siRNA for Targeted Gene Knockdown: From Design Principles to Therapeutic Applications
This article provides a comprehensive resource for researchers and drug development professionals on the application of small interfering RNA (siRNA) for targeted gene silencing.
siRNA for Targeted Gene Knockdown: From Design Principles to Therapeutic Applications
Abstract
This article provides a comprehensive resource for researchers and drug development professionals on the application of small interfering RNA (siRNA) for targeted gene silencing. It covers the foundational mechanism of RNA interference and explores the latest advances in siRNA design, delivery platforms, and optimization strategies to enhance efficacy and minimize off-target effects. A strong emphasis is placed on robust validation methodologies and comparative analysis of different siRNA formats. The content also discusses the translation of siRNA technology from a research tool to a growing class of therapeutics, addressing both current successes and ongoing challenges in the field.
The siRNA Revolution: Understanding the Core Mechanism and Therapeutic Potential
RNA interference (RNAi) is a highly conserved biological mechanism that facilitates post-transcriptional gene silencing across diverse eukaryotic organisms, including plants, insects, and mammals [1] [2]. This pathway utilizes small non-coding RNA molecules to direct the sequence-specific silencing of complementary messenger RNA (mRNA) targets. The most well-studied RNAi molecules are small interfering RNAs (siRNAs) and microRNAs (miRNAs), which operate through related but distinct pathways to regulate gene expression [1]. Since the initial discovery of RNAi in Caenorhabditis elegans in 1998, and the subsequent awarding of the Nobel Prize in Physiology or Medicine in 2006 to the scientists who elucidated this mechanism, RNAi technology has evolved into a powerful tool for genetic research and therapeutic development [3] [2].
Therapeutically, siRNA-based approaches have gained significant momentum following regulatory approval of the first siRNA drug, patisiran, in 2018 for the treatment of hereditary transthyretin-mediated amyloidosis [3]. Since then, several additional siRNA therapeutics have received approval (givosiran, lumasiran, inclisiran, vutrisiran, and nedosiran), with over 260 siRNA drug candidates currently in preclinical or clinical development across therapeutic areas including cancer, infectious diseases, neurological conditions, cardiovascular disorders, and diabetes [3] [1]. The programmable nature of siRNA molecules, where target specificity is determined primarily by the guide strand sequence, makes them exceptionally versatile tools for selectively silencing disease-associated genes previously considered "undruggable" [4].
The Core Mechanism: From siRNA Delivery to mRNA Degradation
siRNA Structure and Cellular Uptake
Synthetic siRNAs are typically 21-25 nucleotide double-stranded RNA duplexes with 3' dinucleotide overhangs on both strands [3] [5]. The duplex consists of two complementary strands: the antisense (guide) strand, which ultimately directs target recognition, and the sense (passenger) strand, which is degraded during RISC activation [5]. For experimental applications, siRNAs are most commonly generated through solid-phase chemical synthesis methods that yield highly pure, stable oligonucleotides that can be readily chemically modified to enhance their properties [5].
A critical challenge in siRNA applications lies in achieving efficient intracellular delivery. Naked, unmodified siRNAs face substantial barriers including rapid degradation by ubiquitous ribonucleases in biological fluids, renal clearance, inefficient cellular uptake due to their negative charge, and potential immunogenicity [3] [2]. Multiple delivery strategies have been developed to overcome these limitations:
- Transfection: Cationic liposomes or polymers form complexes with negatively charged siRNA, facilitating cellular uptake through endocytosis. While effective for many cell types, not all cells are amenable to transfection reagents [5].
- Electroporation: Application of an electrical pulse creates temporary pores in the cell membrane, allowing siRNA entry. This method is effective for difficult-to-transfect cells but may increase cell death [5].
- Viral-Mediated Delivery: Engineered viral vectors (lentivirus, retrovirus, adeno-associated virus) enable efficient delivery and stable expression in difficult-to-transfect cells and in vivo applications, though they may trigger antiviral responses and require specialized facilities [5].
- Chemically Modified siRNA: Incorporation of strategic chemical modifications (e.g., 2'-O-methyl, 2'-fluoro groups) can enhance nuclease resistance and enable passive cellular uptake in some cases without transfection reagents [3] [5].
RISC Assembly and Activation
Once inside the cytoplasm, the siRNA duplex undergoes a carefully orchestrated process of RISC assembly and activation [6]. The double-stranded siRNA is recognized and loaded into the multi-protein RNA-induced silencing complex (RISC). Central to RISC function is the Argonaute 2 (AGO2) protein, an endonuclease that serves as the catalytic engine of the complex [3] [6] [7].
During RISC activation, the siRNA duplex is unwound in an ATP-independent manner, and the passenger strand is selectively ejected and degraded. The guide strand is retained by AGO2 to form the mature, single-stranded siRNA-RISC complex [3] [1]. The selection of which strand serves as the guide is influenced by the relative thermodynamic stability of the duplex ends, with the strand whose 5' end is less stably paired preferentially loaded as the guide [1]. Recent research has highlighted that the specific guide-RNA sequence can significantly impact the kinetics of RISC activation and subsequent target cleavage, with different sequences exhibiting up to 250-fold variations in slicing rates [8].
Target Recognition and mRNA Cleavage
The mature RISC complex, guided by the siRNA strand, scans cellular mRNAs for complementary sequences [6]. When the guide RNA pairs with its target mRNA through Watson-Crick base pairing, predominantly within the seed region (nucleotides 2-8), the AGO2 protein catalyzes site-specific endonucleolytic cleavage of the mRNA [3]. This cleavage occurs between nucleotides corresponding to positions 10 and 11 relative to the 5' end of the guide strand [3].
The cleaved mRNA fragments are then rapidly degraded by cytoplasmic exonucleases, effectively preventing translation and abrogating protein synthesis [3]. The RISC complex itself can subsequently engage in multiple rounds of target identification and cleavage, amplifying the gene silencing effect from a single siRNA-RISC complex [6].
The following diagram illustrates the complete siRNA-mediated gene silencing pathway:
Quantitative Analysis of siRNA Efficacy Factors
Multiple factors influence the efficacy of siRNA-mediated gene silencing. The following table summarizes key sequence and structural features that impact silencing efficiency, based on systematic analyses:
Table 1: Key Features Impacting siRNA Efficacy and Specificity
| Feature | Impact on Efficacy | Optimal Characteristics | Rationale |
|---|---|---|---|
| Guide Strand Sequence | High variation in silencing efficiency [8] | Sequences enabling faster RISC slicing kinetics [8] | Different guide sequences alter slicing rates by >250-fold; faster slicing improves knockdown [8] |
| Central Pairing Stability | Impacts 3'-mismatch tolerance [8] | Moderate stability; AU-rich centers have weak activity and require extensive 3' complementarity [8] | Guides with weak central pairing require extensive 3' complementarity to populate slicing-competent conformation [8] |
| Seed Region (nt 2-8) | Critical for target recognition; major source of off-target effects [5] | Avoid high complementarity to non-target transcripts [5] | Seed region homology can cause unintended silencing of transcripts with partial complementarity [5] |
| GC Content | Moderate impact [4] | <60% [4] | High GC content can negatively impact silencing efficiency and increase off-target effects [4] |
| Chemical Modification Pattern | Significant impact on stability and activity [4] | High 2'-O-methyl content improves nuclease resistance [4] | Modification pattern significantly impacts efficacy more than structural features; essential for in vivo stability [4] |
| Duplex Structure | Limited impact [4] | Asymmetric (3' overhangs) or blunt; sequence-dependent effects [4] | Structural features (symmetric vs. asymmetric) show minimal impact on efficacy compared to sequence and modification [4] |
Experimental Protocol: Assessing siRNA-Mediated Gene Silencing
This protocol describes a standardized methodology for evaluating siRNA efficacy in mammalian cell cultures, incorporating design considerations, delivery optimization, and functional validation.
siRNA Design and Preparation
Target Sequence Selection:
- Identify 19-21 nucleotide target sequences within the mRNA of interest, typically within the open reading frame (ORF) or 3' untranslated region (3' UTR) [4].
- Apply design algorithms (e.g., SMARTselection) that incorporate rules for minimizing off-target effects: avoid sequences with ≥60% GC content, exclude CCCC or GGGG stretches, and avoid high-frequency seed sequences from known mammalian microRNAs [4] [5].
- BLAST potential sequences against the appropriate genome database to ensure specificity for the target gene.
- Recommendation: Design and test 3-4 individual siRNAs targeting different regions of the mRNA to identify the most effective sequence [5].
Control siRNAs:
- Positive Control: A well-characterized siRNA targeting a ubiquitously expressed gene (e.g., GAPDH, ACTB) to validate delivery efficiency and silencing capability [5].
- Negative Control: A scrambled sequence siRNA with no significant homology to any known genes in the target organism, or a siRNA targeting a non-human gene (e.g., GFP) [5].
- Untreated Control: Cells receiving transfection reagent alone to establish baseline gene expression and assess potential cytotoxicity of the transfection process [5].
Cell Seeding and Transfection
Cell Preparation:
- Culture appropriate cell lines (e.g., HeLa, HEK293) under standard conditions.
- Seed cells in 24-well or 12-well plates at 30-50% confluence in antibiotic-free medium 18-24 hours before transfection to ensure active cell division at the time of transfection.
Transfection Complex Formation:
- For a single well of a 24-well plate, dilute 20-50 nM siRNA in 50 µL of serum-free Opti-MEM medium. Gently mix.
- In a separate tube, dilute 1-2 µL of a cationic lipid-based transfection reagent (e.g., Lipofectamine RNAiMAX) in 50 µL of serum-free Opti-MEM. Incubate for 5 minutes at room temperature.
- Combine the diluted siRNA with the diluted transfection reagent. Mix gently and incubate for 20 minutes at room temperature to allow siRNA-lipid complex formation.
Transfection:
- Add the 100 µL siRNA-lipid complex dropwise to cells containing fresh, complete growth medium.
- Gently swirl the plate to ensure even distribution.
- Incubate cells at 37°C in a CO₂ incubator for 24-72 hours before analysis.
Functional Validation and Analysis
mRNA Knockdown Assessment (qRT-PCR):
- Timepoint: Harvest cells 48 hours post-transfection for optimal mRNA knockdown analysis.
- Procedure: Isolate total RNA using a commercial kit. Perform reverse transcription to generate cDNA. Conduct quantitative real-time PCR (qRT-PCR) using primers specific for the target gene and housekeeping genes (e.g., GAPDH, β-actin) for normalization.
- Analysis: Calculate percentage knockdown using the 2^(-ΔΔCt) method relative to negative control siRNA-treated cells.
Protein Knockdown Assessment (Western Blot):
- Timepoint: Harvest cells 72 hours post-transfection for optimal protein-level analysis, accounting for protein half-life.
- Procedure: Lyse cells in RIPA buffer, separate proteins by SDS-PAGE, transfer to a membrane, and probe with antibodies against the target protein and a loading control (e.g., β-tubulin, GAPDH).
- Analysis: Quantify band intensity using densitometry software and normalize to loading controls to determine percentage protein reduction.
Phenotypic Analysis (If Applicable):
- Depending on the target gene, perform functional assays (e.g., proliferation, apoptosis, migration) at relevant timepoints to correlate gene silencing with biological effects.
Table 2: Key Research Reagent Solutions for siRNA Experiments
| Reagent / Resource | Function | Key Features & Considerations |
|---|---|---|
| Predesigned siRNA Libraries (e.g., siGENOME, ON-TARGETplus) [5] | Target-specific gene silencing | Expert-designed sequences; available with chemical modifications to reduce off-target effects; often available in pooled formats (SMARTpools) [5] |
| Cationic Lipid-Based Transfection Reagents [5] | Deliver siRNA into cells | Form complexes with siRNA via electrostatic interactions; suitable for many standard cell lines; optimization of lipid:siRNA ratio is critical for efficiency and minimizing cytotoxicity |
| Accell siRNA [5] | Delivery to difficult-to-transfect cells | Chemically modified for passive uptake without transfection reagents; enables gene silencing in sensitive primary cells and neurons; serum can inhibit delivery efficiency |
| Validated Positive Control siRNAs [5] | Experimental control | Target essential, ubiquitously expressed genes (e.g., GAPDH, Polo-like Kinase 1); verify transfection efficiency and silencing capability in each experiment |
| Scrambled Negative Control siRNAs [5] | Experimental control | No significant sequence identity to any known gene; distinguishes sequence-specific silencing from non-specific effects of transfection or cellular stress |
| qRT-PCR Assays | Quantify mRNA knockdown | Gene-specific primers/probes; require stable housekeeping genes for normalization; critical for validating target engagement at the mRNA level |
| Validated Antibodies | Quantify protein knockdown | Target-specific antibodies with demonstrated specificity for Western blot; required to confirm functional silencing at the protein level |
Troubleshooting Common Experimental Issues
Table 3: Troubleshooting Guide for siRNA Experiments
| Problem | Potential Causes | Solutions |
|---|---|---|
| Low Silencing Efficiency | Ineffective transfection; poor siRNA design; insufficient incubation time | Optimize transfection conditions (reagent concentration, complexation time); test alternative siRNAs targeting different regions; extend time between transfection and analysis (up to 72-96 hours) [5] |
| High Cell Toxicity | Transfection reagent cytotoxicity; excessive siRNA concentration | Titrate down transfection reagent and siRNA concentrations; try less cytotoxic delivery methods (e.g., polymer-based reagents, Accell siRNA) [5] |
| Inconsistent Results Between Replicates | Uneven cell seeding; improper transfection complex formation | Ensure homogeneous cell suspension when seeding; mix transfection complexes thoroughly before addition; add complexes dropwise across the well surface |
| Off-Target Effects | Seed region homology; passenger strand activity | Use siRNAs with validated specificity; employ pooled siRNA designs; utilize siRNAs with chemical modifications that reduce passenger strand loading and seed-mediated off-targets [5] |
| Inefficient Delivery in Difficult Cells | Low division rate; primary cells; complex morphology | Utilize specialized delivery methods (e.g., Accell siRNA, electroporation, viral delivery) optimized for challenging cell types [5] |
The discovery of RNA interference (RNAi) represents a paradigm shift in molecular biology, providing researchers with a powerful tool for targeted gene knockdown. This phenomenon, first identified in the nematode Caenorhabditis elegans, has evolved from a fundamental biological curiosity to a revolutionary therapeutic platform. The translation of this natural gene silencing mechanism into clinically approved medicines marks one of the most significant advancements in modern pharmacotherapy, enabling the precise targeting of previously undruggable genes [3]. This Application Note details the key historical milestones in siRNA therapeutics, providing experimental context and methodological frameworks that have facilitated this remarkable journey from basic research to clinical application, with particular emphasis on the foundational discoveries made in C. elegans.
Historical Timeline and Foundational Discovery in C. elegans
The RNAi timeline began with pivotal basic research in a model organism, which ultimately unlocked a new therapeutic modality. The following table summarizes the major historical milestones:
Table 1: Key Historical Milestones in siRNA Therapeutics
| Year | Milestone | Significance |
|---|---|---|
| 1998 | Discovery of RNAi in C. elegans [3] | Foundational research by Fire and Mello demonstrated that double-stranded RNA could potently and specifically silence gene expression in C. elegans. |
| 2006 | Nobel Prize in Physiology or Medicine [3] | Andrew Fire and Craig C. Mello were awarded the Nobel Prize for their discovery of RNA interference. |
| 2018 | First FDA-approved siRNA drug, Patisiran [9] [10] [11] | Patisiran (ONPATTRO) was approved for hereditary transthyretin-mediated amyloidosis with polyneuropathy, validating siRNA as a human therapeutic. |
| 2019-2023 | Approval of five additional siRNA drugs [9] [10] [11] | Givosiran (2019), Lumasiran (2020), Inclisiran (2021), Vutrisiran (2022), and Nedosiran (2023) were approved, expanding the scope of treatable diseases. |
The initial discovery was made in C. elegans, a small, transparent nematode that is a premier model organism in biological research due to its simplicity, short life cycle, and completely mapped cell lineage [12] [13]. Its anatomical simplicity—about 1000 somatic cells and a fully sequenced genome—made it an ideal system for uncovering fundamental genetic mechanisms [13] [14]. This discovery illuminated a conserved gene regulatory pathway that would later be exploited for therapeutic development.
Currently Approved siRNA Therapeutics
Since the 2018 approval of Patisiran, the siRNA therapeutic landscape has expanded rapidly. All approved agents are double-stranded RNAs designed to target specific mRNA sequences in hepatocytes, facilitated by advanced delivery systems [10] [11]. The following table provides a detailed comparison of the currently approved siRNA drugs.
