-molecular-weight band should be visible.
3. Optimizing Electrophoresis Parameters Improper gel electrophoresis causes band spreading (smearing) and poor resolution, critical when distinguishing between methylated and unmethylated fragments.
Table 2: Optimized Agarose Gel Electrophoresis Parameters for Southern Blotting
| Parameter | Standard Protocol | Optimized Protocol | Rationale |
|---|---|---|---|
| Agarose Type | Standard | High-quality, low-EEO | Reduces electroendosmosis, sharpens bands. |
| Gel Concentration | 0.8% | 0.7% | Better resolution for larger DNA fragments (1-20 kbp). |
| Voltage | 10 V/cm | 1-2 V/cm | Prevents band "smiling" and overheating, which melts agarose. |
| Running Buffer | 1X TAE | 1X TBE | Higher buffering capacity for long runs. |
| Running Temperature | Ambient | 4°C (cold room) | Maintains gel integrity, prevents DNA denaturation. |
| Loading Dye | Standard | Contains 30% glycerol | Prevents diffusion of DNA into wells. |
| Post-run Treatment | Direct staining | Soak in 0.25 M HCl for 15 min (depurination) | Fragments large DNA for efficient transfer. |
Protocol 3.1: Southern Blotting-Compatible Agarose Gel Electrophoresis Objective: To achieve sharp, well-resolved DNA bands for capillary transfer. Procedure:
- Prepare 300 mL of 0.7% high-grade agarose in 1X TBE. Microwave to dissolve, cool to 55°C.
- Pour gel in a horizontal tray with a well comb. Allow to set for 1 hour at 4°C.
- Place gel tank in a cold room (4°C). Add 1X TBE running buffer to submerge gel by 2-3 mm.
- Load digested DNA samples (20-30 µg total) mixed with glycerol-based loading dye.
- Run gel at 1.5 V/cm (e.g., 30 V for a 20 cm gel) for 16-20 hours.
- Post-electrophoresis, soak gel in 0.25 M HCl with gentle agitation for 15 min to depurinate DNA. Rinse with deionized water.
- Proceed to denaturation/neutralization steps per standard Southern blot protocol.
4. The Scientist's Toolkit: Key Research Reagent Solutions Table 3: Essential Materials for High-Integrity DNA Southern Blotting
| Reagent/Material | Function & Importance |
|---|---|
| High-Quality, Low-EEO Agarose | Forms uniform gel matrix with minimal electroendosmosis, critical for sharp band resolution. |
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII) | Key tool for methylation mapping; requires clean DNA for complete digestion. |
| Proteinase K (Molecular Grade) | Digests nucleases and other proteins during DNA isolation, protecting DNA from degradation. |
| RNase A (DNase-free) | Removes RNA contamination that can inflate DNA quantification and interfere with blotting. |
| Nylon Membrane (Positively Charged) | Robust binding of denatured DNA for subsequent hybridization and stripping/re-probing. |
| 32P-dCTP or Chemiluminescent Probe Labeling Kit | Generates high-sensitivity probes for detection of target sequences, even from faint bands. |
| Phosphorimager Screen & Scanner | Enables quantitative, high-dynamic-range detection of hybridization signals versus film. |
5. Visualizing the Workflow and Problem-Solving Logic
Diagram 1 (96 chars): Systematic troubleshooting path for faint/smeared Southern blot bands.
Diagram 2 (96 chars): Core Southern blot workflow for DNA methylation analysis.
Within the context of DNA methylation analysis via Southern blotting, the specificity of probe hybridization is paramount. Non-specific binding (NSB) of probes to non-target sequences or the membrane itself can produce high background noise, obscuring true methylation-specific signals. This application note details protocols for optimizing stringency washes and selecting blocking agents to suppress NSB, thereby enhancing the clarity and reliability of methylation data critical for epigenetic research and drug development.
Key Concepts & Mechanisms
NSB in Southern blotting arises from electrostatic interactions between the negatively charged phosphate backbone of nucleic acid probes and positively charged groups on nitrocellulose/nylon membranes, or from weak complementary base-pairing with non-target DNA. Stringency, controlled by temperature and ionic strength during washes, determines the stability of hybridized duplexes. Blocking agents pre-saturate these non-specific binding sites.
Diagram 1: Factors Influencing Southern Blot Stringency
Research Reagent Solutions Toolkit
| Reagent | Function in Mitigating NSB |
|---|---|
| Salmon Sperm DNA | A classical blocking agent; sheared and denatured, it competes for non-specific membrane binding sites. |
| Denhardt's Solution | A mixture of Ficoll, polyvinylpyrrolidone, and BSA; coats the membrane to prevent probe adhesion. |
| SDS (Sodium Dodecyl Sulfate) | An ionic detergent used in wash buffers and hybridization solutions to disrupt hydrophobic interactions. |
| SSPE or SSC Buffers | Provide consistent ionic strength (Na⁺) during washes; critical for precise stringency control. |
| Formamide | A denaturant added to hybridization buffer; lowers the effective melting temperature, allowing high-stringency hybridization at lower, safer temperatures. |
| Commercial Blocking Mixes | Optimized, standardized formulations (e.g., containing casein, synthetic polymers) offering consistent, high-efficiency blocking. |
Table 1: Effect of Wash Stringency on Signal-to-Noise Ratio in Methylation-Specific Southern Blots
| Stringency Condition | [SSC] | Temperature (°C) | Avg. Target Signal Intensity (AU) | Avg. Background Intensity (AU) | Signal-to-Noise Ratio |
|---|---|---|---|---|---|
| Low (Post-Hybridization) | 2x | Room Temp | 10500 | 2450 | 4.3 |
| Medium (First Wash) | 1x | 42 | 10200 | 980 | 10.4 |
| High (Final Wash) | 0.1x | 65 | 9870 | 210 | 47.0 |
Table 2: Comparison of Blocking Agent Efficacy
| Blocking Agent (Pre-hybridization) | Concentration | Background Reduction (% vs. No Block) | Target Signal Retention (%) | Best For |
|---|---|---|---|---|
| Denatured Salmon Sperm DNA | 100 µg/mL | 85% | 100% | Standard genomic blots |
| Denhardt's Solution | 1x (0.02% each) | 78% | 98% | High-complexity probes |
| Commercial Nucleic Acid-Free Block | 5% (w/v) | 92% | 99% | Methylation-specific probes |
| BSA (Bovine Serum Albumin) | 5% (w/v) | 70% | 101% | General use supplement |
Detailed Experimental Protocols
Protocol 1: Standard Pre-hybridization Blocking and Hybridization for Methylation Analysis
Objective: To effectively block membrane prior to application of methylation-sensitive restriction fragment probes.
- Membrane Preparation: Following Southern transfer, UV-crosslink DNA to a positively charged nylon membrane.
- Pre-hybridization: Place membrane in a hybridization tube or bag. Add pre-warmed hybridization buffer (e.g., 6x SSC, 5x Denhardt's, 0.5% SDS, 100 µg/mL denatured salmon sperm DNA).
- Incubate: Rotate at the pre-determined hybridization temperature (typically 65°C for standard probes, or 42°C with 50% formamide) for 1-4 hours.
- Probe Preparation: Denature radiolabeled or chemiluminescent probe specific to the methylated/unmethylated sequence (95°C, 5 min). Quench on ice.
- Hybridization: Replace pre-hybridization buffer with fresh buffer containing the denatured probe. Incubate overnight with rotation.
Protocol 2: Tiered Stringency Wash Procedure
Objective: To systematically remove non-specifically bound probe while retaining specific hybrids.
- Post-Hybridization Rinse: Remove hybridization buffer. Rinse membrane briefly with Low Stringency Wash Buffer (2x SSC, 0.1% SDS) at room temperature.
- First Wash (Medium Stringency): Wash membrane with 200-300 mL of Medium Stringency Buffer (1x SSC, 0.1% SDS) at 42°C for 15 minutes with gentle agitation. Monitor background with a Geiger counter or appropriate imager.
- Second Wash (High Stringency): Wash membrane with 200-300 mL of High Stringency Buffer (0.1x SSC, 0.1% SDS) at 65°C for 15 minutes. This is critical for methylation analysis where single-base mismatches may exist.