Table 2: FDA-Approved siRNA Therapeutics (as of 2025)
| Brand Name (Generic) | Approval Year | Indication | Molecular Target | Delivery System | Dosing Frequency |
|---|---|---|---|---|---|
| ONPATTRO (Patisiran) [9] [10] [11] | 2018 | Hereditary transthyretin-mediated amyloidosis (hATTR) with polyneuropathy | Transthyretin (TTR) mRNA | Lipid Nanoparticle (LNP) | IV infusion every 3 weeks |
| GIVLAARI (Givosiran) [9] [10] [11] | 2019 | Acute Hepatic Porphyria (AHP) | Aminolevulinate synthase 1 (ALAS1) mRNA | GalNAc conjugate | Subcutaneous injection monthly |
| OXLUMO (Lumasiran) [9] [10] [11] | 2020 | Primary Hyperoxaluria Type 1 (PH1) | Hydroxyacid oxidase 1 (HAO1) mRNA | GalNAc conjugate | Subcutaneous injection (initial 3 monthly, then quarterly) |
| LEQVIO (Inclisiran) [9] [10] [11] | 2021 | Hypercholesterolemia | Proprotein convertase subtilisin/kexin type 9 (PCSK9) mRNA | GalNAc conjugate | Subcutaneous injection (at 0, 3 months, then every 6 months) |
| AMVUTTRA (Vutrisiran) [9] [10] | 2022 | hATTR with polyneuropathy | Transthyretin (TTR) mRNA | GalNAc conjugate | Subcutaneous injection every 3 months |
| RIVFLOZA (Nedosiran) [9] [10] | 2023 | Primary Hyperoxaluria Type 1 (PH1) | Lactate dehydrogenase (LDH) mRNA | GalNAc conjugate | Subcutaneous injection monthly |
A critical differentiator among these therapies is their delivery technology. Patisiran utilizes a lipid nanoparticle (LNP) system for encapsulation and delivery to hepatocytes. In contrast, the other five approved drugs employ N-acetylgalactosamine (GalNAc) conjugation, which facilitates highly efficient uptake by hepatocytes through binding to the asialoglycoprotein receptor (ASGPR) on the cell surface [10] [11] [3]. This targeted delivery approach minimizes systemic exposure and enhances therapeutic efficacy.
Core Mechanism of Action and Experimental Workflow
The RNAi Pathway
The therapeutic action of siRNAs harnesses the endogenous RNA interference pathway. The following diagram illustrates the core mechanism of siRNA-mediated gene silencing after cellular uptake.
The mechanism involves a conserved, multi-step pathway [9] [3]:
- Cytosolic Entry and RISC Loading: The synthetic siRNA duplex is introduced into the cell cytoplasm, aided by its delivery system (LNP or GalNAc). The RNA-induced silencing complex (RISC), particularly the Argonaute-2 (AGO2) endonuclease, incorporates the siRNA.
- Strand Separation and Activation: The siRNA duplex is unwound, the passenger (sense) strand is ejected and degraded, and the guide (antisense) strand remains bound to RISC, forming the active complex.
- Target Recognition and Cleavage: The guide strand directs the activated RISC to its complementary messenger RNA (mRNA) target via Watson-Crick base pairing. The AGO2 enzyme within RISC catalyzes the endonucleolytic cleavage of the target mRNA.
- Gene Silencing: The cleaved mRNA fragments are rapidly degraded by cellular exonucleases, preventing translation into the target protein and effectively silencing gene expression. The activated RISC can catalyze multiple rounds of mRNA cleavage, providing sustained effect.
Protocol: In Vitro Assessment of siRNA Efficacy
This protocol outlines a standard workflow for validating siRNA efficacy and cytotoxicity in cell culture models, a critical step in therapeutic development.
Title: Basic Workflow for In Vitro siRNA Screening
Objective: To transfert siRNA into cultured cells and evaluate its target knockdown efficiency and potential cytotoxicity.
Materials:
- Research Reagent Solutions: See Table 3.
- Equipment: Cell culture hood, CO2 incubator, micropipettes, centrifuge, multi-well plates, real-time PCR system, spectrophotometer/fluorometer.
Table 3: Essential Reagents for In Vitro siRNA Experiments
| Reagent / Material | Function | Example / Note |
|---|---|---|
| Validated siRNA | The active molecule for gene knockdown. | Include both target-specific and non-targeting negative control siRNAs. |
| Transfection Reagent | Facilitates siRNA entry into cells. | Cationic lipids or polymers (e.g., Lipofectamine RNAiMAX [15]). |
| Cell Line | Model system for testing. | Should express the target mRNA; can be a stable overexpression line [15]. |
| Cell Culture Medium | Supports cell growth and health. | Serum-free medium (e.g., Opti-MEM) is often used during transfection. |
| qRT-PCR Reagents | Quantifies mRNA levels post-transfection. | Probes or dyes specific for the target and housekeeping genes. |
| Cell Viability Assay Kit | Measures cytotoxicity. | MTT, MTS, or similar assays [15]. |
Procedure:
- Cell Seeding: Plate cells in a multi-well plate at 30-50% confluence in appropriate growth medium and allow to adhere for 24 hours [15].
- siRNA-Transfection Complex Preparation:
- Dilute the siRNA (e.g., 5-50 nM final concentration) in a serum-free medium (e.g., Opti-MEM).
- Dilute the transfection reagent separately in the same medium.
- Combine the diluted siRNA and transfection reagent, mix gently, and incubate for 15-20 minutes at room temperature to allow complex formation.
- Transfection: Add the siRNA-transfection complexes directly to the cells in a drop-wise manner. Gently swirl the plate to ensure even distribution.
- Incubation: Incubate the cells for 24-48 hours at 37°C in a CO2 incubator.
- Harvest and Analysis:
- Viability Assay: At 24 hours post-transfection, perform an MTT assay. Incubate cells with MTT reagent for several hours, then measure absorbance at 570 nm to determine cell viability relative to controls [15].
- Knockdown Efficiency: At 48 hours post-transfection, harvest cells for RNA extraction. Perform quantitative RT-PCR (qRT-PCR) using primers specific to the target gene. Normalize target mRNA levels to a housekeeping gene (e.g., GAPDH, β-actin) and calculate percentage knockdown relative to cells treated with a non-targeting control siRNA [15].
Key Technological Breakthroughs: Delivery and Stabilization
The clinical translation of siRNA faced significant hurdles, including rapid degradation in serum, inefficient cellular uptake, and potential immunogenicity. Key innovations overcame these barriers:
Chemical Modifications: Strategic modifications to the siRNA backbone dramatically improved stability and pharmacokinetics. Common modifications include [3]:
- Sugar Modifications: 2'-O-methyl (2'-OMe), 2'-fluoro (2'-F), and Locked Nucleic Acid (LNA) in the ribose ring to enhance nuclease resistance and binding affinity.
- Backbone Modifications: Phosphorothioate (PS) linkages, which replace a non-bridging oxygen with sulfur, increasing stability against nucleases and promoting protein binding for improved tissue distribution.
- Terminal Modifications: Conjugation of ligands like GalNAc for targeted delivery.
Advanced Delivery Systems: Two primary delivery platforms have been successfully implemented in the clinic [10] [11] [3]:
- Lipid Nanoparticles (LNPs): As used in Patisiran, LNPs protect the siRNA from degradation, facilitate endosomal escape, and are efficiently taken up by hepatocytes.
- GalNAc Conjugation: A trivalent N-acetylgalactosamine molecule conjugated to the siRNA enables high-affinity binding to the asialoglycoprotein receptor (ASGPR) on hepatocytes, triggering rapid receptor-mediated internalization. This efficient targeting allows for subcutaneous administration and less frequent dosing [10].
The journey of siRNA from its serendipitous discovery in C. elegans to a robust therapeutic platform exemplifies the power of basic biological research to fuel clinical innovation. The six currently approved drugs, with their durable effects and novel targeting mechanisms, have established RNAi as a distinct and valuable drug class, particularly for rare genetic liver disorders [11]. The experimental protocols and tools outlined in this Application Note provide a framework for researchers to explore new applications.
The future of siRNA therapeutics is expansive. Ongoing research focuses on overcoming remaining challenges, such as delivery to tissues beyond the liver. Advances in novel conjugation strategies, lipid and polymer nanoparticles, and innovative chemical modifications are actively being pursued to target the central nervous system, ocular tissues, and tumors [15] [3]. Furthermore, the exploration of siRNA for highly prevalent conditions such as cardiovascular diseases, with Inclisiran as a pioneer, opens the door for a significant expansion of its impact on public health. As delivery technologies mature, the siRNA pipeline is poised to silence previously unreachable genetic drivers of disease, solidifying its role in the next generation of precision medicines.
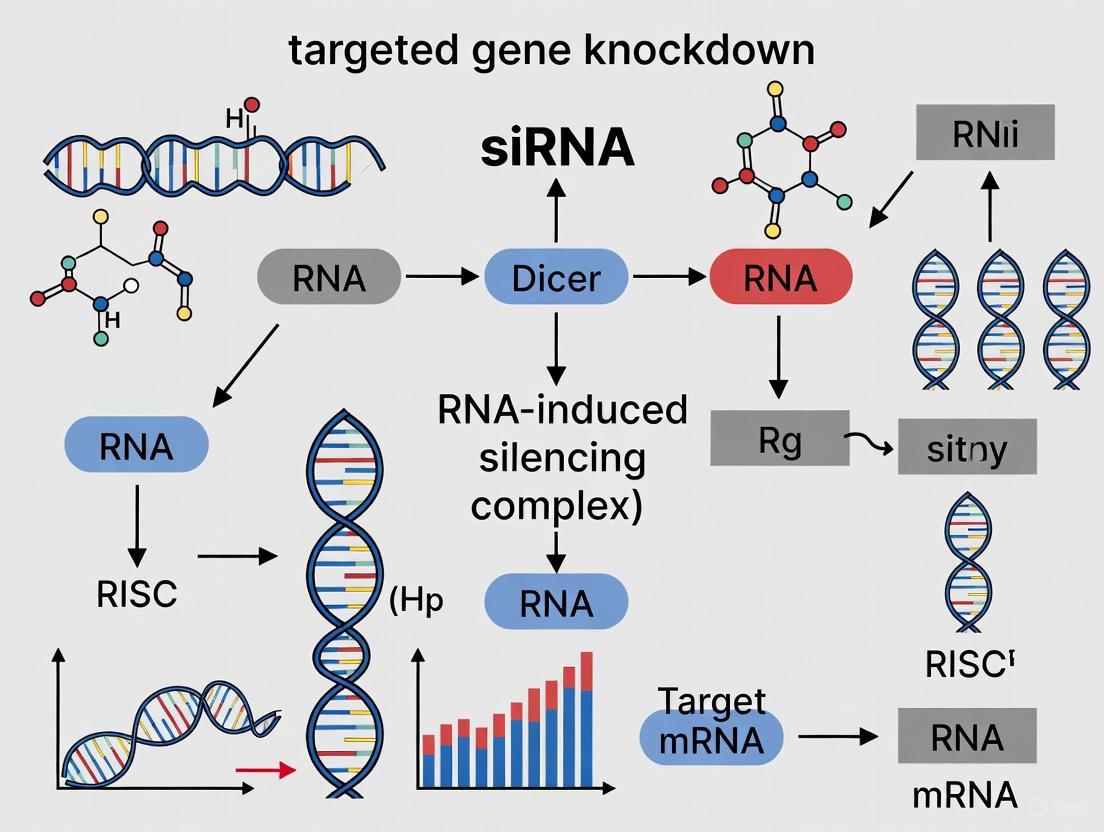
Small interfering RNA (siRNA) is a foundational tool in molecular biology for achieving targeted gene knockdown via the RNA interference (RNAi) pathway. This application note details the core structural components of siRNA—the guide and passenger strands—and elucidates the critical role of the Dicer enzyme in their biogenesis and function. Framed within the context of gene knockdown research, this document provides researchers, scientists, and drug development professionals with structured data, experimental protocols, and key resource information to support the design and implementation of effective siRNA-based experiments.
Core Components of siRNA
Structural Anatomy of siRNA
siRNA is a synthetic RNA duplex typically 21–23 nucleotides (nt) in length, designed to specifically target a particular mRNA for degradation [5]. Its canonical structure consists of several key features:
- Double-Stranded Duplex: Two RNA strands form a duplex 19 to 25 base pairs (bp) in length [5] [16].
- 3' Overhangs: The duplex often features 2-nt 3' overhangs on both ends, a characteristic inherited from its processing by Dicer [16].
- Strand Asymmetry: The two strands are functionally distinct:
- Guide Strand (Antisense Strand): This strand is selectively loaded into the RNA-induced silencing complex (RISC) and serves as the sequence-specific guide for identifying complementary mRNA targets [5].
- Passenger Strand (Sense Strand): This strand is displaced and degraded during RISC activation [17].
Table 1: Key Structural Features of Canonical siRNA
| Feature | Description | Functional Significance |
|---|---|---|
| Length | 21–23 nt duplex; 19–25 bp | Optimal for RISC loading and function [5] [16] |
| Overhangs | 2-nt 3' overhangs | Facilitates recognition by Dicer and Argonaute proteins [16] |
| Strand Identity | Guide (antisense) and Passenger (sense) | Determines which strand is used for target recognition [5] |
| Thermodynamic Asymmetry | Difference in base-pairing stability at the 5' ends of the duplex | A key determinant for guide strand selection; the strand with the less stable 5' end is preferentially loaded [17] |
The Role of Dicer in siRNA Biogenesis and Function
Dicer is a specialized RNase III enzyme that initiates the RNAi pathway. Its primary function is to cleave long double-stranded RNA (dsRNA) precursors into mature siRNA duplexes [17] [18]. In humans, Dicer functions in concert with double-stranded RNA-binding proteins (dsRBPs) like TRBP and PACT [17].
The mechanism of Dicer involves two distinct RNA binding sites:
- A catalytic site for processing long dsRNA substrates into siRNAs.
- A sensing site on its helicase domain that binds the siRNA product and helps sense its thermodynamic asymmetry [17].
Following dicing, the siRNA undergoes repositioning within the Dicer complex. The initial dsRNA substrate binds to the catalytic arm, but after cleavage, the nascent siRNA product re-localizes to the helicase domain of Dicer. This repositioning is crucial for directional binding and subsequent guide strand selection [17]. In organisms like Drosophila, the Dicer-2–R2D2 heterodimer senses siRNA asymmetry, with R2D2 binding the more stable end and Dicer-2 binding the less stable end, which dictates the orientation for RISC loading [18].
siRNA Mechanism and Pathway
The following diagram illustrates the core pathway of siRNA-mediated gene silencing, from dicing to target mRNA degradation.
Diagram 1: The siRNA-Mediated Gene Silencing Pathway. This workflow outlines the key steps from long double-stranded RNA processing to mRNA cleavage by the activated RISC.
Guide Strand Selection Mechanism
A critical step in RNAi is the selective loading of the correct guide strand into RISC. The following diagram details the molecular mechanism of strand selection based on thermodynamic asymmetry.
Diagram 2: Mechanism of Guide Strand Selection. Dicer senses the difference in thermodynamic stability at the 5' ends of the siRNA duplex, determining which strand is loaded into RISC as the guide.
Experimental Protocols and Design Considerations
Protocol for In Vitro siRNA-Mediated Gene Knockdown
This protocol outlines a standard procedure for transient gene knockdown in cultured mammalian cells using synthetic siRNA.
Materials:
- Cultured mammalian cells
- Synthetic siRNA (e.g., ON-TARGETplus, Accell)
- Transfection reagent (e.g., cationic lipid-based)
- Opti-MEM or similar serum-free medium
- Normal growth medium
- RNA extraction kit
- qRT-PCR reagents for knockdown validation
Procedure:
- siRNA Design and Preparation: Design siRNA sequences targeting your gene of interest. Follow design rules in Section 4.2. Resuspend synthetic siRNA in provided buffer to create a stock solution (e.g., 20 µM).
- Cell Seeding: Seed cells in a 24-well plate at 50–70% confluence in normal growth medium without antibiotics. Incubate at 37°C for 24 hours.
- Transfection Complex Formation:
- Tube A: Dilute 5 µL of 20 µM siRNA stock (final 100 pmol) in 50 µL Opti-MEM.
- Tube B: Dilute 1–2 µL of transfection reagent in 50 µL Opti-MEM. Incubate for 5 minutes.
- Combine solutions from Tubes A and B. Mix gently and incubate for 20 minutes at room temperature to allow complex formation.
- Transfection: Add the 100 µL transfection complex dropwise to cells. Gently swirl the plate to distribute evenly.
- Incubation and Analysis: Incubate cells for 48–72 hours at 37°C.
- Knockdown Validation (48-72 hours): Extract total RNA and perform qRT-PCR to assess target mRNA levels.
- Phenotypic Analysis (72+ hours): Proceed with functional assays (e.g., Western blot, viability, apoptosis) to assess the biological impact of knockdown.
Key Design Considerations for Effective siRNA
Effective siRNA design is critical for maximizing gene silencing and minimizing off-target effects. Modern algorithms (e.g., SMARTselection) use rules derived from large-scale functional studies [5].