- Final Rinse: Briefly rinse membrane in 0.1x SSC at room temperature.
- Detection: Proceed to autoradiography or chemiluminescent detection as per probe label.
Diagram 2: Southern Blot Wash Stringency Optimization Workflow
Advanced Optimization for Methylation-Specific Probes
When using probes designed to distinguish methylated from unmethylated alleles following restriction digest (e.g., with HpaII vs. MspI), stringency must be finely tuned to discriminate potentially single-base-pair differences in hybridization stability. Incorporating 10-20% formamide into the hybridization buffer allows for high-stringency conditions at lower temperatures (e.g., 42°C), preserving membrane integrity. For chemiluminescent detection, increase the blocking agent concentration by 1.5x and consider adding 0.5% casein to the blocking solution to further reduce NSB from detection enzymes.
Meticulous optimization of blocking agents and a tiered stringency wash protocol are non-negotiable steps for producing publication-quality Southern blot data in DNA methylation studies. The systematic approach outlined here enables researchers to maximize signal-to-noise ratio, ensuring accurate interpretation of epigenetic states fundamental to gene regulation research and therapeutic development.
Application Notes: Standardization in DNA Methylation Analysis via Southern Blotting
Within DNA methylation research, particularly using Southern blotting, achieving reproducible results across laboratories and over time is a significant hurdle. The technique's multi-step, manually intensive nature introduces variability that can obscure true biological signals. This document outlines standardized protocols and the critical use of control samples to mitigate these reproducibility challenges, framed within a thesis investigating epigenetic alterations in disease models.
Quantitative Data on Reproducibility Variables
Table 1: Key Sources of Variability in Southern Blot Methylation Analysis
| Process Stage | Variable Parameter | Typical Range/Impact | Standardization Goal |
|---|---|---|---|
| DNA Digestion | Restriction Enzyme Activity | +/- 20% batch-to-batch | Use consistent units (U/µg); include complete digest control. |
| Gel Electrophoresis | Agarose Concentration | 0.8% - 1.2% impacts migration | Fix at 1.0% for consistent fragment separation. |
| Blotting | Capillary Transfer Time | 12-24 hours (incomplete transfer) | Standardize 16-hour transfer with fixed buffer volume and stack weight. |
| Hybridization | Probe Specific Activity | 1-5 x 10^9 cpm/µg variability | Normalize signal to reference control on each blot. |
| Washing | Stringency (SSC Conc.) | 0.1x - 2x SSC impacts background/signal | Define exact SSC/Temp combos for high and low stringency. |
| Detection | Film/Imager Exposure | Linear range often exceeded | Use calibrated phosphorimager; multiple exposures. |
Table 2: Essential Control Samples for Interpretation
| Control Sample | Description | Expected Result | Purpose in Analysis |
|---|---|---|---|
| In Vitro Methylated DNA (IVM) | Genomic DNA treated with SssI methylase. | 100% methylation at CpG sites. | Positive control for probe hybridization; defines fully methylated state. |
| Unmethylated DNA | DNA from normal tissue or treated with amplification. | 0% methylation at target loci. | Negative control; defines baseline. |
| No-DNA Blank | Loading buffer only. | No bands. | Controls for reagent contamination. |
| Digestion Control | DNA + Enzyme, known unmethylated site. | Complete digestion to smaller fragments. | Verifies restriction enzyme efficacy. |
| Molecular Weight Marker | Labeled DNA ladder. | Precise size bands. | Accurate fragment sizing. |
Detailed Experimental Protocols
Protocol 1: Standardized Southern Blotting for Methylation Analysis at a Single Locus Objective: To reproducibly assess the methylation status of a specific CpG island promoter region. Materials: High-molecular-weight genomic DNA, Methylation-sensitive restriction enzyme (e.g., HpaII), Its methylation-insensitive isoschizomer (e.g., MspI), Agarose, Gel electrophoresis system, Nylon membrane, Hybridization oven, Radioactive or chemiluminescently labeled locus-specific probe. Procedure:
- DNA Digestion: In parallel reactions, digest 10 µg of each sample DNA with (a) HpaII (sensitive to methylation) and (b) MspI (insensitive). Include IVM and unmethylated DNA controls. Use 5 units enzyme per µg DNA, incubate at 37°C for 16 hours.
- Gel Electrophoresis: Load digested DNA on a 1.0% agarose gel in 1x TAE. Include molecular weight marker. Run at 25V for 16 hours.
- Depurination & Denaturation: Soak gel in 0.25M HCl for 15 min, then in denaturation solution (1.5M NaCl, 0.5M NaOH) for 30 min.
- Capillary Transfer: Perform neutral Southern transfer to a positively charged nylon membrane using 20x SSC buffer for exactly 16 hours.
- Crosslinking: UV-crosslink DNA to membrane.
- Pre-hybridization & Hybridization: Pre-hybridize membrane at 65°C for 1 hour in Church buffer. Add denatured, labeled probe (10^6 cpm/mL) and hybridize at 65°C for 16 hours.
- Washing: Perform two low-stringency washes (2x SSC, 0.1% SDS) at room temp for 15 min each. Perform one high-stringency wash (0.1x SSC, 0.1% SDS) at 65°C for 30 min.
- Detection: Expose to phosphorimager screen for 2, 6, and 24 hours. Quantify band intensity relative to controls.
Protocol 2: Preparation and Use of In Vitro Methylated (IVM) Control DNA Objective: To generate a 100% methylated control for standardization. Procedure:
- Incubate 20 µg of genomic DNA (e.g., from placental tissue) with 4 units of SssI CpG methylase per µg DNA in 1x reaction buffer containing 160µM S-adenosylmethionine (SAM).
- Incubate at 37°C for 4 hours. Add another 2 units/µg enzyme and fresh SAM, incubate for an additional 2 hours.
- Purify DNA via phenol-chloroform extraction and ethanol precipitation.
- Verify complete methylation by performing a parallel HpaII/MspI digest as in Protocol 1. The IVM DNA should show identical, high molecular weight fragments for both enzymes.
Visualization
Diagram Title: Standardized Southern Blot Workflow with Integrated Controls
Diagram Title: Methylation Interpretation Logic for HpaII/MspI
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Reproducible Methylation Southern Blots
| Item | Function & Standardization Rationale |
|---|---|
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII, NotI) | Cleave only at unmethylated recognition sequences. Use consistent supplier and lot; calibrate activity. |
| Isoschizomer Control Enzymes (e.g., MspI, NdeI) | Cut same sequence regardless of methylation. Essential control for DNA quality and digestion efficiency. |
| SssI CpG Methylase | Used to generate 100% methylated positive control DNA (IVM), standardizing the "fully methylated" signal baseline. |
| Positively Charged Nylon Membrane | Binds DNA irreversibly after transfer. Consistent brand/pore size ensures uniform binding capacity. |
| Radioactive (³²P) or Chemiluminescent Probes | High-sensitivity detection. Probe specific activity must be calculated and standardized per blot. |
| Standardized Hybridization Buffer (e.g., Church Buffer) | Provides consistent ionic and compositional environment for probe-target annealing, reducing blot-to-blot variability. |
| Phosphorimaging System & Screens | Quantitative, wide dynamic range detection. Superior to film for reproducible quantification. Calibrate regularly. |
| Internal Control DNA (IVM & Unmethylated) | Most Critical. Provides reference points on every blot for data normalization and quality assurance. |
Application Notes
This protocol details the adaptation of traditional Southern blotting for DNA methylation analysis into a medium-throughput format, enabling the parallel processing of 20-40 samples per week by a single researcher. The core innovation lies in the integration of semi-automated electrophoresis, capillary transfer systems, and multi-channel pipetting to maintain analytical rigor while significantly improving throughput and reproducibility. The method is optimized for the analysis of methylation-sensitive restriction fragment length polymorphisms (MS-RFLP) at specific genomic loci, crucial for biomarker validation in oncology and epigenetic drug development.
Key adaptations include the use of 20- or 30-well combs in agarose gels, simultaneous capillary transfer of multiple membranes in a stackable apparatus, and hybridization with up to three distinct, non-overlapping probes labeled with different fluorochromes (e.g., Cy3, Cy5, Alexa 488) for multiplex detection. This reduces gel runs, transfer steps, and hybridization events by approximately 60-70% compared to classic low-throughput Southern blotting.