Table 2: Key Parameters for Rational siRNA Design
| Parameter | Recommendation | Rationale |
|---|---|---|
| Target Sequence | Select a region within the mRNA coding sequence or 3' UTR. Avoid regions with high homology to other genes. | Maximizes specificity and reduces off-target silencing [5]. |
| GC Content | Maintain between 30–50%. | Extremely high or low GC content can reduce silencing efficiency and specificity [19]. |
| Thermodynamic Profile | Ensure the 5' end of the intended guide strand has lower binding stability than its 3' end. | Promotes preferential loading of the intended guide strand into RISC [17] [5]. |
| Seed Region (nt 2-8) | Avoid complementarity to off-target transcripts. Use chemical modifications if necessary. | The seed region is critical for initial target binding; managing its sequence reduces off-target effects [5]. |
| Chemical Modifications | Incorporate modifications like 2'-O-methyl (2'-OMe) or phosphorothioate (PS). | Enhances nuclease resistance, improves specificity, and reduces immune stimulation [20] [21]. |
Advanced siRNA Structural Variants
While the 21-nt siRNA with 3' overhangs is standard, research has identified potent non-classical structures.
Table 3: Structural Variants of siRNA
| Variant | Structure | Advantages and Applications |
|---|---|---|
| Dicer-Substrate siRNA (dsiRNA) | 27-bp duplex with asymmetric design (e.g., 25-nt sense/27-nt antisense strand). | Increased potency as it is processed by Dicer, leading to more efficient RISC loading [16]. |
| Blunt siRNA | 19–25 bp duplex without overhangs. | Can trigger efficient gene silencing, demonstrating flexibility in structural requirements [16]. |
| Asymmetric Design | Structurally asymmetric overhangs (e.g., DNA substitutions in one strand). | Promotes preferential incorporation of the antisense strand into RISC, enhancing specificity [16]. |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for siRNA Research
| Reagent / Tool | Function | Example Use-Case |
|---|---|---|
| Predesigned siRNA (e.g., ON-TARGETplus) | High-quality, chemically modified siRNAs for high-throughput gene silencing. | Rapid knockdown of a single gene or a gene family with minimized off-target effects [5]. |
| Custom siRNA Synthesis | Synthesis of specific siRNA sequences tailored to unique targets. | Targeting specific SNPs, novel transcripts, or non-coding RNAs not covered by predesigned libraries [5]. |
| Transfection Reagents (Cationic Lipids/Polymers) | Form complexes with siRNA to facilitate cellular uptake. | Delivering siRNA into standard cell lines (e.g., HEK-293, HeLa) [5]. |
| Accell siRNA | Chemically modified siRNA for delivery without transfection reagents. | Gene silencing in difficult-to-transfect cells (e.g., primary neurons, immune cells) [5]. |
| siRNA Libraries | Genome-wide or pathway-focused collections of siRNAs. | Large-scale functional genomic screens to identify novel genes involved in a biological process [5]. |
| Positive & Negative Control siRNAs | Validated siRNA against a housekeeping gene and non-targeting sequence. | Optimizing transfection efficiency and controlling for non-sequence-specific effects [5]. |
Small interfering RNA (siRNA) therapeutics represent a revolutionary class of gene-targeted medicines that operate through the natural cellular process of RNA interference (RNAi). These double-stranded RNA molecules, typically 21–23 nucleotides in length, offer a fundamentally distinct mechanism of action compared to traditional small molecules and antibody-based therapeutics [22] [23]. By specifically targeting and silencing disease-causing genes at the post-transcriptional level, siRNA drugs provide researchers with an unprecedented ability to modulate previously inaccessible biological pathways [24]. The core advantages of this technology—exceptional specificity, modular programmability, and the capacity to target "undruggable" genes—are reshaping therapeutic development across diverse disease areas, from genetic disorders to cancer and viral infections [22] [24] [25].
The molecular specificity of siRNA stems from Watson-Crick base pairing, wherein the antisense (guide) strand directs the RNA-induced silencing complex (RISC) to complementary messenger RNA (mRNA) sequences for precise cleavage and degradation [22] [23]. This mechanism enables researchers to target individual gene isoforms with nucleotide-level precision, a capability that remains challenging for conventional small-molecule drugs that typically target protein active sites [24]. The programmable nature of siRNA design allows for rapid development against new targets simply by modifying the oligonucleotide sequence, significantly accelerating the therapeutic discovery pipeline compared to traditional drug modalities [23].
Key Advantages: Quantitative Comparison
Table 1: Comparative Analysis of siRNA Therapeutics Versus Traditional Modalities
| Feature | siRNA Therapeutics | Small Molecule Drugs | Monoclonal Antibodies |
|---|---|---|---|
| Target Specificity | Gene-level specificity via complementary base pairing | Binds protein active sites; limited by protein structure | Binds specific epitopes; limited to extracellular targets |
| Development Timeline | Shorter R&D cycles due to programmable design [26] | Lengthy optimization of chemical structures | Complex development of biological production |
| Druggable Target Space | Targets "undruggable" genes including transcription factors [24] [25] | ~15% of the human genome [22] | Limited to extracellular and cell surface targets |
| Duration of Action | Sustained effects (weeks to months) due to catalytic RISC activity [26] | Short duration (hours to days) requiring frequent dosing | Moderate duration (days to weeks) |
| Therapeutic Applicability | Broad: genetic disorders, cancers, viral infections, metabolic diseases [22] [26] | Extensive but limited by target chemistry | Primarily inflammatory diseases, oncology |
Table 2: Clinical-Stage Examples Demonstrating siRNA Advantages
| siRNA Drug (Target) | Indication | Key Advantage Demonstrated | Development Status |
|---|---|---|---|
| Patisiran (TTR) | Hereditary transthyretin-mediated amyloidosis | First FDA-approved RNAi therapeutic; targets disease-causing gene [22] | Approved (2018) |
| siG12D-LODER (KRAS G12D) | Pancreatic cancer | Targets "undruggable" KRAS mutation [23] | Clinical trials |
| Dual-targeting anti-KRAS/MYC | Solid tumors | Co-silencing of two undruggable oncogenes [25] | Preclinical development |
| Inclisiran (PCSK9) | Hypercholesterolemia | Sustained effects with infrequent dosing [26] | Approved |
| Cond-siRNA (CaN) | Heart failure | Conditionally activated only in diseased cardiomyocytes [27] | Preclinical development |
Application Note 1: Targeting "Undruggable" Oncogenes
Background and Significance
The ability to target traditionally "undruggable" genes represents one of the most significant advantages of siRNA therapeutics. In oncology, key drivers like KRAS and MYC have eluded successful targeting by small molecules for decades due to their protein structures and intracellular locations [25]. KRAS mutations are present in approximately 25% of all human cancers, while MYC is dysfunctional in 50-70% of cancers, making them highly prioritized but challenging therapeutic targets [25].
Experimental Approach: Dual-Targeting Strategy
Recent breakthroughs demonstrate the power of siRNA to simultaneously silence multiple oncogenes. Pecot and colleagues developed a novel "two-in-one" inverted RNAi molecule capable of co-silencing both mutated KRAS and overexpressed MYC [25]. This approach resulted in a remarkable 40-fold improvement in inhibition of cancer cell viability compared to individual siRNA targeting, demonstrating synergistic effects against these critical oncogenic drivers [25].
Table 3: Research Reagent Solutions for Oncogene-Targeted siRNA Studies
| Reagent/Method | Function/Application | Example in Practice |
|---|---|---|
| Cholesterol-enriched exosomes (Chol/MEs) | Enhanced siRNA delivery via membrane fusion | PLK1 siRNA delivery in colorectal cancer models [24] |
| Inverted RNAi molecules | Enable simultaneous silencing of multiple genes | Dual targeting of KRAS and MYC oncogenes [25] |
| LODER polymers | Localized sustained release of siRNA | siG12D-LODER for KRAS-mutant pancreatic cancer [23] |
| GalNAc conjugates | Hepatocyte-specific siRNA delivery | Approved siRNAs for hepatic conditions [22] |
| Electroporation frameworks | Scalable loading of siRNA into exosomes | Capricor's systematic loading approach [28] |
Protocol: Dual-Targeting siRNA Design and Validation
Step 1: Target Sequence Selection
- Identify unique 21-23 nucleotide sequences within KRAS (e.g., mutation hotspots like G12D, G12V) and MYC mRNA
- Verify specificity using NCBI BLAST against human transcriptome to minimize off-target effects [23]
- Select sequences with ~50% GC content, avoiding problematic motifs (e.g., GGGG repeats) [27]
Step 2: Molecular Assembly
- Design inverted siRNA architecture with complementary regions enabling single-molecule assembly
- Incorporate chemical modifications (2'-O-methyl, LNA) to enhance stability and reduce immunostimulation [27]
- Synthesize and purify using standard oligonucleotide synthesis methods
Step 3: In Vitro Validation
- Transfect cancer cell lines (e.g., HCT116 colorectal, pancreatic cancer lines) using appropriate delivery systems
- Assess gene silencing efficiency via RT-qPCR at 48-72 hours post-transfection
- Evaluate functional effects using cell viability assays (MTT, CellTiter-Glo) and apoptosis detection (annexin V staining)
- Confirm pathway modulation through Western blotting for KRAS, MYC, and downstream effectors
Step 4: In Vivo Evaluation
- Formulate siRNA for in vivo delivery (e.g., lipid nanoparticles, exosomal systems)
- Administer to xenograft mouse models via appropriate routes (IV, localized)
- Monitor tumor growth inhibition and assess target engagement in excised tumors
- Evaluate safety profile through histological examination of major organs
Diagram 1: Dual-targeting siRNA mechanism for undruggable oncogenes.
Application Note 2: Conditional siRNA for Cell-Type Specific Silencing
Background and Significance
A critical challenge in therapeutic siRNA development is achieving cell-type specific silencing to minimize off-target effects in healthy tissues. Conditional siRNA (Cond-siRNA) technology addresses this limitation by creating "smart" therapeutics activated only in diseased cells [27]. This approach represents the pinnacle of siRNA programmability, leveraging disease-specific biomarkers to trigger therapeutic activity precisely where needed.
Experimental Approach: Cardiac Hypertrophy Targeting
Gokulnath and colleagues developed a novel Cond-siRNA construct activated by Nppa mRNA, which is specifically upregulated in cardiomyocytes under pathological stress but exhibits low baseline expression in healthy hearts [27]. This Cond-siRNA silences the key pro-hypertrophic gene calcineurin (CaN) only when activated by the Nppa biomarker, creating a feedback loop that automatically attenuates hypertrophic signaling in stressed cardiomyocytes while sparing other cell types [27].
Protocol: Conditional siRNA Design and Implementation
Step 1: Disease-Specific Sensor Identification
- Screen potential sensor mRNAs using disease models (e.g., phenylephrine treatment in cardiomyocytes)
- Validate biomarker specificity through RT-qPCR in diseased vs. healthy tissues
- Select sensor with maximal disease-to-healthy expression ratio (Nppa demonstrated >10-fold induction) [27]
Step 2: Riboswitch Assembly
- Design sensor strand complementary to 31-33 nucleotide segment of biomarker 3' UTR
- Create core strand with complementary segments (11-12 bases) for sensor interaction
- Incorporate therapeutic guide strand targeting gene of interest (e.g., CaN for hypertrophy)
- Optimize thermodynamic properties using Nupack software for nucleic acid structure prediction [27]
Step 3: Specificity Validation
- Transfert Cond-siRNA into target (cardiomyocytes) and non-target (cardiac fibroblasts, T cells) populations
- Induce disease state (phenylephrine treatment) in target cells
- Assess CaN silencing specifically in activated cardiomyocytes via RT-qPCR and Western blot
- Confirm minimal baseline activity in unstimulated cells and non-target cell types
Step 4: Functional Efficacy Assessment
- Measure hypertrophy markers (cell size, ANP secretion) in disease models
- Evaluate NFATc1 nuclear translocation as indicator of CaN pathway activity
- Validate in tissue-on-chip models simulating pathological stress (pressure overload)
Diagram 2: Conditional siRNA mechanism for tissue-specific gene silencing.
Application Note 3: Advanced Delivery Systems for Enhanced Specificity
Background and Significance
The programmability of siRNA extends beyond sequence design to include delivery system engineering, enabling tissue-specific targeting and improved therapeutic indices. Recent advances in delivery platforms have dramatically enhanced the specificity and efficacy of siRNA therapeutics by facilitating targeted cytosolic delivery while minimizing off-target effects [24] [28].
Experimental Approach: Cholesterol-Enriched Exosomes
Zhang et al. engineered cholesterol-enriched exosomes (Chol/MEs) that enable siRNA to bypass endosomal entrapment and directly enter the cytosol via membrane fusion [24]. This delivery platform demonstrated superior gene silencing efficiency compared to conventional transfection agents (Lipofectamine 2000, RNAiMAX), reducing PLK1 expression and achieving remarkable tumor growth inhibition (0.05-fold of PBS control) in preclinical colorectal cancer models [24].
Protocol: Engineered Exosome Production and Application
Step 1: Exosome Engineering
- Transfert 293F cells with plasmids for exosome production
- Modulate cholesterol content in exosomal membranes through metabolic engineering
- Harvest exosomes from cell culture supernatants via differential centrifugation
- Characterize using nanoparticle tracking analysis and Western blotting for exosomal markers
Step 2: siRNA Loading Optimization
- Implement electroporation parameters optimized for siRNA loading
- Assess both scale-up and scale-out strategies for clinical translation [28]
- Determine loading efficiency using fluorescently labeled siRNA and quantification methods
- Validate siRNA integrity post-loading through gel electrophoresis
Step 3: Functional Delivery Validation
- Test cellular uptake in target vs. non-target cell lines using flow cytometry
- Confirm endosomal bypass and direct cytosolic delivery via confocal microscopy
- Evaluate gene silencing potency across multiple siRNA targets
- Compare to commercial transfection reagents for benchmarking performance
Step 4: In Vivo Application
- Administer siRNA-loaded Chol/MEs via appropriate routes (IV, oral for GI cancers)
- Assess biodistribution using near-infrared imaging
- Quantify target engagement in tumors and major organs
- Evaluate safety profile through comprehensive histopathological analysis
Table 4: Advanced Delivery Platforms for Enhanced siRNA Specificity
| Delivery Platform | Mechanism of Action | Advantages | Research Applications |
|---|---|---|---|
| Cholesterol-enriched Exosomes (Chol/MEs) | Membrane fusion enabling direct cytosolic delivery [24] | Bypasses endosomal trapping; reduced immunogenicity; enables oral delivery [24] | PLK1 silencing in colorectal cancer; oral siRNA delivery |
| GalNAc Conjugates | Receptor-mediated endocytosis via asialoglycoprotein receptor | Hepatocyte-specific targeting; clinical validation [22] | Liver-directed therapies (PCSK9, TTR) |
| Lipid Nanoparticles (LNPs) | Endocytosis and endosomal release | Broad applicability; FDA-approved formulations | Systemic siRNA delivery; vaccine applications |
| Targeted RNAi Molecules (TRiM) | Component-based modular targeting | Tunable pharmacokinetics and biodistribution | Tissue-specific silencing beyond liver |
Technical Considerations and Troubleshooting
Optimizing Specificity and Reducing Off-Target Effects
Despite the inherent specificity of siRNA, off-target effects remain a significant consideration in experimental design. These primarily occur through two mechanisms: immune stimulation through interferon response and miRNA-like effects due to partial complementarity to untargeted transcripts [22]. To minimize these effects:
- Seed Region Analysis: Utilize BLAST and other computational tools to identify and avoid 7-nucleotide matches within the seed region (positions 2-8) of the guide strand to untargeted mRNAs [23]
- Chemical Modifications: Incorporate 2'-O-methyl modifications particularly in the guide strand seed region to reduce off-target silencing while maintaining on-target activity [27]
- Asymmetric Design: Favor asymmetric duplexes with thermodynamically less stable 5' end of the antisense strand to ensure proper RISC loading [22]
- Concentration Optimization: Identify the lowest effective concentration that induces target gene silencing to minimize nonspecific effects [22]
Protocol: Specificity Validation Workflow
Step 1: In Silico Specificity Screening
- Perform comprehensive BLAST analysis against human transcriptome
- Identify and redesign siRNAs with significant off-target potential
- Utilize prediction algorithms (e.g., Smith-Waterman) for enhanced specificity profiling
Step 2: Transcriptomic Assessment
- Conduct RNA-seq of treated vs. untreated cells
- Analyze differential expression beyond target gene
- Validate potential off-targets through RT-qPCR
- Compare to non-targeting control siRNA treatments
Step 3: Functional Confirmation
- Employ rescue experiments with modified target sequences
- Validate phenotype specificity using orthogonal approaches (CRISPR, antibodies)
- Assess immune activation through interferon-responsive gene expression panels
The unique advantages of siRNA therapeutics—exceptional specificity, modular programmability, and access to previously "undruggable" targets—position this technology as a transformative modality in biomedical research and therapeutic development. The experimental approaches and protocols detailed herein provide researchers with robust frameworks for leveraging these advantages across diverse applications, from oncology to cardiology.
Future directions in siRNA research will likely focus on expanding tissue targeting beyond the liver, enhancing conditional activation strategies for unprecedented specificity, and developing multi-targeting approaches for complex disease pathways. The continued evolution of delivery platforms, combined with increasingly sophisticated siRNA design principles, promises to unlock new therapeutic possibilities and accelerate the translation of siRNA discoveries from bench to bedside.
As the field advances, the integration of siRNA tools with other emerging technologies—including CRISPR-based screening for target identification [29] and organ-on-chip systems for functional validation [27]—will further enhance our ability to precisely modulate disease-relevant genes with minimal off-target effects, ultimately enabling more effective and safer therapeutic interventions for challenging diseases.
Designing and Delivering siRNA: From Rational Sequence Selection to Advanced Delivery Systems
Small interfering RNA (siRNA) technology has emerged as a powerful tool for targeted gene knockdown in research and therapeutic development. The efficacy of siRNA-mediated silencing is not uniform; it critically depends on the intelligent selection of the target site within the mRNA and the inherent biochemical properties of the siRNA molecule itself. Among these properties, the accessibility of the target mRNA region and the guanine-cytosine (GC) content of the siRNA are two of the most pivotal factors determining success. This application note details evidence-based protocols for identifying accessible mRNA regions and optimizing siRNA GC content, framed within the broader context of a research thesis on siRNA for targeted gene knockdown. The guidance is structured to provide researchers, scientists, and drug development professionals with actionable methodologies to enhance the efficiency and specificity of their gene silencing experiments.
The fundamental mechanism of RNA interference (RNAi) begins with the loading of the siRNA guide strand into the RNA-induced silencing complex (RISC). The activated RISC then scans the cytosolic mRNA pool to find and cleave a perfectly complementary target sequence [21]. However, the inherent structure of the mRNA presents a significant barrier. Stable secondary structures, such as hairpins and stem-loops, can shield potential target sites, making them inaccessible to RISC binding [30]. Furthermore, the GC content of the siRNA duplex influences its thermodynamic stability and its correct loading into RISC. An overly stable duplex (high GC content) can impede strand separation, a necessary step for RISC activation, while a very unstable duplex (low GC content) may not form properly [31] [32].
The following workflow diagram illustrates the critical decision points and their relationships in the optimal siRNA design process.
Core Design Parameters and Quantitative Criteria
A systematic approach to siRNA design requires adherence to a set of well-established, quantitative sequence and thermodynamic parameters. The table below summarizes the key criteria that should be evaluated for each candidate siRNA sequence.
Table 1: Key Criteria for Effective siRNA Design
| Parameter | Optimal Range/Feature | Rationale & Impact |
|---|---|---|
| GC Content | 30% - 52% [33] | Balances duplex stability; >60% GC negatively impacts silencing [4]. |
| Sequence Length | 21-23 nucleotides [19] | Standard length for RISC incorporation and target recognition. |
| Thermodynamic Asymmetry | Unstable 5' end (A/U-rich) of the guide strand [30] | Promotes correct guide strand loading into RISC, improving efficiency by 2-5 fold. |
| Position-Specific Nucleotides | 'A' at position 19, 'U' at position 10 [33] | Position-specific preferences correlated with high silencing efficacy. |
| Avoidance of Repeats | No CCCC or GGGG stretches [4] | Prevents synthetic challenges and potential structural issues. |
| Off-Target Filtering | < 78% query coverage with other genes via BLAST [33] | Minimizes homology-driven off-target effects. |
Adhering to these parameters during the initial design phase significantly increases the probability of identifying highly functional siRNAs. It is recommended to design and synthesize multiple siRNA candidates targeting different regions of the same mRNA to empirically confirm the robustness of the knockdown [31].
Protocol: Computational Screening for Optimal siRNA
This protocol provides a step-by-step methodology for the in-silico design and selection of siRNA candidates, integrating the criteria from Table 1.
Materials and Software Requirements
Table 2: Research Reagent Solutions and Computational Tools
| Item Name | Function/Application |
|---|---|
| NCBI Nucleotide Database | Repository for retrieving the target mRNA sequence in FASTA format [19]. |
| siRNA Design Software (e.g., siDirect, Ambion/Invitrogen Designer, GenScript's tool) | Algorithms to generate candidate siRNA sequences based on established rules [30] [33]. |
| Secondary Structure Prediction Tool (e.g., RNAfold, OligoWalk) | Predicts mRNA target site accessibility and local free energy (ΔG) [30] [34]. |
| BLAST (Basic Local Alignment Search Tool) | Checks sequence specificity to minimize off-target effects [31] [33]. |
| Molecular Docking Software (e.g., AutoDock, GROMACS) | Models siRNA interaction with Argonaute-2 (AGO2) to predict binding affinity and silencing potential [19] [34]. |
Step-by-Step Procedure
mRNA Sequence Retrieval:
- Obtain the complete coding DNA sequence (CDS) of your target gene in FASTA format from a reliable database such as the NCBI Nucleotide Database (e.g., Accession Number: NM_004248.3) [19].
Initial siRNA Candidate Generation:
- Input the FASTA sequence into a specialized siRNA design software.
- The software will scan the mRNA, typically identifying 21-nucleotide sequences that start with an "AA" dinucleotide and filtering them based on GC content (aim for 30-52%) [31] [33].
- The output will be a list of potential siRNA candidate sequences.
Refinement Based on Target Site Accessibility:
- To prioritize candidates, analyze the predicted accessibility of their target sites on the mRNA.
- Use tools like OligoWalk (part of the RNAstructure package) or RNAfold.
- Action: Calculate the hybridization energy (ΔG) between the siRNA guide strand and its target mRNA. Favorable (more negative) ΔG values (e.g., -31.1 to -37.3 kcal/mol) indicate stronger and more accessible binding [34].
- Action: Visually inspect the predicted secondary structure of the mRNA. Prioritize target sites located in regions with low local free energy, which are likely to be open and unstructured, rather than within stable hairpins or stem-loops [30].
Specificity and Off-Target Assessment:
- Perform a BLAST search of both the sense and antisense strands of each candidate siRNA against the appropriate genome database (e.g., human, mouse, rat).
- Action: Exclude any sequences with >16-17 contiguous base pairs of homology to other coding sequences or a query coverage of 78% or higher with non-target genes [31] [33].
Advanced Computational Validation (Optional but Recommended):
- For high-confidence leads, use molecular docking to simulate the interaction between the siRNA guide strand and the human Argonaute-2 (AGO2) protein.
- Action: Retrieve the 3D structure of AGO2 from a database like the Protein Data Bank (PDB).
- Action: Docking scores (e.g., between -330 and -351 kcal/mol) indicate efficient RISC loading. Follow this with molecular dynamics (MD) simulations (e.g., using GROMACS) to confirm the structural stability of the siRNA-AGO2 complex over time, with stable Root-mean-square deviation (RMSD) values (e.g., 2.1–2.6 Å) being a positive indicator [19] [34].
Protocol: Experimental Validation of siRNA Efficacy
After in-silico selection, experimental validation is essential to confirm silencing efficiency and specificity.
Materials
- Cell Line: Relevant to your research (e.g., A549 for lung cancer studies [33]).
- siRNA Transfection Reagent: Such as RNAiMax or other lipid-based agents [35].
- Validated siRNA Sequences: Synthesized candidate siRNAs, a non-targeting scrambled negative control siRNA, and a positive control siRNA (e.g., against a housekeeping gene) [35] [33].
- qRT-PCR Kit: For quantifying mRNA knockdown.
- Western Blot Equipment: For assessing protein-level knockdown.
Step-by-Step Procedure
Cell Culture and Transfection:
- Plate cells at 40-80% confluency in appropriate growth media without antibiotics 24 hours before transfection [35].
- Complex the candidate siRNAs with the transfection reagent according to the manufacturer's instructions. A reverse transfection protocol, where cells are plated directly onto the siRNA-reagent complexes, can save time and sometimes improve efficiency [35].
- Critical: Transfect multiple siRNA candidates (2-4) targeting different regions of the same mRNA to ensure consistent biological effects and confirm on-target activity [31].
- Include both negative control (scrambled sequence) and positive control siRNAs in every experiment.
Optimization of Transfection Conditions:
- Titrate the siRNA concentration (e.g., 1-50 nM) to find the lowest dose that achieves maximal knockdown, thereby minimizing potential off-target effects and cytotoxicity [35].
- If cytotoxicity is observed, consider replacing the transfection media with fresh growth media 8-24 hours post-transfection.
Efficiency Measurement:
- mRNA Knockdown: Harvest cells 24-48 hours post-transfection. Isolate total RNA and perform quantitative RT-PCR (qRT-PCR) to measure remaining target mRNA levels, normalized to a housekeeping gene. Effective siRNAs should achieve ≥70% knockdown of the target mRNA [35].
- Protein Knockdown: Harvest cells 48-72 hours post-transfection. Analyze protein expression levels via Western blot or other immunoassays. The time to maximal protein knockdown depends on the protein's half-life [35].
The relationship between optimal design, successful RISC loading, and mRNA cleavage is summarized in the following pathway diagram.
The strategic selection of accessible mRNA target sites and the careful management of siRNA GC content are foundational to successful gene silencing. By following the detailed protocols and adhering to the quantitative design criteria outlined in this application note, researchers can systematically enhance the efficacy and reliability of their siRNA-based experiments. This structured approach, which integrates robust computational screening with rigorous experimental validation, is essential for generating high-quality data, advancing functional genomics research, and accelerating the development of siRNA therapeutics.
Within the realm of targeted gene knockdown research, small interfering RNAs (siRNAs) represent a powerful therapeutic tool. However, their application is contingent upon overcoming inherent challenges related to stability and bioavailability. Nuclease degradation and off-target effects can significantly hinder their efficacy. Consequently, strategic chemical modification is not merely an enhancement but a fundamental requirement for developing viable siRNA-based therapeutics. This Application Note delineates the critical roles of three cornerstone chemical modifications—2'-O-Methyl (2'-OMe), 2'-Fluoro (2'-F), and phosphorothioate (PS) linkages—in optimizing siRNA performance. We provide a synthesized overview of quantitative findings, detailed protocols for introducing these modifications, and essential resources for the scientific practitioner, all framed within the context of a robust research and development workflow.
Modification Profiles and Quantitative Impact
Extensive research has been dedicated to understanding how specific chemical modifications influence the stability, potency, and specificity of siRNAs. The data summarized in the following tables provide a comparative overview of the key modifications discussed in this note.
Table 1: Comparison of Key siRNA Chemical Modifications
| Modification | Primary Function | Impact on Stability | Impact on Potency | Key Considerations |
|---|---|---|---|---|
| 2'-O-Methyl (2'-OMe) | Ribose modification; enhances nuclease resistance [36]. | Significantly improves serum stability; increasing 2'-OMe content enhances potency and duration in vivo [37]. | Can negatively impact activity at specific positions (e.g., guide strand position 14, 3' terminus of 20-mer guides) [37]. | Tolerability is highly position-dependent [37] [38]. |
| 2'-Fluoro (2'-F) | Ribose modification; confers nuclease resistance [36]. | Improves stability against nucleases [37]. | Generally well-tolerated; can partially compensate for negative effects of 3' terminal 2'-OMe in 20-mer guides when placed at position 5 [37]. | Often used in an alternating pattern with 2'-OMe [37]. |
| Phosphorothioate (PS) | Backbone modification; replaces non-bridging oxygen with sulfur [36]. | Improves resistance to nucleases, particularly exonucleases; enhances cellular uptake via hydrophobicity [39] [36]. | Extensive modification can reduce gene-silencing activity and increase cytotoxicity [36]. | Chirality matters: Rp at 5' end and Sp at 3' end of guide strand improve Ago2 loading and pharmacokinetics [39]. |
The quantitative impact of these modifications is critical for rational design. The table below consolidates key experimental findings on their effect on siRNA stability.
Table 2: Quantitative Data on Modification Efficacy
| Modification / Combination | Experimental Context | Key Quantitative Outcome | Source |
|---|---|---|---|
| 3' Terminal 2'-OMe (vs. 2'-F) | Fully modified, asymmetric siRNAs with 20-mer guide strands. | Reduced activity for >60% of sequences tested; IC50 increased up to 7.3-fold [37]. | Davis et al., 2020 |
| 2'-OMe + Cationic Oligosaccharide (ODAGal4) | Serum degradation assay with HPRT1-targeting siRNA. | Half-life increased from 5.50 h (unmodified) to 9.98 h [36]. | Sasaki et al., 2020 |
| PS + Cationic Oligosaccharide (ODAGal4) | Serum degradation assay with fully PS-modified siRNA and ODAGal4. | Half-life >15 times longer than unmodified siRNA without ODAGal4 [36]. | Sasaki et al., 2020 |
| Combined 2'-OMe & PS (MS) | CRISPR gRNA for co-electroporation with Cas9 mRNA. | Enabled efficient gene editing where unmodified gRNAs failed; increased nuclease resistance [40]. | Basila et al., 2017 |
Detailed Experimental Protocols
Protocol: Introducing 2'-O-Methyl and 2'-Fluoro Modifications via Solid-Phase Synthesis
This protocol outlines the procedure for synthesizing oligonucleotides with site-specific 2'-OMe and 2'-F modifications using solid-phase phosphoramidite chemistry, which is suitable for producing siRNAs and guide RNAs up to approximately 100 nucleotides [41] [40].
I. Materials and Reagents
- Phosphoramidites: Standard 2'-ACE-protected ribo-, 2'-O-methyl-, and 2'-fluoro-phosphoramidites (e.g., from Glen Research or ChemGenes).
- Solid Support: Controlled-pore glass (CPG) or polystyrene support with the first nucleoside attached.
- Activating Agent: 0.25 M 5-Benzylthio-1H-tetrazole (BTT) in acetonitrile (ACN).
- Oxidizer Solution: 0.02 M Iodine in THF/Pyridine/Water.
- Capping Solutions: Cap A (Acetic Anhydride/Pyridine/THF) & Cap B (1-Methylimidazole/Pyridine/THF).
- Deprotection Reagent: Methylamine in aqueous ethanol or AMA (Ammonium Hydroxide / 40% Aqueous Methylamine).
- Solvents: Anhydrous acetonitrile, Dichloromethane (DCM).
- Synthesis Equipment: Automated DNA/RNA synthesizer.
II. Step-by-Step Procedure
- Preparation: Prime the synthesizer and reagent lines with anhydrous acetonitrile. Load the appropriate phosphoramidites and the solid support containing the 3'-terminal nucleoside into the instrument.
- Detritylation: Flush the column with 3% trichloroacetic acid (TCA) in DCM for 25-35 seconds to remove the 5'-DMT protecting group, then wash with ACN.
- Coupling: Deliver the designated phosphoramidite (standard, 2'-OMe, or 2'-F) and the activating agent (BTT) simultaneously to the column. Couple for 2-5 minutes depending on the specific phosphoramidite and sequence.
- Oxidation: Flush the column with the standard iodine oxidizer for 15-30 seconds to convert the phosphite triester to a phosphate triester. For a Phosphorothioate (PS) linkage, substitute the oxidizer with a sulfurization reagent (e.g., 0.05 M solution of DDTT in ACN).
- Capping: Flush the column with Cap A and Cap B solutions simultaneously for 10-15 seconds each to block unreacted 5'-OH groups from further elongation.
- Cycle Completion: Repeat steps 2-5 for each subsequent nucleotide until the full sequence is assembled.
- Cleavage and Deprotection: Cleave the oligonucleotide from the support and remove the base and 2'-ACE protecting groups by treating the controlled-pore glass with methylamine/AMM at a specific temperature (e.g., 65°C for 15-30 minutes) [40].
- Purification and Analysis: Desalt the crude oligonucleotide (e.g., via size-exclusion chromatography). For higher purity, purify by preparative anion-exchange HPLC or reverse-phase HPLC. Analyze the final product by LC-MS or MALDI-TOF for identity and purity confirmation [41] [40].
Protocol: Serum Stability Assay for Modified siRNAs
This protocol describes a standard method for evaluating the nuclease resistance of chemically modified siRNAs in serum, a critical test for predicting in vivo performance [36].
I. Materials and Reagents
- Test siRNA: Chemically modified siRNA duplex, resuspended in nuclease-free buffer.
- Control siRNA: Unmodified or differently modified siRNA duplex of the same sequence.
- Serum: Mouse, human, or fetal bovine serum (FBS).
- Stop Solution: 8 M Urea, 50 mM EDTA, 0.05% Bromophenol Blue, 0.05% Xylene Cyanol.
- Proteinase K
- Equipment: Water bath or incubator (37°C), gel electrophoresis system, gel imager.
II. Step-by-Step Procedure
- Reaction Setup: Prepare a mixture containing serum (e.g., 90% final concentration) and pre-warm it to 37°C. Add the siRNA duplex to a final concentration of 0.5-1 µM to initiate the degradation reaction. Incubate at 37°C.
- Time-Point Sampling: At predetermined time points (e.g., 0, 1, 3, 6, 12, 24 hours), withdraw an aliquot of the reaction mixture.
- Reaction Termination: Immediately mix the aliquot with an equal volume of Stop Solution. Add Proteinase K (e.g., 1 mg/mL final concentration) and incubate at 37°C for 30 minutes to digest serum proteins.
- Analysis by Gel Electrophoresis: Load the processed samples onto a non-denaturing polyacrylamide gel (e.g., 15%) or an agarose gel. Run the gel at a constant voltage until sufficient separation of intact siRNA from degradation products is achieved.
- Visualization and Quantification: Stain the gel with a nucleic acid stain (e.g., SYBR Gold) and image using a gel documentation system. Quantify the band intensity of the intact siRNA duplex.