Quantitative Performance Data
Table 1: Throughput and Performance Metrics Comparison
| Parameter | Classic Southern Blot | Adapted Medium-Throughput Protocol | Improvement Factor |
|---|---|---|---|
| Samples per Gel Run | 6-8 | 20-30 | ~3.75x |
| Total Hands-on Time (for 24 samples) | ~22 hours | ~14 hours | ~1.6x reduction |
| Assay Time (from digest to result) | 7-10 days | 4-5 days | ~1.8x reduction |
| DNA Required per Locus | 5-10 µg | 2-5 µg | ~2x reduction |
| Inter-Assay CV (for fragment sizing) | 8-12% | 5-7% | Improved reproducibility |
| Probe Multiplexing Capacity | 1 (typically radioisotope) | 3 (fluorescent) | 3x data per membrane |
Table 2: Recommended Restriction Enzyme Cocktails for MS-RFLP
| Locus Type | Methylation-Sensitive Enzyme | Methylation-Insensitive Companion | Purpose |
|---|---|---|---|
| CpG Island Promoter | HpaII (C*CGG) | MspI (C*CGG) | Cleavage depends on methylation of internal cytosine. |
| Imprinted Region | NotI (GC*GGCCGC) | EcoRV (GAT*ATC) | Maps methylation at rare, informative GC-rich sites. |
| Repetitive Element (Control) | None used singly | HinfI (G*ANTC) | Assesses global DNA quality and completeness of digestion. |
Detailed Experimental Protocol
I. DNA Digestion and Quantification (Semi-Automated)
- Setup: In a 96-well PCR plate, prepare master mixes for each restriction enzyme cocktail using a multi-channel pipette. Each reaction contains: 2.5 µg genomic DNA, 1X restriction enzyme buffer, 20 U of each enzyme, and nuclease-free water to 50 µL.
- Digestion: Seal plate and incubate at 37°C for 16 hours (overnight) in a thermal cycler with a heated lid to prevent evaporation.
- Clean-up & Quantification: Purify digested DNA using a 96-well silica-membrane binding plate per manufacturer's protocol. Elute in 50 µL TE buffer. Quantify DNA using a fluorescent DNA-binding dye (e.g., PicoGreen) in a microplate reader. Normalize all samples to 50 ng/µL.
II. Medium-Throughput Gel Electrophoresis
- Casting: Prepare a 0.8% agarose gel in 1X TAE buffer in a large (20x25 cm) casting tray. Use a 30-well comb. Allow to set fully.
- Loading: Mix 25 µL (1.25 µg) of each purified digest with 5 µL of 6X loading dye. Load entire volume per well. Include a molecular weight ladder (e.g., DIG-labeled or fluorescent) in the first and last lanes.
- Run: Electrophorese at 35V constant voltage for 16 hours (overnight) in a cooled (4°C) or thermostatically controlled (14°C) tank with buffer circulation.
III. Simultaneous Capillary Transfer
- Membrane Stack Assembly: Cut one nylon membrane and four sheets of absorbent blotting paper to the exact gel dimensions. Cut 20 sheets of Whatman 3MM paper slightly larger.
- Depurination & Denaturation: Sequentially submerge the gel in: 0.25 M HCl (15 min), denaturation solution (0.5 M NaOH, 1.5 M NaCl; 2 x 15 min), and neutralization solution (0.5 M Tris-HCl, pH 7.5, 1.5 M NaCl; 2 x 15 min).
- Transfer Stack: Place a glass plate over a tray containing 20X SSC. Drape a wick of 3MM paper over the plate, immersed at both ends. Place the gel face down on the wick. Place the pre-wetted (in 20X SSC) membrane on the gel. Add four sheets of blotting paper, then a 5cm stack of dry 3MM paper, a glass plate, and a 500g weight.
- Parallel Processing: Assemble up to three identical stacks separated by plastic mesh spacers in the same tray. Transfer for 6-8 hours.
IV. Crosslinking, Probing, and Multiplex Detection
- Immobilization: UV-crosslink DNA to each membrane (1200 J/cm²).
- Multiplex Probe Labeling: Label up to three PCR-amplified probes (150-500 bp) with different fluorophores using a Nick Translation or Random Priming kit (e.g., using Cy3-dUTP, Cy5-dUTP).
- Hybridization: Pre-hybridize membrane in Church's buffer at 65°C for 1 hr. Add all labeled probes simultaneously (20 ng/mL each). Hybridize at 65°C for 16 hours.
- Washing & Imaging: Wash stringently: 2X SSC/0.1% SDS at 65°C (5 min), then 0.2X SSC/0.1% SDS at 65°C (2 x 15 min). Image using a fluorescence laser scanner with appropriate excitation/emission filters for each fluorophore.
Visualizations
Medium-Throughput Southern Blot Workflow
Multiplex Probe Detection Strategy
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Item | Function in Protocol |
|---|---|
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII, NotI) | Specifically cleave unmethylated CpG sites, creating RFLPs dependent on methylation status. |
| Fluorescent dNTPs (Cy3-dUTP, Cy5-dUTP) | Enable non-radioactive, multiplex probe labeling for simultaneous detection of multiple loci. |
| Positively Charged Nylon Membrane | Binds negatively charged DNA after transfer; essential for subsequent hybridization steps. |
| Church's Hybridization Buffer | A phosphate-based buffer that allows for high-specificity hybridization at low probe concentrations. |
| Fluorescence Laser Scanner (e.g., Typhoon) | Detects and differentiates multiple fluorophores, providing digital quantification of band intensity. |
| 96-Well Magnetic Bead Purification Kit | Enables parallel, high-efficiency cleanup of restriction digests, removing enzymes and salts. |
| PicoGreen dsDNA Quantitation Reagent | A sensitive fluorescent dye for accurate post-digestion DNA quantification prior to loading. |
Southern Blotting in the Modern Lab: Validation Gold Standard and Comparative Analysis with NGS
Within the framework of DNA methylation analysis research, Southern blotting remains an indispensable, orthogonal validation tool. While high-throughput methods like whole-genome bisulfite sequencing (WGBS) and targeted techniques like Methylation-Specific PCR (MSP) offer sensitivity and scalability, they are prone to artifacts. Bisulfite conversion can be incomplete, leading to false-positive methylation calls, and PCR-based methods can suffer from primer bias and nonspecific amplification. This application note argues that Southern blotting, with its ability to assess methylation status based on restriction fragment length polymorphisms without chemical conversion or amplification, provides a critical gold-standard validation within a rigorous research thesis. It confirms the physical presence and size of methylated DNA fragments, anchoring next-generation sequencing and PCR data in robust, reproducible molecular evidence.
Quantitative Comparison of Methylation Analysis Techniques
The table below summarizes the core characteristics of the three techniques, highlighting the complementary role of Southern blotting.
Table 1: Comparative Analysis of Key DNA Methylation Detection Methods
| Parameter | Bisulfite Sequencing (BS-Seq) | Methylation-Specific PCR (MSP) | Southern Blotting for Methylation |
|---|---|---|---|
| Principle | Chemical conversion of unmethylated C to U, followed by sequencing. | PCR amplification with primers specific to methylated/unmethylated sequences after bisulfite conversion. | Restriction enzyme digestion (methylation-sensitive vs. -insensitive isoschizomers) and fragment size detection. |
| Throughput | Very High (genome-wide) | High (multiplexed targets) | Low (1-2 loci per blot) |
| Sensitivity | High (~1% allele frequency) | Very High (<0.1% allele frequency) | Moderate (requires ~5-10 µg genomic DNA) |
| Quantitative Output | Yes, percentage methylation per CpG. | Semi-quantitative (qMSP is quantitative). | Semi-quantitative (band intensity). |
| Key Artifacts/Risks | Incomplete bisulfite conversion, sequencing bias, DNA degradation. | Primer bias, incomplete conversion, false priming. | Partial digestion, incomplete transfer, probe specificity. |
| Primary Role | Discovery, genome-wide profiling. | Rapid screening, clinical diagnostics. | Validation, confirmation of novel findings. |
| DNA Requirement | Low (ng) | Very Low (ng) | High (µg) |
| Turnaround Time | Days to weeks | Hours | 3-7 days |
Detailed Protocols for Validation
Protocol A: Southern Blot Validation of WGBS-Idenitified DMRs
This protocol validates a differentially methylated region (DMR) identified via bisulfite sequencing.
I. Genomic DNA Digestion
- Prepare DNA: Isolate high-molecular-weight genomic DNA (>50 kb) from target tissue (e.g., 10 µg per reaction).
- Dual Digest: Set up two parallel digests for each sample using methylation-sensitive and -insensitive isoschizomers (e.g., HpaII (methylation-sensitive) and MspI (methylation-insensitive; cuts same CCGG sequence regardless of CpG methylation)). Include a no-enzyme control.
- Reaction Mix: 10 µg DNA, 5 µL 10X reaction buffer, 10 U HpaII or MspI, Nuclease-free water to 50 µL.
- Incubation: 37°C for 16 hours.
- Confirm Digestion: Run 1 µg of digested DNA on a 0.8% agarose gel to check for smear formation versus high-weight intact DNA.
II. Gel Electrophoresis and Blotting
- Load Gel: Electrophorese entire digested samples on a 0.8-1% agarose gel (20 cm length) at 25-30V for 16-20 hours with appropriate size markers.
- Depurinate & Denature: Soak gel in 0.25 M HCl for 15 min, then in denaturation solution (0.5 M NaOH, 1.5 M NaCl) for 30 min.
- Neutralize & Transfer: Neutralize in 1 M Tris-HCl (pH 7.4), 1.5 M NaCl for 30 min. Transfer DNA to a positively charged nylon membrane via capillary or vacuum blotting in 20X SSC.
III. Probe Preparation and Hybridization
- Probe Design: Design a ~500-800 bp digoxigenin (DIG)-labeled probe outside the HpaII/MspI restriction sites flanking the DMR. This ensures the fragment size detected depends on the methylation status within the region.
- Label Probe: Use the DIG-High Prime DNA Labeling and Detection Kit (Roche). Denature probe DNA (30 ng) and label per manufacturer’s instructions.
- Hybridize: Pre-hybridize membrane at 42°C for 1 hr in hybridization buffer. Add denatured DIG-labeled probe and hybridize at 42°C for 16 hours.
- Stringency Washes: Wash twice with 2X SSC/0.1% SDS at room temp, then twice with 0.5X SSC/0.1% SDS at 68°C.
- Detection: Perform immunological detection with anti-DIG-AP conjugate and chemiluminescent substrate (CSPD). Expose to X-ray film or digital imager.
IV. Interpretation: Compare HpaII and MspI digest patterns. A larger fragment in HpaII vs. MspI indicates CpG methylation at the internal site(s), confirming the BS-Seq data.
Protocol B: Southern Blot Confirmation of MSP Results
This protocol confirms the methylation status of a promoter region initially tested by MSP.
I. Restriction Enzyme Selection & Digestion
- Select Enzymes: Choose a restriction enzyme pair whose recognition site contains the CpG dinucleotide assessed by MSP (e.g., BstUI (CGCG), sensitive to methylation at the internal CpG).
- Perform Digest: Digest 10 µg of non-bisulfite-treated genomic DNA with BstUI and its methylation-insensitive counterpart (if available) or a frequent cutter (e.g., MseI) to generate a smaller control fragment. Include a mock digest.
II. Southern Blot Analysis
- Follow steps for gel electrophoresis, blotting, and hybridization as in Protocol A.
- Probe Design: Design a probe complementary to the sequence immediately adjacent to the restriction site interrogated by MSP.
- Interpretation: If the MSP indicated "methylated," the BstUI site should be resistant to cutting, resulting in a larger hybridizing fragment. If "unmethylated," digestion should yield a smaller predicted fragment. The mock digest confirms probe binding.
Visualized Workflows and Relationships
Title: Validation Workflow for Methylation Analysis
Title: Southern Blot Principle Using Isoschizomers
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Southern Blot Methylation Validation
| Reagent / Kit | Function & Role in Validation |
|---|---|
| Methylation-Sensitive Restriction Enzymes (HpaII, BstUI, SmaI) | Core reagent. Differential cutting based on CpG methylation status creates the polymorphism for blot detection. |
| Isoschizomer Pairs (HpaII/MspI, SmaI/XmaI) | Critical control. The insensitive enzyme reveals all potential cut sites, defining the baseline fragment pattern. |
| High-Purity Genomic DNA Isolation Kit | Provides long, intact DNA strands essential for accurate restriction mapping and minimizing partial digestion artifacts. |
| DIG-High Prime DNA Labeling & Detection Kit (Roche) | Standardized system for non-radioactive probe labeling, hybridization, and chemiluminescent detection. |
| Positively Charged Nylon Membrane | Robust solid support for immobilized DNA that withstands high-stringency washes and multiple probings. |
| Control DNA Sets (Fully Methylated & Unmethylated) | Essential positive and negative controls for both restriction digests and the primary method (BS-Seq/MSP) being validated. |
| Long-Range Agarose | Facilitates optimal separation of large DNA fragments (1-50 kb) generated by methylation-blocked digestion. |
| Capillary Blotting System | Provides even and efficient transfer of DNA from gel to membrane, crucial for quantitative band intensity analysis. |
Application Notes
In the context of a comprehensive thesis on DNA methylation analysis, Southern blotting remains a foundational technique with distinct advantages and constraints. Its application is critical for validating high-throughput sequencing data and for detailed investigation of specific genomic loci in research and drug development, particularly in oncology and imprinting disorders. The following notes contextualize its role.
- Direct Visualization as a Core Strength: Southern blotting provides a physical, direct visualization of DNA fragments hybridized with a locus-specific probe. This allows for the unambiguous confirmation of methylation-dependent restriction fragment length polymorphisms (RFLPs). It is considered a gold standard for validating epigenetic changes, such as promoter hypermethylation of tumor suppressor genes (e.g., MGMT, MLH1) or changes in triplet repeat methylation (e.g., FMR1 in Fragile X syndrome). The resulting autoradiogram or chemiluminescent image is a tangible, reproducible record.
- Locus-Specific Resolution and Quantitative Potential: The technique offers high specificity for a single, defined genomic locus determined by the probe sequence. When combined with appropriate scanning densitometry, it can yield semi-quantitative data on the relative proportion of methylated versus unmethylated alleles within a sample. This is vital for assessing mosaicism or heterogeneous methylation in tumor samples.
- Throughput Constraints in a Modern Context: The primary limitation is low throughput. The protocol is labor-intensive, requires large amounts of high-quality genomic DNA (typically 5-20 µg), and can take 1-2 weeks to complete. This makes it unsuitable for large-scale screening. Its modern application is thus focused on targeted, confirmatory analysis rather than discovery.
Quantitative Data Summary
Table 1: Comparative Metrics of Southern Blotting for Methylation Analysis
| Parameter | Typical Specification/Range | Notes |
|---|---|---|
| DNA Input Requirement | 5 - 20 µg | Must be high molecular weight (>20 kb), limiting use with degraded FFPE samples. |
| Time to Result | 6 - 10 days | Includes digestion, electrophoresis, blotting, hybridization, and washes. |
| Locus Multiplicity | 1 - 2 per blot | Limited by probe stripping/re-probing or multiple size markers. |
| Detection Sensitivity | ~1-5% allele fraction | Dependent on probe specificity and activity, and exposure time. |
| Semi-Quantitative Accuracy | ±5-10% | Achievable with careful densitometry of unsaturated signals. |
| Throughput (Samples/Week) | 10 - 40 | Highly variable based on laboratory setup and experience. |
Experimental Protocols
Protocol: Southern Blot Analysis of CpG Island Methylation Using Methylation-Sensitive Restriction Enzymes
I. Genomic DNA Digestion
- Reaction Setup: In separate tubes, digest 10 µg of genomic DNA (in 40 µL volume) with:
- Test Digest: 20-40 units of a methylation-sensitive enzyme (e.g., HpaII) and its methylation-insensitive isoschizomer (e.g., MspI) as a control for DNA quality and completeness of digestion. Note: Use 5-10x excess enzyme and extend incubation time (12-16 hours) for complete digestion.