- Data Analysis: Plot the percentage of remaining intact siRNA against time. Calculate the half-life of the siRNA by fitting the data to a first-order decay model or an exponential decay function.
Workflow and Pathway Visualization
The following diagram illustrates the logical workflow for designing, creating, and testing chemically modified siRNAs, as detailed in the protocols above.
Diagram 1: Workflow for developing chemically modified siRNAs.
The strategic placement of modifications is critical for success. The subsequent diagram outlines a decision pathway for selecting the appropriate modification based on the desired outcome, incorporating findings on positional tolerability.
Diagram 2: Decision pathway for selecting chemical modifications.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of the protocols and strategies outlined in this note relies on access to high-quality, specialized reagents. The following table lists key resources for siRNA modification research.
Table 3: Essential Research Reagents for siRNA Modification
| Reagent / Resource | Function / Application | Example / Source |
|---|---|---|
| 2'-OMe & 2'-F Phosphoramidites | Building blocks for solid-phase synthesis of modified RNA oligonucleotides. | Commercially available from vendors like Glen Research, ChemGenes, and Sigma-Aldrich. |
| Cationic Oligosaccharide (ODAGal4) | Binds major groove of siRNA duplex; synergistically enhances stability, especially with PS modifications [36]. | Synthesized as described in Sasaki et al., 2020 [36]. |
| Chiral (stereodefined) PS Reagents | For introducing phosphorothioate linkages with specific Rp or Sp configuration to improve Ago2 loading and pharmacokinetics [39]. | Specialized phosphoramidites or sulfurizing agents (e.g., DDTT). |
| Automated Oligonucleotide Synthesizer | Instrument for automated solid-phase synthesis of modified and unmodified oligonucleotides. | Instruments from vendors like Biolytic, GE Healthcare, and K&A Labs. |
| Stability Assay Components | For evaluating nuclease resistance of modified siRNAs in biologically relevant conditions. | Commercial sera (e.g., FBS from Gibco), Proteinase K (e.g., from Roche). |
The therapeutic application of small interfering RNA (siRNA) for targeted gene knockdown represents a paradigm shift in biomedical research and drug development. siRNA operates by harnessing the endogenous RNA interference (RNAi) pathway, where the RNA-induced silencing complex (RISC) is guided by the siRNA to complementary mRNA sequences, resulting in their cleavage and degradation, thereby preventing translation of the target protein [42] [43]. However, the major hurdle confronting siRNA therapeutics is the efficient and specific delivery of these nucleic acids to target cells and tissues. Naked siRNA is susceptible to rapid nuclease degradation, suffers from poor cellular uptake, and can elicit unintended immune responses [44] [43] [45]. Consequently, the development of sophisticated delivery platforms is paramount to the success of RNAi-based therapies. This application note details the core delivery strategies—Lipid Nanoparticles (LNPs), GalNAc conjugates, and Viral Vectors—framed within the context of siRNA research for targeted gene knockdown.
The three primary delivery platforms offer distinct mechanisms for siRNA delivery, each with unique advantages and limitations. Lipid Nanoparticles (LNPs) are multi-component, spherical nanoscale carriers that encapsulate and protect siRNA, facilitating cellular uptake and endosomal escape [42] [43]. GalNAc (N-acetylgalactosamine) conjugates represent a ligand-based approach, where siRNA is directly conjugated to a trivalent GalNAc moiety that selectively targets the Asialoglycoprotein Receptor (ASGPR) highly expressed on hepatocytes [44]. Viral Vectors, such as those based on Adeno-Associated Virus (AAV), are engineered viruses designed to deliver genetic material encoding for short hairpin RNAs (shRNAs) that are processed into siRNAs inside the cell [46].
Table 1: Comparative Analysis of Key siRNA Delivery Platforms
| Feature | Lipid Nanoparticles (LNPs) | GalNAc Conjugates | Viral Vectors (e.g., AAV) |
|---|---|---|---|
| Mechanism | Nanoparticle encapsulation and cellular endocytosis [43] | Receptor-mediated endocytosis via ASGPR [44] | Viral infection and transduction leading to sustained shRNA expression [46] |
| Primary Application | Hepatocyte & non-hepatocyte targets (e.g., HSCs); vaccines [42] [47] | Highly specific hepatocyte delivery [44] [48] | Long-term gene silencing; research applications [46] |
| Key Advantage | Versatility in targeting; high payload capacity; proven clinical success [42] [43] | Exceptional hepatocyte specificity; simple, well-defined chemistry; subcutaneous administration [44] | Potentially durable, long-lasting silencing effect from a single dose [46] |
| Key Limitation | Potential for off-target accumulation; complex formulation [42] [49] | Restricted primarily to liver hepatocytes [49] [45] | Risk of immunogenicity; limited payload capacity; potential for genomic integration [43] [46] |
| Clinical Status | Multiple approved drugs (e.g., Patisiran) and vaccines [42] [43] | Multiple approved drugs (e.g., Givosiran) and late-stage candidates [44] [42] | Widely used in gene therapy; some concerns for RNAi applications (e.g., oncogenesis) [50] |
Table 2: Quantitative Performance Metrics of Delivery Platforms from Preclinical Studies
| Platform / Specific Technology | Target Gene / Model | Key Efficacy Metric | Result |
|---|---|---|---|
| LNP (AA-T3A-C12 lipidoid) | HSP47 / CCl4-induced mouse liver fibrosis model [47] | Gene Silencing (HSP47 protein) | ~65% knockdown [47] |
| LNP (AA-T3A-C12 lipidoid) | HSP47 / CCl4-induced mouse liver fibrosis model [47] | Collagen Deposition Reduction | Significant reduction vs. MC3 LNP [47] |
| GalNAc-LNP (GL6 Design) | ANGPTL3 / LDLR-deficient NHP [48] | Liver Editing Efficiency (CRISPR) | 61% editing (vs. 5% without ligand) [48] |
| GalNAc-LNP (GL6 Design) | ANGPTL3 / Wild-type NHP [48] | Protein Reduction (ANGPTL3) | 89% reduction at 6 months [48] |
| Galactose-Liposome (Gal-LipoNP) | Fas / ConA-induced mouse hepatitis model [50] | Hepatoprotection (Serum ALT/AST) | Significant reduction vs. controls [50] |
Key Principles and Mechanisms
Lipid Nanoparticles (LNPs) and Targeted Delivery
LNPs are complex systems typically composed of four key lipid components: an ionizable lipid, phospholipid, cholesterol, and a PEG-lipid [42] [47]. The ionizable lipid is crucial for encapsulating the negatively charged siRNA and facilitating endosomal escape due to its ability to become positively charged in the acidic environment of the endosome [42]. Recent advances focus on modifying these components, particularly the ionizable lipid, to achieve cell-specific targeting beyond hepatocytes.
For instance, to treat liver fibrosis, researchers have developed ligand-tethered lipidoids. A combinatorial library of anisamide (AA)-tethered lipidoids was synthesized and screened to identify candidates (e.g., AA-T3A-C12) that selectively deliver siRNA to activated hepatic stellate cells (HSCs) via the overexpressed sigma receptor [47]. This LNP demonstrated superior silencing of the Hsp47 gene and greater reduction of collagen deposition compared to non-targeted LNPs [47].
GalNAc-Conjugates and ASGPR Biology
The GalNAc-siRNA conjugate platform leverages the high-affinity interaction between synthetic GalNAc ligands and the Asialoglycoprotein Receptor (ASGPR) [44]. ASGPR is a C-type lectin abundantly expressed on the sinusoidal surface of hepatocytes (~500,000 to 1,000,000 receptors per cell) [44]. Its natural function is to clear desialylated glycoproteins from circulation. The receptor exhibits a preference for multivalent GalNAc presentations (tri > di > mono), which enhances binding avidity and triggers rapid clathrin-mediated endocytosis [44] [48]. Upon endocytosis, a small fraction of the siRNA conjugate escapes the endosomal compartment to reach the cytoplasm and load into the RISC, while the remainder is degraded in the lysosome. The receptor itself recycles back to the cell membrane [44]. This efficient targeting system allows for robust gene silencing in hepatocytes after subcutaneous administration with minimal off-target effects.
Diagram 1: GalNAc-siRNA Conjugate Mechanism
Viral Vectors for shRNA Delivery
Viral vectors deliver genetic material that encodes short hairpin RNAs (shRNAs). Inside the cell nucleus, this genetic cassette is transcribed, and the shRNA transcript is exported to the cytoplasm and processed by the enzyme Dicer into functional siRNA, which then enters the RISC pathway [43] [46]. While this platform can achieve long-lasting gene silencing, its application is tempered by potential risks, including sustained overexpression of shRNAs leading to cytotoxicity, and immune responses against the viral capsid [46] [50].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for siRNA Delivery
| Reagent / Material | Function / Application | Key Characteristics |
|---|---|---|
| Ionizable Lipids (e.g., MC3, C12-200) | Core component of LNPs; enables siRNA encapsulation and endosomal escape [42] [48] | Positively charged at low pH; biodegradable; defines LNP tropism and potency [42] |
| Targeting Ligands (e.g., GalNAc, Anisamide) | Confers cell-specific targeting to nanoparticles or conjugates [44] [47] | High affinity for target receptor (e.g., ASGPR, Sigma receptor); compatible with conjugation chemistry |
| Chemically Modified siRNA | The active therapeutic agent; designed for specific mRNA target [44] [43] | 2'-fluoro (2'-F) or 2'-O-methyl (2'-OMe) modifications to enhance stability and reduce immunogenicity [44] |
| Polyethylene Glycol (PEG)-Lipid | Component of LNPs; stabilizes particle formation and modulates pharmacokinetics [42] [47] | Short acyl chains promote rapid dissociation in vivo; influences particle size and opsonization |
| AAV Vectors (e.g., AAV8, AAV9) | Viral delivery system for shRNA genes; provides long-term expression [46] | Serotype determines tissue tropism; limited packaging capacity (~4.7 kb) |
Detailed Experimental Protocols
Protocol 1: Formulating Targeted LNPs for Hepatic Stellate Cell Delivery
This protocol details the synthesis of anisamide-targeted lipidoids and the formulation of siRNA-LNPs for targeting activated HSCs in liver fibrosis models, based on the work of Wang et al. [47].
A. Synthesis of AA-T3A-C12 Lipidoid
- Reaction Setup: In a single pot, conjugate anisoyl-N-hydroxysuccinimide (anisoyl-NHS, 1 equiv) to the primary amine of the branched polyamine core T3A (1 equiv) in dimethyl sulfoxide (DMSO). Stir at room temperature for 10-12 hours under an inert atmosphere.
- Tail Addition: Without purification, add an excess of the epoxide tail 1,2-Epoxydecane (C12, ~8 equiv) to the reaction mixture. Heat to 60-70°C and stir vigorously for 48-72 hours to allow the ring-opening reaction to proceed to completion.
- Product Isolation: After cooling, precipitate the crude AA-T3A-C12 lipidoid in diethyl ether. Re-dissolve and dialyze the product against molecular grade water using a 3.5 kDa molecular weight cut-off (MWCO) membrane for 48 hours to remove unreacted precursors and solvents. Lyophilize the purified lipidoid to obtain a solid for long-term storage at -20°C.
B. Formulation of AA-T3A-C12/siRNA LNPs via Microfluidic Mixing
- Lipid Phase Preparation: Dissolve the synthesized AA-T3A-C12 lipidoid, cholesterol, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), and PEG-lipid (e.g., C14-PEG) at a molar ratio of 50:38.5:10:1.5 in pure ethanol to a final lipid concentration of 10-20 mg/mL.
- Aqueous Phase Preparation: Dilute the siRNA (e.g., against Hsp47) in a citrate buffer (25 mM, pH 4.0) to a concentration of 0.1-0.2 mg/mL.
- Nanoparticle Formation: Use a microfluidic mixer (e.g., NanoAssemblr, Precision NanoSystems). Set the flow rate ratio (aqueous:organic) to 3:1 and a total flow rate of 12 mL/min. Combine the two phases in the mixing chamber to form stable LNPs.
- Buffer Exchange and Characterization: Dialyze the formulated LNPs extensively against phosphate-buffered saline (PBS, 1X, pH 7.4) at 4°C to remove residual ethanol and adjust the pH. Characterize the final formulation for particle size (Dynamic Light Scattering, target ~65 nm), polydispersity index (PDI, target <0.2), and siRNA encapsulation efficiency (RiboGreen assay, target >85%) [47].
Diagram 2: Targeted LNP Formulation Workflow
Protocol 2: Evaluating GalNAc-Conjugated siRNA In Vivo
This protocol outlines the steps for testing the efficacy of a GalNAc-siRNA conjugate in an animal model of liver disease, derived from established preclinical studies [44] [48].
A. Animal Model and Dosing
- Model Selection: Select an appropriate mouse model (e.g., C57BL/6 for general studies, or specific disease models like
Ldlr-/-for hypercholesterolemia studies). - Conjugate Administration: Prepare the GalNAc-siRNA conjugate in a sterile, physiological buffer such as PBS. Administer the conjugate via subcutaneous (s.c.) injection into the interscapular region or dorsal skinfold. A typical dose for a robust silencing effect in mice ranges from 1 to 10 mg/kg of conjugate. Include control groups receiving a non-targeting siRNA conjugate or PBS.
B. Tissue Collection and Analysis
- Sample Collection: At the experimental endpoint (e.g., 3-7 days post-injection), euthanize the animals and collect blood via cardiac puncture. Perfuse the liver with cold PBS via the portal vein to remove blood. Excise the liver, snap-freeze a portion in liquid nitrogen for molecular analysis, and preserve another portion in formalin for histology.
- Efficacy Assessment:
- mRNA Knockdown: Isolate total RNA from homogenized liver tissue using a commercial kit. Perform quantitative reverse transcription PCR (qRT-PCR) to quantify the levels of the target mRNA, normalized to a housekeeping gene (e.g., Gapdh). Calculate percentage knockdown relative to the control group.
- Protein Knockdown: Analyze the target protein level by Western blot or enzyme-linked immunosorbent assay (ELISA) from liver lysates or serum samples, depending on the protein.
- Phenotypic Assessment: For disease models, include relevant histological staining (e.g., H&E for general morphology, Sirius Red for collagen/fibrosis) and measure serum biomarkers (e.g., ALT, AST for hepatotoxicity) to correlate gene silencing with functional improvement.
The choice of delivery platform is a critical determinant in the success of any siRNA-based research or therapeutic program. LNPs offer versatility and high potency, with emerging capabilities for targeting specific liver cell types beyond hepatocytes. GalNAc conjugates provide an elegant, simple, and exceptionally efficient solution for hepatocyte-specific gene silencing. Viral vectors remain a powerful tool for long-term silencing in research but require careful consideration of safety profiles. By understanding the principles, advantages, and practical protocols associated with each platform, researchers can strategically select and optimize the right delivery system to advance their siRNA gene knockdown projects from the bench toward the clinic.
Small interfering RNA (siRNA) therapeutics have emerged as a powerful class of drugs that leverage the natural RNA interference (RNAi) pathway to achieve targeted gene knockdown. By mediating sequence-specific degradation of messenger RNA (mRNA), siRNA molecules can precisely silence disease-causing genes, offering a transformative approach for treating conditions with genetic underpinnings [3]. The field has matured significantly since the Nobel Prize-winning discovery of RNAi in 1998, with the first FDA-approved siRNA therapeutic (patisiran) gaining regulatory clearance in 2018 and numerous additional drugs receiving approval in subsequent years [51] [3]. This application note details experimental protocols and key applications of siRNA technology across genetic disorders, oncology, and viral infections, providing researchers with practical methodologies for therapeutic development.
The fundamental mechanism of siRNA action involves synthetic double-stranded RNA duplexes, typically 21-23 nucleotides in length with 3' dinucleotide overhangs [5]. Upon introduction into the cytosol, the antisense (guide) strand loads into the RNA-induced silencing complex (RISC), directing it to complementary target mRNA sequences. The Argonaute 2 (AGO2) endonuclease within RISC then catalyzes site-specific cleavage of the target mRNA, effectively preventing translation of the pathogenic protein [3]. This exquisite specificity enables targeting of previously "undruggable" proteins, positioning siRNA therapeutics as a versatile modality for precision medicine across diverse disease areas [24].
siRNA Applications in Genetic Disorders
Clinical Successes and Therapeutic Approach
siRNA therapeutics have demonstrated remarkable success in treating hereditary genetic disorders, particularly those affecting the liver. The paradigm for this application involves silencing dominant-negative mutant genes or modulating pathological pathways through precise gene knockdown. Approved therapies primarily utilize either lipid nanoparticles (LNPs) or GalNAc conjugates for hepatocyte-directed delivery, capitalizing on the liver's role in synthesizing many pathogenic proteins [51] [3].