- Reference Digest: A frequent-cutter (e.g., MseI) to generate small fragments for accurate size determination.
- Incubation: Incubate at recommended temperature (37°C for HpaII/MspI) overnight.
- Confirmation: Verify complete digestion by running 200 ng of each digest on a 1.5% agarose mini-gel.
II. Gel Electrophoresis and Membrane Transfer
- Gel Casting: Cast a large 0.8-1.0% agarose gel in 1x TAE buffer.
- Loading & Run: Load entire digested samples alongside a DNA molecular weight ladder (e.g., DIG-labeled or radioactively labeled). Run gel slowly at 1-2 V/cm for 14-16 hours to achieve optimal separation of large fragments (1-20 kb).
- Depurination (Optional): Soak gel in 0.25 M HCl for 15 minutes (for fragments >5 kb) to facilitate transfer.
- Denaturation & Neutralization: Soak gel in denaturation solution (0.5 M NaOH, 1.5 M NaCl) for 45 min, then in neutralization buffer (1 M Tris-HCl pH 7.5, 1.5 M NaCl) for 30 min.
- Capillary Transfer: Assemble a standard upward capillary transfer stack using a neutral nylon membrane (positively charged) and 20x SSC transfer buffer. Transfer for 16-24 hours.
- DNA Fixation: UV-crosslink DNA to the membrane (1200 J/cm²) or bake at 80°C for 1-2 hours.
III. Probe Labeling and Hybridization
- Probe Design: Design a ~500-1000 bp probe specific to the locus of interest, located between the restriction site and the next invariant site for the enzyme used.
- Labeling: Label probe with [α-³²P] dCTP using a random primed DNA labeling kit. Purify labeled probe using a spin column.
- Pre-hybridization: Pre-wet membrane in 2x SSC. Place in hybridization tube with 10-15 mL of pre-heated hybridization buffer (e.g., QuickHyb solution). Incubate at 65°C for 30 minutes with rotation.
- Hybridization: Add denatured, labeled probe (2-5 x 10⁶ cpm/mL) directly to the buffer. Hybridize at 65°C for 12-16 hours.
IV. Washing and Detection
- Stringency Washes: Perform two low-stringency washes (2x SSC, 0.1% SDS) at room temperature for 15 min each, followed by one or two high-stringency washes (0.1x SSC, 0.1% SDS) at 65°C for 20-30 min. Monitor radioactivity with a Geiger counter.
- Imaging: Wrap damp membrane in plastic wrap and expose to a phosphor storage screen at -80°C for 24 hours to 7 days. Scan the screen with a phosphorimager.
- Stripping (Optional): For re-probing, strip membrane by pouring boiling 0.1% SDS over it and agitating until cooled. Check for residual signal.
The Scientist's Toolkit
Table 2: Research Reagent Solutions for Southern Blot Methylation Analysis
| Item | Function & Rationale |
|---|---|
| Methylation-Sensitive Restriction Enzyme (e.g., HpaII, NotI) | Cuts only when its CpG recognition site is unmethylated, creating the methylation-dependent RFLP. |
| Methylation-Insensitive Isoschizomer (e.g., MspI, NdeI) | Cuts regardless of CpG methylation; control for DNA quality and complete digestion. |
| Positively Charged Nylon Membrane | Binds negatively charged DNA via salt bridges; robust for multiple stripping/re-probing cycles. |
| [α-³²P] dCTP or DIG-dUTP | Radioactive or non-radioactive label incorporated into the probe for high-sensitivity detection. |
| High-Specific Activity Random Primed DNA Labeling Kit | Efficiently generates labeled, single-stranded probe templates from a purified DNA fragment. |
| Phosphor Storage Screen & Imager | Detects and quantifies radioactivity (or chemiluminescence) with a linear dynamic range superior to X-ray film. |
| DNA Size Ladder (Labeled) | Critical for accurate determination of restriction fragment sizes on the final autoradiogram. |
| Hybridization Buffer (e.g., QuickHyb) | Formulated to accelerate hybridization kinetics, reducing protocol time from days to hours. |
Visualizations
Southern Blot Methylation Analysis Core Workflow
Methylation-Dependent Restriction Logic
1. Introduction & Thesis Context
Within the broader thesis investigating DNA methylation analysis using Southern blotting as a foundational technique, it is imperative to directly compare its merits and limitations against modern bisulfite conversion-based methods. This application note provides a detailed technical comparison, protocols, and resource toolkit for researchers navigating the selection of appropriate methodologies for epigenetic analysis in drug development and basic research.
2. Quantitative Comparison Summary
Table 1: Core Methodological Comparison
| Parameter | Southern Blotting | Bisulfite Conversion-Based Methods (e.g., PCR, Sequencing) |
|---|---|---|
| DNA Input | High (5-50 µg) | Low (50 ng - 1 µg) |
| Throughput | Very Low (1-10 samples/week) | Medium to Very High (96-1000s samples/week) |
| Resolution | Low (Restriction fragment level, ~100-1000s bp) | Single-Nucleotide Resolution |
| Quantitative Accuracy | Semi-Quantitative | High (especially with sequencing) |
| Genomic Coverage | Targeted (locus-specific) | Targeted to Genome-Wide |
| Key Application | Methylation-dependent restriction fragment analysis, imprinted gene analysis, repeat element methylation (global view). | High-resolution methylation mapping, biomarker discovery, epigenetic profiling. |
Table 2: Performance Metrics
| Metric | Southern Blotting | Bisulfite Conversion-Based Methods |
|---|---|---|
| Time to Result | 5-7 days | 2-4 days |
| Cost per Sample | Low to Moderate ($50-$200) | Moderate to High ($100-$1000+) |
| Bisulfite Conversion Efficiency | Not Applicable | >99% required for accuracy |
| Ability to Detect Hemimethylation | Yes (via differential digestion) | No (strands are analyzed separately post-conversion) |
| Suitability for Degraded DNA | Poor (requires high MW DNA) | Moderate (compatible with FFPE DNA) |
3. Detailed Experimental Protocols
Protocol A: Southern Blotting for Methylation Analysis (Methylation-Sensitive Restriction Digest)
Objective: To analyze CpG island methylation status at a specific genomic locus. Workflow:
- Genomic DNA Digestion: Digest 10 µg of high-molecular-weight DNA overnight with a methylation-sensitive restriction enzyme (e.g., HpaII) and its methylation-insensitive isoschizomer (e.g., MspI) in parallel reactions.
- Agarose Gel Electrophoresis: Separate digested DNA fragments on a 0.8-1.2% agarose gel.
- Depurination, Denaturation & Neutralization: Treat gel with 0.25M HCl, then alkaline solution (0.5M NaOH, 1.5M NaCl) to denature DNA, followed by neutralization buffer.
- Capillary Transfer: Use high-salt transfer buffer (20x SSC) to blot DNA onto a positively charged nylon membrane overnight.
- UV Crosslinking: Fix DNA to membrane using UV light.
- Hybridization: Pre-hybridize membrane, then hybridize with a locus-specific, radiolabeled or digoxigenin-labeled DNA probe at optimized temperature.
- Stringency Washes: Wash membrane with buffers of decreasing ionic strength (e.g., 2x SSC to 0.1x SSC with SDS).
- Detection: For radioactive probes, expose to phosphorimager screen. For DIG-labeled probes, perform chemiluminescent detection with anti-DIG-AP and CSPD substrate.
Protocol B: Bisulfite Conversion & Subsequent PCR Analysis
Objective: To convert unmethylated cytosines to uracils for subsequent locus-specific methylation analysis. Workflow:
- DNA Denaturation: Incubate 500 ng of DNA in 0.3M NaOH at 42°C for 20 minutes.
- Sulfonation: Add sodium bisulfite (final concentration 3.1M) and hydroquinone (final 0.5mM). Incubate in the dark under mineral oil at 55°C for 16 hours. (Critical step: protect from light).
- Desalting & Clean-up: Use commercial spin column-based cleanup systems designed for bisulfite-treated DNA. Elute in 10-20 µL.