Table 1: Approved siRNA Therapeutics for Genetic Disorders
| Therapeutic | Target Gene | Indication | Delivery System | Key Clinical Outcome |
|---|---|---|---|---|
| Patisiran | TTR | Hereditary transthyretin-mediated amyloidosis | LNP (DLin-MC3-DMA) | Improved neuropathy scores, reduced TTR protein [51] |
| Vutrisiran | TTR | Hereditary transthyretin-mediated amyloidosis | GalNAc conjugate | Significantly lower side effects vs. patisiran [51] |
| Givosiran | ALAS1 | Acute hepatic porphyria | GalNAc conjugate | 74% reduction in frequency of main symptoms [51] |
| Lumasiran | HAO1 | Primary hyperoxaluria type 1 | GalNAc conjugate | Reduced oxalate production [3] |
The following diagram illustrates the mechanistic differences between LNP and GalNAc-conjugated siRNA delivery to hepatocytes:
Protocol: Development of siRNA Therapeutics for Genetic Disorders
Phase 1: Target Selection and siRNA Design
- Identify pathogenic gene: Select genes with validated disease association through genetic studies (e.g., TTR for hATTR amyloidosis, ALAS1 for acute hepatic porphyria) [51].
- Design siRNA sequences: Utilize bioinformatics tools (e.g., Cm-siRPred) to predict optimal siRNA sequences targeting conserved regions of the mRNA [52]. Focus on regions with minimal homology to other human transcripts to reduce off-target effects.
- Incorporate chemical modifications: Include 2'-O-methyl (2'-OMe), 2'-fluoro (2'-F) substitutions, and phosphorothioate (PS) bonds to enhance nuclease resistance, reduce immunogenicity, and improve pharmacokinetic properties [3].
Phase 2: Delivery System Optimization
- Select delivery platform: For liver targets, choose between LNP and GalNAc-conjugation based on intended administration route and safety profile [51].
- Formulate LNP systems: Combine ionizable lipids (e.g., DLin-MC3-DMA), helper phospholipids (e.g., DSPC), cholesterol, and PEGylated lipids (e.g., PEG2000-C-DMG) using microfluidic mixing technology [51].
- Conjugate GalNAc ligands: Synthesize triantennary GalNAc structures for covalent attachment to siRNA sense strand, enabling selective uptake via asialoglycoprotein receptor (ASGPR) on hepatocytes [3].
Phase 3: In Vitro and In Vivo Validation
- Cell-based screening: Transfect hepatocyte cell lines (e.g., HepG2, primary hepatocytes) using appropriate transfection reagents. Measure target mRNA knockdown via qRT-PCR and protein reduction via Western blot at 48-72 hours post-transfection [5].
- Animal efficacy studies: Administer formulated siRNA to disease-relevant animal models (e.g., transgenic mice for hATTR). Use dose range of 0.1-10 mg/kg via intravenous (LNP) or subcutaneous (GalNAc) routes. Monitor target protein reduction in plasma and tissues over 2-8 weeks [51].
- Toxicology assessment: Evaluate histopathological changes, immune activation, and liver enzyme elevations in rodent and non-rodent species following repeat dosing [53].
siRNA Applications in Oncology
Novel Approaches for Cancer Therapy
Oncology represents a promising frontier for siRNA therapeutics, with applications focusing on silencing oncogenes, genes involved in synthetic lethal interactions, and targets implicated in drug resistance. Recent advances have enabled more efficient delivery to tumor sites through improved nanoparticle designs and targeting ligands [24] [54]. siRNA's programmability allows for simultaneous targeting of multiple pathways, offering opportunities for combination approaches that address cancer heterogeneity and evolution.
Table 2: Promising siRNA Targets and Approaches in Oncology
| Molecular Target | Cancer Type | Therapeutic Approach | Development Status |
|---|---|---|---|
| PLK1 | Colorectal cancer | Synthetic lethality via cholesterol-enriched exosomes | Preclinical [24] |
| STAT3 | Multiple solid tumors | siRNA targeting signaling pathway | Clinical trials (DCR-STAT3) [54] |
| PD-L1 | Immuno-oncology | Immune checkpoint blockade | Clinical trials (DCR-PDL1) [54] |
| KRAS | Pancreatic, lung | Targeting undruggable oncogenes | Preclinical development |
The synthetic lethality approach using siRNA is particularly promising for targeting cancer-specific vulnerabilities:
Protocol: siRNA-Based Cancer Therapeutic Development
Phase 1: Target Identification and Validation
- Genetic dependency screening: Perform high-throughput siRNA screens against candidate targets in cancer cell lines using arrayed siRNA libraries (e.g., ON-TARGETplus). Identify genes essential for cancer cell survival but dispensable in normal cells [5].
- Synthetic lethal partner identification: For cancers with known mutations (e.g., BRCA1/2), screen for synthetic lethal interactions using combinatorial siRNA approaches [24].
- Biomarker development: Identify predictive biomarkers for patient stratification using gene expression profiling and functional genomic approaches.
Phase 2: Tumor-Targeted Delivery System Development
- Exosome engineering: Isolate exosomes from appropriate cell sources and enrich membrane cholesterol content (Chol/MEs) to enhance stability and promote membrane fusion-mediated cytosolic delivery [24].
- Active targeting strategies: Functionalize nanoparticles with tumor-targeting ligands (e.g., antibodies, peptides, aptamers) that recognize tumor-specific antigens [52].
- Formulation optimization: Develop stable formulations suitable for various administration routes, including intravenous, intratumoral, and oral delivery for gastrointestinal cancers [24].
Phase 3: Preclinical Efficacy Assessment
- In vitro potency assays: Treat cancer cell lines with formulated siRNA and assess:
- Gene silencing efficiency (qRT-PCR, Western blot)
- Functional consequences (cell proliferation, apoptosis, cell cycle analysis)
- Combination effects with standard therapies [5]
- In vivo tumor models: Evaluate antitumor activity in subcutaneous and orthotopic xenograft models, as well as genetically engineered mouse models:
- Administer siRNA formulations at 1-5 mg/kg dose, 2-3 times weekly
- Monitor tumor growth, metastasis, and animal survival
- Assess target engagement in tumor tissue [24]
- PD/PK studies: Characterize pharmacokinetics, biodistribution, and pharmacodynamic effects of siRNA in tumor and normal tissues.
siRNA Applications in Viral Infections
Antiviral Strategies and Targets
siRNA therapeutics offer a promising approach for combating viral infections by directly targeting essential viral genes or host dependency factors. This strategy has been successfully applied against various viruses, including hepatitis B virus (HBV), SARS-CoV-2, and human immunodeficiency virus (HIV) [55]. The sequence-specific nature of siRNA enables rapid adaptation to emerging viral variants, making it particularly valuable for addressing antiviral resistance and pandemic preparedness.
Table 3: siRNA Applications in Viral Infections
| Viral Pathogen | Target Strategy | Key Targets | Development Status |
|---|---|---|---|
| HBV | Viral gene silencing | HBsAg, polymerase | Clinical trials [55] |
| SARS-CoV-2 | Conserved viral regions | RNA-dependent RNA polymerase, spike | Preclinical/Clinical development [55] |
| HIV | Viral and host targets | Viral structural genes, host co-receptors | Preclinical development [55] |
| Respiratory viruses | Host dependency factors | Viral entry receptors, replication machinery | Research phase |
The following workflow outlines the development process for antiviral siRNA therapeutics:
Protocol: Developing siRNA Therapeutics for Viral Infections
Phase 1: Antiviral siRNA Design and Screening
- Target selection: Identify highly conserved regions in viral genomes or essential host factors through sequence alignment and dependency factor screening [55].
- siRNA design: Design multiple siRNAs targeting different regions of the viral genome to address potential viral escape mutants. Utilize algorithms that minimize off-target effects while maximizing antiviral potency.
- Chemical optimization: Incorporate 2'-fluoro, 2'-O-methyl, and phosphorothioate modifications to enhance stability in biological fluids and reduce immune stimulation [3].
Phase 2: Delivery to Infected Tissues
- Tissue-specific delivery: For respiratory viruses (e.g., SARS-CoV-2), develop inhalable LNP formulations for direct lung delivery. For hepatotropic viruses (e.g., HBV), utilize GalNAc conjugation or LNPs for liver targeting [55].
- Formulation for intracellular delivery: Optimize LNP compositions containing ionizable lipids (e.g., DLin-MC3-DMA) that promote endosomal escape and cytosolic release of siRNA in infected cells [51].
- Biodistribution assessment: Evaluate tissue distribution of radiolabeled or fluorescently tagged siRNA formulations in animal models to verify delivery to sites of viral replication.
Phase 3: Antiviral Efficacy Assessment
- In vitro antiviral activity:
- Infect permissive cell lines with relevant viruses
- Treat with siRNA formulations at various time points (pre-, co-, or post-infection)
- Quantify viral replication (plaque assay, qPCR for viral RNA)
- Assess cytopathic effect protection [55]
- In vivo efficacy studies:
- Utilize established animal models of viral infection (e.g., humanized mice for HBV, hamster or mouse models for SARS-CoV-2)
- Administer siRNA prophylactically or therapeutically at doses of 1-10 mg/kg
- Monitor viral loads in target tissues, clinical disease parameters, and histopathology
- Evaluate potential for viral escape mutations through sequencing [55]
- Immune response characterization: Assess innate immune activation (e.g., interferon response) and adaptive immunity to both the viral pathogen and siRNA formulation.
Technical Considerations and Research Reagent Solutions
Essential Research Tools and Reagents
Successful development of siRNA therapeutics requires carefully selected reagents and methodologies to address challenges in stability, delivery, and specificity. The following table outlines key solutions for siRNA research and development:
Table 4: Research Reagent Solutions for siRNA Therapeutic Development
| Reagent Category | Specific Products/Technologies | Function and Application |
|---|---|---|
| siRNA Design Tools | Cm-siRPred, eSkip-Finder, SMARTselection | Bioinformatics platforms for predicting optimal siRNA sequences and modification patterns [52] [5] |
| Chemical Modifications | 2'-OMe, 2'-F, PS bonds, GalNAc conjugation | Enhance nuclease resistance, reduce immunogenicity, enable tissue-specific delivery [3] |
| Delivery Systems | LNPs (DLin-MC3-DMA), GalNAc conjugates, cholesterol-enriched exosomes | Protect siRNA during circulation, facilitate cellular uptake, promote endosomal escape [51] [24] |
| In Vitro Screening | ON-TARGETplus siRNA, Accell siRNA, siRNA libraries | High-quality reagents for gene silencing in various cell types, including difficult-to-transfect cells [5] |
| Analytical Methods | RACE analysis, next-generation sequencing | Confirm target engagement, identify potential off-target effects [5] |
Critical Experimental Considerations
Optimizing Specificity and Reducing Off-Target Effects:
- Asymmetric modifications: Heavily modify the passenger strand while minimally modifying the guide strand to promote correct RISC loading [3].
- Seed region optimization: Incorporate thermodynamic-based design considerations or chemical modifications in the seed region (nucleotides 2-8) to discourage microRNA-like off-target interactions [5].
- Pooling strategies: Use pools of multiple siRNAs targeting the same gene to reduce concentration-dependent off-target effects while maintaining potent on-target silencing [5].
- Bioinformatic filtering: Exclude siRNA sequences with high complementarity to off-target transcripts, particularly in the seed region [5].
Addressing Delivery Challenges:
- Endosomal escape enhancement: Incorporate ionizable lipids in LNP formulations that become protonated in acidic endosomes, promoting membrane disruption and cytosolic release [51].
- Tissue-specific targeting: Employ ligand conjugation (e.g., GalNAc for hepatocytes, antibody fragments for tumor antigens) to improve tissue selectivity and reduce dose-limiting toxicities [52].
- Stability optimization: Utilize comprehensive chemical modification patterns including 2'-O-methyl, 2'-fluoro, and phosphorothioate linkages to resist nucleases and extend half-life [3].
The field of siRNA therapeutics continues to evolve rapidly, with ongoing advances in delivery technologies, chemical modifications, and target validation strategies enabling expansion into new disease areas. By applying the methodologies and considerations outlined in this application note, researchers can accelerate the development of innovative siRNA-based treatments for genetic disorders, cancer, viral infections, and beyond.
Small interfering RNA (siRNA) represents a powerful class of therapeutic agents that harness the natural cellular process of RNA interference (RNAi) to achieve targeted gene knockdown. This mechanism, first discovered in plants and later in C. elegans and mammalian cells, enables sequence-specific silencing of disease-causing genes at the mRNA level [56]. The year 2018 marked a transformative milestone in this field with the first-ever FDA approval of an siRNA therapeutic, patisiran (ONPATTRO), for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis (hATTR) in adults [57] [58]. This approval heralded the arrival of an entirely new class of medicines that operate upstream of traditional protein-targeting drugs by preventing the synthesis of disease-causing proteins [58]. This case study examines the development, mechanisms, and experimental approaches for two pioneering siRNA drugs—patisiran and inclisiran—providing a framework for researchers investigating siRNA for targeted gene knockdown.
Drug Profiles and Clinical Efficacy
The translation of siRNA from a research tool to a clinical reality is exemplified by patisiran and inclisiran. While both utilize the core RNAi mechanism, they target different genes and diseases, showcasing the versatility of this platform.
Patisiran (ONPATTRO)
Patisiran is a double-stranded siRNA encapsulated in a lipid nanoparticle (LNP) that targets the mRNA of both mutant and wild-type transthyretin (TTR) [59]. It is indicated for the treatment of polyneuropathy in patients with hATTR amyloidosis, a progressive, debilitating, and often fatal genetic disease caused by mutations in the TTR gene [58]. The efficacy of patisiran was demonstrated in the APOLLO Phase 3 clinical trial, a global, randomized, double-blind, placebo-controlled study.
Table 1: Key Efficacy Results from the Patisiran APOLLO Phase 3 Trial [58]
| Clinical Measure | Baseline to 18 Months (Mean Change) | Treatment Difference vs. Placebo |
|---|---|---|
| Modified Neuropathy Impairment Score +7 (mNIS+7) | Patisiran: -6.0 point improvement | 34.0 points (p < 0.001) |
| Placebo: +28.0 point worsening | ||
| Norfolk Quality of Life-Diabetic Neuropathy (QoL-DN) Score | Patisiran: -6.7 point improvement | 21.1 points (p < 0.001) |
| Placebo: +14.4 point worsening | ||
| Patients with Reversal of Neuropathy Impairment (per mNIS+7) | 56% of patisiran patients vs. 4% of placebo patients | Not applicable |
Inclisiran (Leqvio)
Inclisiran is a synthetic, chemically modified siRNA conjugated to N-acetylgalactosamine (GalNAc) that targets the mRNA of proprotein convertase subtilisin/kexin type 9 (PCSK9) [60] [61]. It is approved as an adjunct to diet and statin therapy for the treatment of adults with hypercholesterolemia or mixed dyslipidemia to lower low-density lipoprotein cholesterol (LDL-C) [62]. Its long-acting effect allows for a dosing regimen of just two injections per year after the initial doses [63].
Table 2: Key Efficacy Results from Inclisiran Phase 3 Clinical Trials [60] [63] [62]
| Trial Population | LDL-C Reduction | Durability |
|---|---|---|
| Heterozygous Familial Hypercholesterolemia (ORION-9) | ~48% reduction | Sustained with twice-yearly dosing |
| Atherosclerotic Cardiovascular Disease (ORION-10 & ORION-11) | ~50% reduction | Sustained with twice-yearly dosing |
| Long-Term Extension (ORION-8, beyond 6 years) | Consistent ~50% reduction | Maintained; 78.4% of patients reached LDL-C target |
Comparative Mechanism of Action
A detailed understanding of the distinct mechanisms and delivery strategies of patisiran and inclisiran is crucial for research and development. The following diagram illustrates the core pathway and key differences in cellular uptake and trafficking.
Mechanism Workflow Description
The siRNA therapeutic mechanism involves several critical stages:
- Administration and Delivery: Patisiran is administered via intravenous infusion as a lipid nanoparticle (LNP) complex, while inclisiran is delivered via subcutaneous injection as a GalNAc conjugate [59] [61].
- Hepatocyte Uptake: The LNP of patisiran is opsonized by apolipoprotein E (ApoE) in the bloodstream, directing it to ApoE receptors on hepatocytes. Inclisiran's GalNAc moiety specifically binds to the asialoglycoprotein receptor (ASGPR) highly expressed on hepatocytes [59] [60] [61]. Both pathways lead to receptor-mediated endocytosis.
- Endosomal Escape and RISC Loading: Following endocytosis, the siRNA must escape the endosome into the cytosol. The LNP and GalNAc technologies are designed to facilitate this release. In the cytosol, the double-stranded siRNA is loaded into the RNA-induced silencing complex (RISC). The passenger strand is degraded, and the guide strand is retained [59] [56].
- Target mRNA Cleavage: The RISC, guided by the siRNA, binds to the complementary target mRNA sequence—TTR mRNA for patisiran and PCSK9 mRNA for inclisiran. The endonuclease Argonaute 2 (AGO2) within RISC cleaves the target mRNA, rendering it non-functional [59] [61].
- Gene Silencing and Phenotypic Effect: The cleaved mRNA is degraded, preventing its translation into the disease-causing protein. For patisiran, this reduces the production of both mutant and wild-type TTR protein, halting the formation of amyloid deposits in tissues [59] [58]. For inclisiran, reduced PCSK9 protein leads to increased recycling of LDL receptors (LDL-R) on the hepatocyte surface, enhancing clearance of LDL-C from the blood and lowering cholesterol [60] [61].
Experimental Protocols for siRNA Research
This section outlines key methodologies for evaluating siRNA therapeutics, from in vitro screening to in vivo efficacy and safety assessment, providing a template for preclinical research.
Protocol 1: In Vitro Screening of siRNA Efficacy and Off-Target Effects
Objective: To identify potent siRNA lead candidates and profile their potential off-target effects in a relevant cell line.
Materials:
- Research Reagents: siRNA library targeting gene of interest, non-targeting siRNA control (scrambled sequence), lipid-based transfection reagent, relevant cell line (e.g., HepG2 for liver targets), qRT-PCR reagents, RNA sequencing library prep kit.
- Equipment: Cell culture incubator, biosafety cabinet, nanodrop spectrophotometer, real-time PCR system, next-generation sequencer.
Methodology:
- Cell Seeding and Transfection: Seed appropriate cells in 96-well plates to reach 60-80% confluency at transfection. Using a transfection reagent, introduce a range of concentrations (e.g., 1 nM, 10 nM, 100 nM) of siRNA candidates and controls into the cells. Include replicates for statistical power.
- RNA Extraction and qRT-PCR: 48 hours post-transfection, lyse cells and extract total RNA. Synthesize cDNA and perform quantitative RT-PCR (qRT-PCR) using primers specific for the target mRNA (e.g., TTR or PCSK9) and housekeeping genes (e.g., GAPDH, β-actin). Calculate percentage target mRNA knockdown using the 2^(-ΔΔCt) method relative to the non-targeting control.
- Off-Target Analysis via Transcriptomics: For lead candidates, transfer the in vitro transfection to a larger scale (6-well plate). 48 hours post-transfection, extract high-quality total RNA. Prepare an RNA sequencing (RNA-Seq) library and perform sequencing on a next-generation platform. Bioinformatic analysis should map sequencing reads to the reference genome and identify genes that are differentially expressed in siRNA-treated cells compared to the non-targeting control, with a specific focus on seed-region-mediated off-target effects [56].
Protocol 2: In Vivo Assessment of Pharmacodynamics and Efficacy
Objective: To demonstrate target engagement and phenotypic effect in an animal model of disease.
Materials:
- Research Reagents: Formulated siRNA (e.g., LNP or GalNAc), vehicle control, animal disease model (e.g., transgenic for human TTR for hATTR amyloidosis), equipment for blood collection, ELISA kits for target protein (TTR/PCSK9) and biomarker (LDL-C) analysis.
- Equipment: Microsyringe for dosing, clinical chemistry analyzer.
Methodology:
- Dosing Regimen: Randomize animals into groups (e.g., vehicle control, siRNA-treated, positive control). Administer the formulated siRNA at a therapeutically relevant dose (e.g., 0.3 mg/kg for patisiran in mice) via the appropriate route (intravenous for LNP, subcutaneous for GalNAc) [59]. The control group receives an equivalent volume of vehicle.
- Longitudinal Blood Collection: Collect blood samples at predefined timepoints (e.g., pre-dose, Day 3, 7, 14, 21, and 28 post-dose). Process samples to isolate serum or plasma.
- Pharmacodynamic (PD) Analysis: Quantify the levels of the target protein (e.g., serum TTR for patisiran, serum PCSK9 for inclisiran) and relevant functional biomarkers (e.g., serum LDL-C for inclisiran) in the collected samples using ELISA or other immunoassays [59] [61]. Plot the percentage reduction over time to determine the depth and duration of the effect.
- Efficacy Endpoint Assessment: At the end of the study, evaluate disease-specific endpoints. For a neuropathy model, this could involve functional scoring or histopathological analysis of nerves. For a cardiovascular model, this could involve measuring aortic plaque burden. Compare results between the siRNA-treated and control groups to establish therapeutic efficacy.
The Scientist's Toolkit: Essential Research Reagents
The development and analysis of siRNA therapeutics require a specific set of reagents and tools. The following table details key components for a research pipeline.
Table 3: Essential Research Reagents for siRNA Therapeutic Development
| Reagent/Tool | Function | Example Application |
|---|---|---|
| siRNA Design Algorithms | In silico design of siRNA sequences with high potency and minimized off-target risk. | Selection of lead candidates targeting a novel gene; identification of sequences with perfect complementarity to target mRNA [56]. |
| Chemically Modified Nucleotides | Enhance siRNA stability against nucleases, reduce immunogenicity, and improve pharmacokinetics. | Incorporation of 2'-O-methyl, 2'-fluoro modifications to increase half-life in vivo [56]. |
| Delivery Vehicles (LNPs, GalNAc) | Facilitate cellular uptake and endosomal escape of siRNA into target cells (e.g., hepatocytes). | Formulating patisiran in LNPs for hepatic TTR knockdown; conjugating inclisiran to GalNAc for ASGPR-mediated liver delivery [59] [60]. |
| In Vitro Transcription/Translation Systems | Study the direct functional consequence of mRNA cleavage in a cell-free environment. | Confirming specific silencing of target mRNA protein product. |
| RNA-Induced Silencing Complex (RISC) Loading Assays | Measure the efficiency with which an siRNA is incorporated into the functional RISC machinery. | In vitro assessment of the intrinsic activity of chemically modified siRNA variants [56]. |
| Anti-Dicer/PKR Antibodies | Detect and quantify activation of innate immune responses by siRNA. | Monitoring potential off-target immune activation in treated cells or animal models [56]. |
The successful clinical translation of patisiran and inclisiran validates siRNA as a powerful therapeutic modality for targeted gene knockdown. Their development showcases two effective delivery paradigms—LNP and GalNAc-conjugation—for achieving potent and durable silencing of hepatocyte-expressed disease genes. The structured experimental protocols for in vitro screening and in vivo validation provide a roadmap for researchers aiming to develop the next generation of RNAi therapeutics. As the field progresses, overcoming challenges related to extra-hepatic delivery and long-term safety monitoring will further expand the reach of siRNA drugs, solidifying their role in the future of precision medicine.
Overcoming siRNA Hurdles: Strategies for Enhancing Efficacy and Ensuring Safety
Small interfering RNA (siRNA) functions as a powerful tool for precision gene silencing, harnessing the natural RNA interference (RNAi) pathway to degrade target messenger RNA (mRNA) and prevent protein translation. A key challenge, however, lies in the phenomenon of off-target effects, where siRNAs silence unintended genes. This application note details the mechanisms behind these effects, with a specific focus on seed region interactions, and provides validated protocols and strategies to mitigate them, ensuring the integrity of gene knockdown research and therapeutic development.
The core of the off-target problem often originates from a specific segment of the siRNA guide strand known as the seed region (nucleotides 2-8). When this region exhibits partial complementarity to non-target mRNAs, particularly in their 3' untranslated regions (3' UTRs), the siRNA can mimic the behavior of endogenous microRNA (miRNA). This leads to the translational repression or degradation of off-target transcripts [64] [65]. Furthermore, off-target effects can also be driven by the passenger strand if it is inadvertently loaded into the RNA-induced silencing complex (RISC) or through homology-based cross-hybridization outside the seed region [66].
Key Strategies for Mitigation
Multiple strategies have been developed to minimize off-target effects, which can be employed individually or in combination.
Chemical Modifications
Strategic chemical modification of siRNA is a primary method for enhancing specificity. Introducing specific moieties at critical positions can destabilize off-target binding without compromising on-target activity. Table 1 summarizes common modifications and their roles.
Table 1: Common Chemical Modifications for Reducing siRNA Off-Target Effects
| Modification | Common Placement | Primary Function | Impact on Off-Target Effects |
|---|---|---|---|
| 2'-O-Methyl (2'-OMe) [20] | Guide strand, especially seed region | Increases nuclease stability, reduces immunogenicity | Disrupts stable duplex formation with off-target mRNAs [65] |
| 2'-Fluoro (2'-F) [20] | Ribose sugar | Enhances binding affinity to target mRNA, improves stability | Can be combined with 2'-OMe to fine-tune specificity and potency |
| Phosphorothioate (PS) Linkage [20] | Backbone (replaces oxygen with sulfur) | Increases resistance to nucleases, improves pharmacokinetics | Improves overall drug properties, allowing for lower doses |
| Formamide [67] | Guide strand seed region | Inhibits hydrogen bonding in seed region | Selectively destabilizes interactions with off-target mRNAs |
A notable advance is the use of formamide modifications in the seed region. This group inhibits hydrogen bond formation, selectively destabilizing the short base-pairing with off-target mRNAs while having minimal impact on the stable duplex formed with the fully complementary on-target mRNA [67]. Research shows that introducing this modification at a single location in the seed region can suppress off-target effects more efficiently than existing chemical modifications [67].
Another critical approach involves asymmetric design to promote correct RISC loading. The RISC complex preferentially loads the strand with a less thermodynamically stable 5' end. Designs can exploit this by chemically destabilizing the 5' end of the passenger strand or by using a shorter passenger strand, thereby ensuring the intended guide strand is loaded and reducing off-target effects caused by the passenger strand [65].
siRNA Pooling
Using complex pools of siRNAs (d-siRNAs or e-siRNAs) targeting different regions of the same mRNA gene is a highly effective empirical strategy. While a single siRNA might have a potent but problematic seed sequence, a pool dilutes the concentration of any individual siRNA seed. This high complexity means that while hundreds of different siRNAs are active against the target, the concentration of any single problematic siRNA is too low to cause significant off-target silencing [66]. Studies have demonstrated that diced siRNA pools against p38α MAPK showed almost no off-target effects, whereas individual synthetic siRNAs caused hundreds of off-target mRNA changes [66].
Computational Design and Validation
Modern siRNA design leverages sophisticated algorithms and machine learning models that go beyond basic BLAST homology checks. These tools predict efficacy and off-target potential by analyzing sequence features, thermodynamic properties, and other factors during the design phase [30] [65]. Furthermore, a novel parameter called siRMSD (small interfering RNA root-mean-square deviation) has been introduced to quantify structural distortions caused by chemical modifications. A strong correlation has been demonstrated between deviations from the canonical A-form RNA structure and a reduction in off-target effects, providing a predictive framework for rational siRNA design [68].
Experimental Protocols for Validation
Protocol: Validating Specificity Using Microarray or RNA-Seq
This protocol assesses genome-wide off-target effects following siRNA treatment.
1. Sample Preparation:
- Transfert cells with the candidate siRNA, a scrambled negative control siRNA, and a positive control siRNA known to silence a specific gene.
- Use at least two biological replicates per condition.
- Critical: Use the lowest effective siRNA concentration (e.g., ≤20 nM) to minimize saturation of the RNAi machinery and reduce sequence-agnostic off-target effects [66].
2. RNA Isolation and Analysis:
- Harvest cells 24-48 hours post-transfection and isolate total RNA using a standard kit (e.g., Qiagen RNeasy).
- Assess RNA integrity and purity (e.g., via Bioanalyzer; RIN > 8.0 is desirable).
3. Expression Profiling:
- Proceed with either microarray analysis or next-generation RNA sequencing (RNA-Seq). RNA-Seq is generally preferred for its broader dynamic range and ability to detect novel transcripts.
- Follow the manufacturer's instructions for library preparation and sequencing.
4. Data Analysis:
- Map sequencing reads to the reference genome/transcriptome.
- Identify differentially expressed genes (DEGs) between the test siRNA and the scrambled control. A typical cutoff is |log2(fold change)| > 1 and an adjusted p-value < 0.05.
- Key Analysis Step: To distinguish true seed-driven off-targets from secondary effects, compare the list of DEGs to a list of genes predicted to have complementarity to the siRNA's seed region (nucleotides 2-8 of the guide strand). An enrichment of seed-matched genes among the DEGs indicates a classic miRNA-like off-target effect [66] [65].
Protocol: Assessing RISC Strand Bias with Asymmetric Design
This biochemical protocol confirms preferential loading of the guide strand into RISC.
1. siRNA Design and Synthesis:
- Design the siRNA with a thermodynamically unstable 5' end on the guide strand (e.g., richer in A/U bases) and a more stable 5' end on the passenger strand (e.g., richer in G/C bases) [30].
- Synthesize the siRNA duplex with the desired asymmetric stability profile.
2. RISC Loading Assay:
- Incubate the siRNA with a purified, recombinant RISC loading complex or a cytoplasmic cell lysate under conditions that support RISC assembly.
- Use a native gel electrophoresis mobility shift assay to visualize the formation of siRNA-RISC complexes.
3. Strand Detection:
- To directly quantify which strand is loaded, a more advanced method involves using northern blotting or quantitative RT-PCR with strand-specific probes after immunoprecipitation of Argonaute 2 (Ago2), the core RISC protein.
- A significant enrichment of the guide strand in the Ago2 immunoprecipitate compared to the passenger strand confirms successful asymmetric loading.
Visualization of Strategies and Workflow
The following diagram illustrates the core problem of seed-mediated off-target effects and the primary strategies to mitigate them.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Resources for siRNA Specificity Research
| Item | Function/Description | Example Use Case |
|---|---|---|
| Chemically Modified siRNA | Custom siRNAs with modifications (2'-OMe, 2'-F, PS, Formamide) to enhance stability and specificity. | Testing the efficacy of novel modifications in reducing seed-driven off-target effects [67] [20]. |
| Diced siRNA (d-siRNA) Pool | A complex pool of siRNAs generated by enzymatic cleavage (Dicer/RNase III) of a long dsRNA template. | Comparing off-target profiles of pooled vs. single siRNAs to validate the pooling strategy [66]. |
| Computational Design Tools | Algorithms and machine learning models for predicting siRNA efficacy and off-target potential. | Initial in silico screening of candidate siRNA sequences to filter out those with high risk for off-target effects [30] [65]. |
| Strand-Specific Detection Probes | Probes (for Northern blot or qRT-PCR) that uniquely bind to the guide or passenger strand of the siRNA. | Experimentally verifying which strand is loaded into RISC in an asymmetric design assay. |
| Argonaute 2 (Ago2) Antibody | Antibody for immunoprecipitating the RISC complex. | Isolating the active RISC to analyze which siRNA strand it contains [64]. |
The therapeutic application of small interfering RNA (siRNA) is often challenged by its potential to activate the innate immune system. Mammalian cells recognize double-stranded RNA (dsRNA) as a signature of viral infection, triggering a potent immune response that can confound experimental results and compromise therapeutic safety [69]. This application note provides a detailed framework for managing siRNA immunogenicity through strategic sequence design and chemical modifications, enabling researchers to achieve specific gene silencing without unintended immune activation.
Mechanisms of siRNA-Induced Immune Activation
The innate immune system primarily detects siRNA through pattern recognition receptors (PRRs) that identify foreign nucleic acids. Understanding these pathways is fundamental to designing immunologically silent siRNAs.
Key Immune Recognition Pathways
Table 1: Pattern Recognition Receptors for siRNA Immune Recognition
| Receptor | Ligand Characteristics | Cellular Location | Primary Immune Response |
|---|---|---|---|
| TLR7 | GU-rich sequences, ssRNA, specific motifs (e.g., 5'-UGU-3', 5'-GUCCUUCAA-3') | Endosome/Lysosome | IFN-α, IFN-β, TNF-α, IL-6, IL-12 |
| TLR8 | ssRNA, UR-rich sequences | Endosome/Lysosome | TNF-α, IL-1, IL-6, IL-12 |
| RIG-I | Blunt-ended dsRNA, uncapped 5'-triphosphate RNA | Cytoplasm | IFN-α, IFN-β |
| PKR | Long dsRNA (>30 bp) | Cytoplasm | IFN-α, IFN-β, inhibition of protein translation |
The visualization below outlines the primary signaling pathways through which siRNA can activate the innate immune response:
Strategic Sequence Design to Minimize Immunogenicity
Identification and Avoidance of Immunostimulatory Motifs
Specific nucleotide sequences within siRNA strands act as potent triggers for immune activation, particularly through endosomal Toll-like receptors (TLRs). The table below summarizes key immunostimulatory motifs to avoid during siRNA design:
Table 2: Immunostimulatory Sequence Motifs and Mitigation Strategies
| Immunostimulatory Motif | Recognition Receptor | Immune Response | Recommended Modification |
|---|---|---|---|
| 5'-UGU-3' | TLR7 | IFN-α production | Replace uridine with adenosine |
| 5'-GUCCUUCAA-3' | TLR7/TLR8 | Cytokine production | Eliminate motif or incorporate 2'-O-Me modifications |
| Multiple uridine residues in close proximity | TLR7/TLR8 | Strong cytokine induction | Replace U with A or dT (deoxythymidine) |
| GU-rich sequences | TLR7 | IFN-α, IL-6, TNF-α | Reduce GU content through sequence redesign |
| Blunt-ended dsRNA | RIG-I | IFN-α, IFN-β | Incorporate 3' overhangs (2-nt) |
Sequence-specific effects can be profound; for instance, substituting guanosine with adenosine significantly reduces TNF-α production, while replacing uridine with adenosine decreases IFN-α production in plasmacytoid dendritic cells [70] [69]. These substitutions represent the first line of defense against sequence-dependent immune activation.
Chemical Modification Strategies
Chemical modifications to the siRNA backbone represent the most effective approach for mitigating immune recognition while maintaining gene silencing activity.