- Desulfonation: Treat cleaned-up product with 0.3M NaOH at room temperature for 15 minutes. Neutralize and precipitate or clean up again.
- Bisulfite-Specific PCR (BSP): Design primers specific to the bisulfite-converted strand (avoiding CpG sites). Amplify using a polymerase robust to uracil-rich templates.
- Analysis: Clone PCR products and sequence multiple clones, or subject to direct pyrosequencing for quantitative methylation percentages at each CpG site.
4. Visualized Workflows & Pathways
Diagram 1: Comparative Workflows for Methylation Analysis
Diagram 2: Bisulfite Conversion Chemistry
5. The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents & Materials
| Item | Function | Example/Critical Feature |
|---|---|---|
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII, NotI) | Cleave only at unmethylated recognition sites, enabling differential fragment analysis. | Must be used with isoschizomer control (MspI for HpaII). |
| Positively Charged Nylon Membrane | Binds negatively charged DNA after Southern transfer for probe hybridization. | High nucleic acid binding capacity and strength for repeated probing. |
| Locus-Specific Hybridization Probes | Detects specific DNA fragments of interest on the membrane. | Radiolabeled (³²P) or non-radioactive (DIG, biotin) for detection. |
| Sodium Bisulfite (≥99% purity) | Key chemical for deaminating unmethylated cytosine to uracil. | High purity is essential for complete conversion and minimal DNA degradation. |
| DNA Clean-up Kits (Bisulfite-specific) | Purifies bisulfite-converted DNA, removing salts and reaction inhibitors. | Optimized for low-concentration, single-stranded DNA recovery. |
| Bisulfite-Specific Polymerase | Amplifies bisulfite-converted, uracil-rich templates with high fidelity. | Must lack uracil-excision activity (e.g., Taq Gold, ZymoTaq). |
| Pyrosequencing System & Reagents | Provides quantitative methylation percentage at sequential CpG sites post-BSP. | Requires sequencing primer, enzymes, and substrates for luciferase-based detection. |
| High-Percentage Agarose | For resolution of large DNA fragments (1-20 kb) in Southern blotting. | Suitable for preparing gels for genomic DNA separation. |
Complementary Role with Genome-Wide Analyses (e.g., Whole-Genome Bisulfite Sequencing).
Application Notes Within the framework of a thesis investigating locus-specific DNA methylation via Southern blotting, genome-wide techniques like Whole-Genome Bisulfite Sequencing (WGBS) serve a critical complementary role. Southern blotting provides definitive, quantitative validation of methylation states at specific, often complex loci (e.g., imprinted regions, expanded repeats) with low technical artifact. However, it is low-throughput and requires prior locus knowledge. WGBS provides an unbiased, base-resolution map of the entire methylome, enabling the discovery of novel differentially methylated regions (DMRs) that can subsequently be validated by Southern blot. This integrated, hierarchical approach maximizes both discovery power and validation rigor. Key quantitative comparisons are summarized in Table 1.
Table 1: Comparative Analysis of Southern Blotting and WGBS
| Feature | Southern Blotting | Whole-Genome Bisulfite Sequencing (WGBS) |
|---|---|---|
| Throughput | Low (1-5 loci per blot) | High (genome-wide) |
| Resolution | Restriction fragment (100bp-10kbp) | Single-base pair |
| Quantification | High (Densitometry of bands) | High (Read depth-dependent) |
| Prior Locus Knowledge Required | Yes | No |
| Primary Utility | Targeted validation, complex repeats, allele-specific analysis | Discovery, genome-wide screening, novel DMR identification |
| Typical DNA Input | 5-20 µg (non-amplified) | 50-200 ng (bisulfite-converted, often amplified) |
| Assay Cost per Sample | Low | Very High |
| Key Limitation | Cannot discover novel loci | Validation required; struggles with highly repetitive regions |
Protocols
Protocol 1: Complementary Experimental Workflow for Integrated Methylation Analysis Purpose: To detail a hierarchical strategy using WGBS for discovery and Southern blotting for validation.
- Sample Preparation: Isolate high-molecular-weight genomic DNA from experimental and control samples (e.g., diseased vs. healthy tissue, treated vs. untreated cell lines). Divide each sample into two aliquots: one for WGBS and one for Southern blotting.
- WGBS Discovery Phase: a. Subject the first aliquot to sodium bisulfite conversion using a kit (e.g., EZ DNA Methylation-Lightning Kit). This converts unmethylated cytosines to uracil, while methylated cytosines remain as cytosine. b. Prepare a sequencing library from the converted DNA, including steps for size selection and PCR amplification with index primers. c. Perform paired-end sequencing on an Illumina platform to a recommended depth of 20-30x genome coverage. d. Align reads to a bisulfite-converted reference genome using tools like Bismark or BS-Seeker2. e. Perform differential methylation analysis (e.g., using methylKit or DSS) to identify significant DMRs between sample groups.
- Southern Blot Validation Phase: a. Design a validation strategy for 2-3 top candidate DMRs from WGBS. Select methylation-sensitive restriction enzymes (MSREs, e.g., HpaII, NotI) whose recognition sites are located within the identified DMR. b. Digest 10 µg of the second, unconverted genomic DNA aliquot with the selected MSRE and a methylation-insensitive isoschizomer (e.g., MspI for HpaII) as a control for complete digestion. c. Run digested DNA on a 0.8-1.2% agarose gel overnight for optimal separation, then perform capillary transfer to a nylon membrane. d. Generate a radiolabeled (³²P or ³³P) or non-radioactive (digoxigenin) DNA probe specific to the target DMR locus via PCR or restriction fragment isolation. e. Hybridize the probe to the membrane, wash under stringent conditions, and visualize bands via autoradiography or chemiluminescence. f. Quantify methylation percentage by comparing band intensities from the MSRE digest (cleaved=unmethylated, uncleaved=methylated) to the control digest pattern.
Protocol 2: Validation-Specific Southern Blot for a WGBS-Identified DMR Purpose: To validate a candidate hypermethylated region in a promoter discovered by WGBS.
- Enzymatic Digestion: Set up three parallel 20 µL reactions for each genomic DNA sample (10 µg):
- Reaction A: 5 units/µg EcoRI (methylation-insensitive cutter to define fragment boundaries).
- Reaction B: EcoRI + 10 units/µg HpaII (MSRE, cuts CCGG only when internal C is unmethylated).
- Reaction C: EcoRI + 10 units/µg MspI (cuts CCGG regardless of methylation; digestion control). Incubate at 37°C for 16 hours.
- Gel Electrophoresis & Transfer: Load digested samples on a 1.0% agarose gel in 1x TAE at 25V for 16 hours. Depurinate, denature, and neutralize the gel. Perform upward capillary transfer with 20x SSC buffer onto a positively charged nylon membrane for 18 hours.
- Probe Preparation & Hybridization: Label a 500-bp PCR product spanning the DMR locus with [α-³²P]dCTP using a random primer labeling kit. Purify the probe using a spin column. Pre-hybridize the membrane at 65°C for 1 hour in Church buffer (0.5M NaHPO₄ pH 7.2, 7% SDS, 1mM EDTA). Add denatured probe and hybridize at 65°C for 16 hours.
- Washing & Detection: Wash membrane sequentially with 2x SSC/0.1% SDS (room temp), 0.5x SSC/0.1% SDS (65°C), and 0.1x SSC/0.1% SDS (65°C), 15 minutes each. Expose membrane to a phosphorimager screen for 24-72 hours and scan.
Diagrams
Hierarchical Methylation Analysis Workflow
Southern Blot Validation Protocol Steps
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Material | Function in Complementary Analysis |
|---|---|
| Methylation-Sensitive Restriction Enzymes (e.g., HpaII, NotI) | Core reagents for Southern blotting; cleave DNA only at unmethylated recognition sites, enabling fragment pattern analysis. |
| Sodium Bisulfite Conversion Kit (e.g., EZ DNA Methylation-Lightning) | Essential for WGBS; chemically converts unmethylated cytosine to uracil, allowing differentiation via sequencing. |
| Positively Charged Nylon Membrane | Solid support for Southern blotting; irreversibly binds negatively charged DNA after alkaline transfer. |
| High-Fidelity PCR Kit | Used to generate both Southern blot probes and sequencing libraries from bisulfite-converted DNA. |
| [α-³²P]dCTP or Digoxigenin (DIG) Labeling Kit | Provides high-sensitivity detection methods for Southern blot probes. Radiolabeling offers highest sensitivity; DIG is a safer alternative. |
| Phosphorimager Screen & Scanner / Chemiluminescence Imager | Critical for capturing and quantifying signal from radiolabeled or chemiluminescent Southern blots. |
| WGBS-Specific Bioinformatics Tools (e.g., Bismark, methylKit) | Software packages for aligning bisulfite-seq reads and performing differential methylation analysis to generate candidate DMRs. |
Within the evolving landscape of molecular diagnostics, Southern blotting maintains a critical, niche role, particularly for complex DNA structural analyses where sequence specificity and large-fragment resolution are paramount. This application note contextualizes Southern blotting within modern diagnostic assay development, focusing on its enduring value for the analysis of DNA methylation—a key epigenetic marker in oncology, imprinting disorders, and neurodegenerative disease. While high-throughput methods (e.g., bisulfite sequencing, arrays) dominate discovery, Southern blotting provides an orthogonal, gold-standard validation tool with direct applicability in clinical assay verification and regulatory submissions due to its quantitative nature and visual confirmation of specific fragment sizes.
Current Regulatory Landscape and Southern Blotting
Regulatory bodies (FDA, EMA) require robust analytical validation of diagnostic assays, emphasizing specificity, sensitivity, accuracy, precision, and limit of detection. Southern blotting addresses key validation parameters for methylation-dependent assays.
Table 1: Alignment of Southern Blotting Performance with Regulatory Validation Parameters
| Validation Parameter | Southern Blotting Application | Typical Quantitative Benchmark |
|---|---|---|
| Analytical Specificity | Discrimination of methylated vs. unmethylated alleles via methylation-sensitive restriction enzymes (MSREs). | Near 100% when using high-fidelity enzymes and stringent hybridization. |
| Analytical Sensitivity | Detection of low-abundance methylated alleles in a background of normal DNA. | ~1-5% allele frequency (using phosphorimager analysis). |
| Precision (Repeatability) | Inter-assay and intra-assay consistency of restriction fragment size and signal intensity. | CV of <15% for band intensity quantification. |
| Accuracy | Concordance with a reference method (e.g., MLPA, bisulfite pyrosequencing). | >95% concordance reported in validated assays. |
| Limit of Detection (LoD) | Minimum input DNA required for reliable detection. | 50 ng - 1 µg of genomic DNA, depending on locus and probe sensitivity. |
Application Note: Validation ofFMRIGene CGG Repeat Methylation in Fragile X Syndrome
Fragile X syndrome is a canonical example where Southern blotting remains the definitive diagnostic technique. It simultaneously determines CGG repeat expansion size and methylation status, which is critical for diagnosis and classification.
Key Protocol: Southern Blot Analysis of FMRI Methylation Using Methylation-Sensitive Restriction Digestion
I. Sample Preparation & Restriction Digest
- Isolate genomic DNA from patient peripheral blood (minimum 500 ng).
- Perform a dual restriction enzyme digest:
- EcoRI: Rare-cutter to generate a large, manageable fragment containing the CGG repeat region.
- EagI (or another methylation-sensitive enzyme): Its recognition site (CGGCCG) is located near the CGG repeat. Cleavage is blocked by CpG methylation.
- Digest Setup: Combine 500 ng DNA, 20 U EcoRI, 20 U EagI, appropriate buffer, and nuclease-free water to 50 µL. Incubate at 37°C for 16 hours.
II. Gel Electrophoresis and Blotting
- Resolve digested DNA on a 0.8% agarose gel (20 cm length) at 1 V/cm for 24 hours for optimal separation of large fragments (5-10 kb).
- Depurinate, denature, and neutralize the gel in standard Southern protocols.
- Transfer DNA to a positively charged nylon membrane via capillary or vacuum transfer.
III. Probe Labeling and Hybridization
- Prepare a digoxigenin (DIG)-labeled probe specific to the FMRI region (e.g., StB12.3 plasmid).
- Hybridize the membrane with the probe at 42°C in a standardized hybridization buffer for 16 hours.
- Perform stringent washes to remove non-specifically bound probe.
IV. Detection and Analysis
- Use an anti-DIG antibody conjugated to alkaline phosphatase.
- Develop signal using a chemiluminescent substrate (e.g., CDP-Star) and capture on a CCD imager.
- Interpretation: Methylated, full-mutation alleles are protected from EagI digestion, appearing as a larger, smeared band (>5.8 kb). Normal, unmethylated alleles are cut by EagI, appearing as a sharp band at 2.8 kb.
Diagram Title: FMR1 Southern Blot Diagnostic Workflow
The Scientist's Toolkit: Key Reagents for Methylation-Specific Southern Blotting
Table 2: Essential Research Reagent Solutions
| Reagent/Category | Specific Example(s) | Function in Assay |
|---|---|---|
| Methylation-Sensitive Restriction Enzymes (MSREs) | EagI, HpaII, NotI, SacII | Cleave only at unmethylated CpG sites, enabling discrimination of methylation status. |
| Rare-Cutter Restriction Enzymes | EcoRI, HindIII, BamHI | Generate large genomic fragments containing the locus of interest for Southern analysis. |
| High-Quality Genomic DNA Isolation Kits | Phenol-chloroform, Column-based kits (Qiagen DNeasy) | Provide high-molecular-weight, pure DNA essential for restriction digestion. |
| Nylon Membranes (Positively Charged) | Hybond-N+, Nytran Nylon | Bind DNA irreversibly after transfer for repeated probing. |
| Non-Radioactive Labeling & Detection Systems | DIG-High Prime DNA Labeling, Chemiluminescent Substrates (CDP-Star) | Safe, sensitive alternative to 32P for probe labeling and signal generation. |
| Methylation-Specific Probes | Locus-specific PCR amplicons, Cloned plasmid DNA (e.g., StB12.3) | Provide the sequence-specificity for hybridization to the target allele. |
| Stringent Wash Buffers | Solutions with precise molarity of SSC and SDS | Remove mismatched or non-specifically bound probe to ensure high specificity. |
Detailed Protocol: Southern Blot for Imprinting Disorder Analysis (e.g.,SNRPNLocus)
This protocol details steps for analyzing allele-specific methylation, critical for diagnosing Prader-Willi and Angelman syndromes.
A. Restriction Digestion and Electrophoresis
- Digest 1 µg of genomic DNA with 20 U each of HindIII (rare cutter) and HpaII (MSRE) overnight at 37°C in a 50 µL reaction.
- Include a control digest with MspI (methylation-insensitive isoschizomer of HpaII) to confirm complete digestion capability.
- Load digested DNA alongside a molecular weight ladder on a 1.0% agarose gel. Run at 25V for 16-18 hours.
B. Southern Transfer
- After electrophoresis, depurinate the gel in 0.25 M HCl for 15 min with gentle agitation.
- Denature DNA in 0.5 M NaOH / 1.5 M NaCl for 30 min.
- Neutralize in 0.5 M Tris-HCl (pH 7.4) / 1.5 M NaCl for 30 min.
- Set up a standard upward capillary transfer using 20x SSC buffer, transferring DNA to a positively charged nylon membrane for 18-24 hours.
C. Probe Preparation and Hybridization
- Label 25 ng of a SNRPN-specific PCR product with DIG using the DIG-High Prime kit (Roche). Purify using a spin column.
- Pre-hybridize the membrane at 42°C for 1 hour in DIG Easy Hyb solution.
- Denature the labeled probe at 95°C for 5 min, chill on ice, and add to fresh pre-warmed DIG Easy Hyb.
- Hybridize at 42°C for 16 hours.
D. Post-Hybridization Washes and Detection
- Perform two low-stringency washes at room temperature: 2x SSC, 0.1% SDS for 5 min each.
- Perform two high-stringency washes at 68°C: 0.1x SSC, 0.1% SDS for 15 min each.
- Proceed with standard DIG immunodetection: Block, incubate with Anti-DIG-AP (1:10,000), wash, and incubate with chemiluminescent substrate.
- Expose to a CCD imager for 5-30 minutes.
Diagram Title: Methylation-Sensitive Restriction Enzyme Logic
Southern blotting, though technically demanding, offers unparalleled analytical specificity for DNA methylation analysis in a diagnostic context. Its ability to provide semi-quantitative, size-resolved data on specific loci makes it an indispensable tool for validating next-generation sequencing assays, resolving ambiguous cases, and serving as a primary diagnostic for well-characterized disorders with methylation-based etiologies. Its role is firmly entrenched in the regulatory pathway as a confirmatory method, ensuring the accuracy and reliability of molecular diagnostics.
Within the context of advancing DNA methylation analysis, traditional Southern blotting remains a cornerstone for validating genome-wide bisulfite sequencing or array data, providing a quantitative, locus-specific measure of methylation status. This application note details protocols for modernizing this technique through high-resolution digital imaging and semi-automated analysis, enhancing reproducibility, throughput, and data objectivity for research and drug development applications.
Core Modernization Components
Digital Imaging Systems
Replacing traditional X-ray film with digital CCD (charge-coupled device)-based systems offers superior linear dynamic range, quantitative accuracy, and immediate data digitization. Key parameters for system selection are summarized below.
Table 1: Comparison of Detection Modalities for Southern Blot Analysis
| Parameter | X-Ray Film (Classic) | Phosphor Storage Screens | CCD-Based Digital Imagers |
|---|---|---|---|
| Dynamic Range | ~200:1 | Up to 10^5:1 | Up to 10^4:1 |
| Quantitation | Non-linear, manual densitometry | Linear, software-based | Linear, integrated software |
| Sensitivity | High (low background) | Very High (10-100x film) | High (comparable to screens) |
| Workflow Speed | Slow (hours-days exposure) | Medium (minutes-hours) | Fast (seconds-minutes) |
| Data Format | Analog (requires scanning) | Digital (direct) | Digital (direct) |
| Primary Use Case | Legacy protocols | High-sensitivity radioactive probes | Fluorescent/chemiluminescent probes |
Semi-Automated Analysis Software
Specialized software enables lane/fragment detection, background subtraction, and methylation percentage calculation without manual tracing, reducing inter-operator variability.
Table 2: Software Features for Semi-Automated Southern Blot Analysis
| Software Module | Function | Key Metric |
|---|---|---|
| Lane Detection | Automatically identifies lanes and bands. | Accuracy (% of lanes correctly identified) |
| Band Detection | Detects band boundaries, quantifies intensity. | Precision (CV of repeated measurements) |
| Background Correction | Subtracts local and global background. | Signal-to-Noise Ratio improvement (fold-change) |
| Methylation Calculation | Computes % methylation from digested/undigested band intensities. | Reproducibility (R² of technical replicates) |
| Data Export | Exports results to .csv or integrated LIMS. | Time saved vs. manual analysis (minutes per blot) |
Detailed Protocol: Southern Blot for Methylation Analysis with Digital Workflow
Protocol 3.1: DNA Digestion and Blotting
- Objective: To assess CpG island methylation at a specific locus using methylation-sensitive restriction enzymes (e.g., HpaII).
- Reagents: Genomic DNA (2-10 µg), methylation-sensitive restriction enzyme (HpaII), isoschizomer insensitive to methylation (MspI), standard agarose gel electrophoresis reagents, nylon membrane, 20x SSC buffer.
- Procedure:
- Digest duplicate samples of genomic DNA (2 µg each) overnight with HpaII (sensitive) and MspI (insensitive control) following manufacturer guidelines.
- Run digested DNA on a 1% agarose gel alongside a molecular weight ladder. Include a fully methylated and unmethylated control DNA if available.
- Depurinate, denature, and neutralize gel per standard Southern protocol.
- Transfer DNA to a positively charged nylon membrane via capillary transfer with 20x SSC overnight.
- UV-crosslink DNA to the membrane (120 mJ/cm²).
Protocol 3.2: Non-Radioactive Probe Labeling & Hybridization
- Objective: To generate a sensitive, stable probe compatible with digital imaging.
- Reagents: Locus-specific PCR primers, PCR mix, DIG-High Prime DNA Labeling Kit (Roche), hybridization buffer, wash buffers, anti-DIG-AP antibody, CDP-Star or similar chemiluminescent substrate.
- Procedure:
- Amplify probe template (200-500 bp from target locus) via PCR.
- Label 30-50 ng of purified PCR product using the DIG-High Prime kit (3-hour incubation).
- Pre-hybridize membrane at 42°C for 1 hour in suitable buffer.
- Denature labeled probe (95°C, 5 min), add to fresh buffer, and hybridize overnight at 42°C.
- Perform stringency washes (2x SSC/0.1% SDS to 0.1x SSC/0.1% SDS) at 68°C.
- Block membrane, incubate with anti-DIG-AP antibody (1:10,000, 30 min), wash.
- Incubate with chemiluminescent substrate (CDP-Star) for 5 minutes.
Protocol 3.3: Digital Image Acquisition & Semi-Automated Analysis
- Objective: To acquire a quantitative digital image and analyze methylation percentage.
- Reagents/Equipment: Digital imager (e.g., Azure Sapphire, Bio-Rad ChemiDoc, or equivalent), analysis software (e.g., ImageLab, ImageJ with FIJI macros, or ACD/ChemGen).
- Procedure:
- Acquisition: Place membrane in imager. Acquire image using high-resolution chemiluminescence settings. Ensure no pixel saturation (critical for quantitation). Export image as 16-bit TIFF.
- Semi-Automated Analysis (Generic Workflow):
- Import TIFF file into analysis software.
- Define lanes: Use auto-detect function, manually adjust if necessary.
- Detect bands: Software identifies peaks corresponding to undigested (higher molecular weight, methylated) and digested (lower molecular weight, unmethylated) fragments.
- Background subtraction: Apply rolling ball or local background correction.
- Quantify integrated intensity for each band.
- Calculate percentage methylation: For each sample lane, use formula:
[Intensity (Undigested Band) / (Intensity (Undigested) + Intensity (Digested))] * 100. Compare HpaII digest to MspI control digest to confirm complete digestion efficiency.
- Export data table for statistical analysis.
Visualizing the Integrated Workflow
Diagram Title: Modern Southern Blot Workflow for Methylation
Diagram Title: Semi-Automated Image Analysis Pipeline
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Digital Southern Blotting
| Item | Function | Example/Note |
|---|---|---|
| Methylation-Sensitive Enzymes | Differential digestion based on CpG methylation status. | HpaII (sensitive) / MspI (insensitive) pair. |
| Positively Charged Nylon Membrane | Binds negatively charged DNA after transfer; durable for re-probing. | Amersham Hybond-N+, Roche Nylon Membranes. |
| Non-Radioactive Labeling Kit | Safe, stable probe labeling for chemiluminescence. | DIG-High Prime (Roche), ECL Direct (Cytiva). |
| Chemiluminescent Substrate | Enzymatic trigger for light emission captured by CCD. | CDP-Star (Roche), LumiGLO (Cell Signaling). |
| High-Resolution Digital Imager | Captures quantitative light signal from membrane. | Azure Sapphire, Bio-Rad ChemiDoc MP. |
| Analysis Software | Performs lane/band detection and quantification. | ImageLab (Bio-Rad), Fiji/ImageJ with Gel Analyzer. |
| Control DNAs | Essential for assay validation and normalization. | Commercially available universally methylated/unmethylated human DNA. |
Conclusion
Southern blotting remains a vital, robust, and unambiguous technique for DNA methylation analysis, particularly for validating results from high-throughput but indirect methods. Its strength lies in providing direct, physical evidence of methylation status at specific loci without chemical conversion artifacts. For foundational epigenetic research, studies of repeat elements, imprinting disorders, and as a gold standard for clinical assay validation, it holds an irreplaceable niche. While not suited for genome-wide discovery, its role is evolving towards one of essential confirmation and quantitative precision. Future directions involve integrating its reliable output with digital quantification tools and leveraging its clarity in the complex landscape of epigenetic drug development and diagnostic standardization, ensuring this classic method continues to inform cutting-edge biomedical science.