Ribose Modifications
Table 3: Chemical Modifications for Reducing siRNA Immunogenicity
| Modification Type | Position | Impact on Immunogenicity | Effect on RNAi Activity | Notes |
|---|---|---|---|---|
| 2'-O-methyl (2'-O-Me) | Positions 2-5 (seed region) | Significant reduction in TLR7/8 and RIG-I recognition | Maintained with proper positioning | Acts as TLR7 antagonist; position 9 on sense strand interferes with RISC |
| 2'-fluoro (2'-F) | Both strands | Reduces immune stimulation | Maintained or improved | Number and position critical for effect |
| 2'-deoxy (2'-H) | Both strands | Mimics DNA, reduces recognition | Maintained with proper design | Particularly effective in thymidine/uridine |
| Locked Nucleic Acid (LNA) | Both strands | Reduces immune recognition | Variable (position-dependent) | Can compromise potency if incorrectly positioned |
| Unlocked Nucleic Acid (UNA) | Seed region | Reduces off-target effects | Maintained with proper design | Immunostimulatory capacity not fully characterized |
The siRMSD (small interfering RNA root-mean-square deviation) parameter has emerged as a valuable predictive tool that quantifies structural distortions induced by chemical modifications. Research demonstrates a strong correlation between deviations from the canonical A-form RNA structure and reduction in siRNA off-target effects, providing a framework for rational siRNA design [68] [71].
Modifications at positions 2-5 in the seed region are particularly effective as they disrupt the A-form RNA duplex on argonaute 2, preventing stable binding to non-target mRNAs [71]. In contrast, modifications at positions 6-8 have minimal impact on off-target effects resulting from changes in thermodynamic stability.
Experimental Protocol for Assessing siRNA Immunogenicity
Comprehensive Immune Profiling Workflow
Detailed Methodology
Step 1: siRNA Design and Modification
- Design siRNA sequences using established algorithms (e.g., SMARTselection) with attention to GC content (30-60%) and length (19-23 bp)
- Incorporate appropriate chemical modifications based on Table 3, focusing on seed region modifications
- Include positive controls (known immunostimulatory sequences) and negative controls (extensively modified sequences)
- Synthesize siRNAs using solid-phase chemical synthesis with quality control (HPLC purification)
Step 2: Immune Cell Preparation
- Isolate primary peripheral blood mononuclear cells (PBMCs) from healthy donors using Ficoll density gradient centrifugation
- Plate PBMCs at 1×10^6 cells/mL in RPMI-1640 complete medium
- Alternatively, use commercially available cryopreserved PBMCs or specialized immune cell lines (e.g., plasmacytoid dendritic cell lines)
Step 3: Transfection Protocol
- Complex siRNAs with appropriate transfection reagents (e.g., lipofectamine) according to manufacturer's instructions
- Use serum-free conditions during transfection complex formation
- Transfer complexes to cells at final siRNA concentrations ranging from 10-100 nM
- Include mock transfection (reagent only) and untreated controls
- Incubate cells for 24-48 hours at 37°C, 5% CO₂
Step 4: Sample Collection
- Collect cell culture supernatants at 6-8 hours (early cytokines) and 24 hours (late cytokines) post-transfection
- Preserve aliquots at -80°C until analysis
- For gene expression analysis, harvest cells in appropriate RNA stabilization reagents
Step 5: Immune Marker Analysis
- Cytokine profiling: Measure IFN-α, IFN-β, TNF-α, IL-6, and IL-12 using ELISA or multiplex bead-based assays
- Gene expression: Quantify interferon-stimulated gene (ISG) expression (e.g., MX1, OAS1) via qRT-PCR
- Cell activation: Assess surface activation markers (e.g., CD80, CD86, CD40) on antigen-presenting cells using flow cytometry
Step 6: Data Interpretation
- Normalize cytokine levels to positive and negative controls
- Establish acceptable thresholds for immune activation (typically <2-fold increase over negative control)
- Correlate immune activation with siRNA sequence features and modification patterns
- Select lead candidates showing minimal immune activation while maintaining target gene silencing efficacy
Table 4: Research Reagent Solutions for siRNA Immunogenicity Assessment
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Specialized siRNA | ON-TARGETplus, Accell siRNA, siGENOME | Pre-designed siRNAs with chemical modifications to reduce immunogenicity |
| Chemical Modification Reagents | 2'-O-methyl RNA phosphoramidites, 2'-F RNA monomers, LNA amidites | Custom synthesis of modified siRNA strands |
| Immune Cell Systems | Primary human PBMCs, plasmacytoid dendritic cells (pDCs), monocyte-derived DCs | In vitro models for immunogenicity screening |
| Detection Assays | Human IFN-α ELISA kit, LEGENDplex Human Inflammation Panel, ISG qRT-PCR panels | Quantification of immune activation markers |
| Transfection Reagents | Lipofectamine RNAiMAX, TransIT-TKO, Dendritic Cell Nucleofector kits | Efficient delivery of siRNA to immune cells |
| Bioinformatics Tools | siRMSD analysis software, TLR motif screening algorithms, RNA secondary structure predictors | In silico prediction of immunostimulatory potential |
Effective management of siRNA immunogenicity requires a multi-faceted approach combining computational design, strategic chemical modifications, and rigorous experimental validation. By implementing the sequence design principles, modification strategies, and assessment protocols outlined in this application note, researchers can significantly reduce off-target immune activation while maintaining potent gene silencing activity. These methodologies provide a robust framework for developing research-grade and therapeutic siRNAs with improved safety profiles.
Small interfering RNA (siRNA) therapeutics represent a transformative approach for targeted gene knockdown, with the potential to silence virtually any disease-causing gene. The RNA interference (RNAi) pathway, first discovered in Caenorhabditis elegans and awarded the Nobel Prize in 2006, operates through a conserved biological mechanism where double-stranded RNA molecules induce sequence-specific suppression of gene expression [72] [73]. Despite this elegant mechanism and significant clinical advances, efficient cytosolic delivery of siRNA remains the primary bottleneck in therapeutic development.
The fundamental challenge lies in navigating multiple biological barriers. Systemically administered siRNAs face rapid enzymatic degradation and clearance before reaching target cells. Their inherent negative charge and hydrophilic nature prevent passive diffusion across cell membranes. Most critically, even when internalized, the vast majority of siRNA molecules become trapped within endolysosomal compartments, unable to reach the cytosol where the RNA-induced silencing complex (RISC) resides [74] [75]. Overcoming these barriers, particularly achieving efficient endosomal escape, represents the central challenge in siRNA therapeutic development. This Application Note provides detailed strategies and protocols to address these critical limitations, enabling researchers to develop more effective siRNA-based gene silencing approaches.
Understanding the Biological Barriers
The RNAi Mechanism and Cellular Uptake Hurdles
The canonical RNAi pathway begins when exogenous double-stranded RNA is processed by the ribonuclease Dicer, which cleaves long dsRNA into 21-23 nucleotide siRNA duplexes with 2-nucleotide overhangs at the 3' ends [72]. One strand of the siRNA (the guide strand) is then loaded into the RNA-induced silencing complex (RISC), where the Argonaute 2 (AGO2) protein facilitates sequence-specific recognition and cleavage of complementary messenger RNA (mRNA), preventing translation of the target protein [3] [76].
Table 1: Key Barriers to Effective siRNA Delivery
| Barrier Category | Specific Challenge | Impact on siRNA Delivery |
|---|---|---|
| Extracellular Barriers | Serum Nuclease Degradation | Rapid cleavage of unmodified siRNA in biological fluids |
| Renal Clearance | Rapid elimination of low molecular weight siRNA | |
| Immune Recognition | Unmodified siRNA can trigger innate immune responses | |
| Cellular Uptake Barriers | Cell Membrane Transit | Negative charge prevents passive diffusion across lipid bilayers |
| Non-Specific Tissue Distribution | Lack of targeting leads to accumulation in non-target tissues | |
| Intracellular Barriers | Endosomal Entrapment | >99% of internalized siRNA remains trapped in endosomes [75] |
| Lysosomal Degradation | Acidic pH and hydrolytic enzymes degrade siRNA | |
| Cytoplasmic Dispersion | Released siRNA must access RISC machinery |
For functional gene silencing, siRNA duplexes must be introduced into the target cell's cytosol. Naked, unmodified siRNAs are highly susceptible to degradation by ubiquitous ribonucleases in biological fluids and cannot cross cell membranes due to their polyanionic nature [3]. Furthermore, without specific targeting moieties, systemically administered siRNAs exhibit poor biodistribution to desired tissues and are rapidly cleared renally. These challenges necessitate the use of sophisticated delivery systems to protect siRNA payloads and facilitate cellular internalization.
The Endosomal Escape Bottleneck
Endosomal entrapment constitutes the most significant efficiency-limiting step in siRNA delivery. Most delivery strategies facilitate cellular uptake through endocytosis, but the internalized siRNA remains membrane-bound within vesicles. As endosomes mature into lysosomes, the increasingly acidic environment and activated nucleases degrade the siRNA payload. Only a minute fraction (typically <2%) of internalized siRNA successfully escapes into the cytosol to engage with RISC [74] [75].
Recent research using live-cell microscopy has visualized this bottleneck, revealing that lipid nanoparticles (LNPs) trigger endosomal membrane disruptions detectable by galectin sensors, particularly galectin-9 [74] [75]. However, these studies also demonstrated that only a subset of damaged endosomes actually contain siRNA cargo, and even when damage occurs, only a fraction of the nucleic acid is released. This inefficiency highlights the critical need for strategies that enhance both the frequency and productivity of endosomal escape events.
Diagram Title: siRNA Intracellular Trafficking and Endosomal Escape Challenge
Formulation Strategies for Enhanced Cellular Uptake
Advanced Nanocarrier Systems
Nanoparticle-based delivery systems have emerged as the most promising approach for protecting siRNA and facilitating cellular internalization. These systems complex with siRNA through electrostatic interactions, creating stable nanoparticles that shield the payload and promote endocytic uptake.
Table 2: Comparison of Major siRNA Nanocarrier Systems
| Carrier Type | Key Components | Mechanism of Action | Advantages | Limitations |
|---|---|---|---|---|
| Lipid Nanoparticles (LNPs) | Ionizable lipid, Helper lipids, Cholesterol, PEG-lipid [74] [3] | pH-dependent membrane disruption in endosomes | Clinical validation (Patisiran), Scalable production | Primarily liver tropism, Potential immunogenicity |
| Polymeric Nanoparticles | Polyethylenimine (PEI), Chitosan, PAMAM dendrimers [77] [3] | Proton sponge effect: buffering capacity causes osmotic swelling and endosome rupture | Tunable properties, High siRNA loading capacity | Higher cytotoxicity compared to LNPs |
| Hybrid Systems | Lipid-polymer hybrids, Peptide-based carriers [77] | Combine advantages of multiple materials | Enhanced stability and efficiency, Modular design | Complex manufacturing and characterization |
| Inorganic Nanoparticles | Mesoporous silica, Gold nanoparticles [78] [3] | Pore loading with controlled release profiles | Tunable pore size, Surface functionalization | Potential long-term toxicity concerns |
| Ligand-Conjugated siRNA | GalNAc, Cholesterol, Antibodies, Aptamers [3] [75] | Receptor-mediated endocytosis | Targeted delivery, Simplified formulation | Still requires endosomal escape enhancement |
Among these, lipid nanoparticles represent the most clinically advanced platform. The first FDA-approved siRNA therapeutic, patisiran, utilizes an LNP formulation containing an ionizable lipid (DLin-MC3-DMA) that becomes protonated in the acidic endosomal environment, facilitating membrane disruption and siRNA release [74] [3]. Recent research has focused on optimizing LNP composition by screening novel ionizable lipids with improved endosomal escape efficiency and reduced toxicity profiles.
siRNA Chemical Modifications
Strategic chemical modifications to the siRNA molecule itself significantly enhance stability and pharmacokinetics without compromising RNAi activity.
Protocol 3.2.1: Design of Chemically Modified siRNA
Objective: To enhance siRNA metabolic stability and reduce immunogenicity while maintaining potent gene silencing activity.
Materials:
- Unmodified siRNA sequence targeting gene of interest
- 2'-O-methyl (2'-OMe), 2'-fluoro (2'-F), or 2'-O-methoxyethyl (2'-MOE) nucleoside phosphoramidites
- Phosphorothioate (PS) backbone modification reagents
- Solid-phase synthesizer for oligonucleotide production
- Purification and analysis equipment (HPLC, mass spectrometry)
Procedure:
- Sequence Design: Identify optimal target sequence within mRNA (typically 21-23 nt with 2-nt 3' overhangs).
- Modification Strategy:
- Apply 2'-OMe, 2'-F, or 2'-MOE modifications to ribose sugars at alternating positions, particularly in the guide strand seed region (nucleotides 2-8) [3].
- Incorporate phosphorothioate (PS) linkages at 5' and 3' ends to enhance nuclease resistance and plasma protein binding.
- Consider pattern-based modifications with heavier modification on passenger strand to promote correct RISC loading.
- Synthesis and Purification: Execute solid-phase synthesis using appropriate phosphoramidite chemistry. Purify using reverse-phase HPLC.
- Quality Control: Verify molecular weight by mass spectrometry and assess purity by analytical HPLC (>95% purity recommended).
- Functional Validation: Test gene silencing efficacy and duration in relevant cell culture models compared to unmodified controls.
Troubleshooting Tip: Excessive modification, particularly in the guide strand seed region or cleavage site (positions 10-11), can impair RISC loading and catalytic activity. Balance stability enhancements with functional preservation through empirical testing.
Methodologies for Enhancing Endosomal Escape
Endosomolytic Agents and Their Mechanisms
Several classes of endosomolytic agents can be co-administered or incorporated into delivery systems to enhance endosomal escape. Research using galectin-9 as a sensitive sensor of membrane damage has demonstrated that small molecule compounds can substantially improve siRNA release from endosomal compartments [75].
Table 3: Endosomolytic Agents for Enhancing siRNA Delivery
| Agent Class | Representative Compounds | Mechanism of Action | Experimental Conditions | Knockdown Enhancement |
|---|---|---|---|---|
| Cationic Amphiphilic Drugs (CADs) | Chloroquine, Siramesine, Amitriptyline [75] | Accumulate in acidic compartments, inducing membrane damage through lipidosis | 10-50 μM, 24h treatment | Up to 47-fold improvement with chloroquine [75] |
| Cell-Penetrating Peptides (CPPs) | Penetratin, TAT, MAP | Form transient pores or inverted micelles in endosomal membranes | Varies by peptide; typically 5-20 μM | 5-20 fold improvement (varies by cell type) |
| pH-Sensitive Polymers | Poly(alkyl acrylic acids), PEI | "Proton sponge" effect: buffering causes osmotic swelling and rupture | Varies by polymer; N/P ratio 5-20 | 10-30 fold improvement |
| Photosensitizers | Tetraphenylporphine (TPP), Verteporfin | Light-induced ROS generation disrupts endosomal membranes | 1-10 μg/mL + light exposure | Spatiotemporally controlled release |
Protocol 4.1.1: Evaluating Small Molecule-Enhanced Endosomal Escape
Objective: To assess and quantify the enhancement of siRNA-mediated gene knockdown using membrane-disrupting small molecules.
Materials:
- Cholesterol-conjugated siRNA (chol-siRNA) targeting reporter gene (e.g., eGFP)
- Small molecule compounds (chloroquine, siramesine, amitriptyline)
- Target cells expressing reporter gene (e.g., HeLa-d1eGFP)
- Live-cell imaging setup with confocal capability
- Fluorescently tagged galectin-9 construct
- Flow cytometer for knockdown quantification
Procedure:
- Cell Preparation: Seed HeLa-d1eGFP cells in 24-well plates at 70% confluence 24h before experiment.
- siRNA Uptake Phase: Incubate cells with chol-siGFP (50-200 nM) in serum-free medium for 6h to allow cellular internalization.
- Small Molecule Treatment: Replace medium with complete medium containing titrated concentrations of small molecule compounds (e.g., 10-100 μM chloroquine, 5-50 μM siramesine, 10-75 μM amitriptyline).
- Live-Cell Imaging (Optional):
- Transfer cells expressing mCherry-galectin-9 to imaging chamber.
- Image every 30-60s for 24h to track de novo galectin-9 recruitment to damaged endosomes.
- Quantify number of galectin-9 foci per cell over time.
- Knockdown Assessment: After 24-48h, harvest cells and analyze eGFP fluorescence by flow cytometry.
- Viability Assessment: Perform parallel MTT assays to ensure enhancements are not due to cytotoxicity.
Calculation:
- Calculate knockdown enhancement factor as: (% Knockdown with compound - % Knockdown without compound) / % Knockdown without compound
- Normalize to cell viability to identify optimal therapeutic window
Safety Note: Small molecule enhancers may exhibit cell-type specific toxicity. Always include viability controls and test multiple concentrations to establish a therapeutic window.
Advanced LNP Formulation for Improved Escape
Recent mechanistic studies have revealed that current LNPs suffer from multiple inefficiencies, including segregation of ionizable lipid and RNA payload during endosomal sorting, with only a fraction of damaged endosomes containing detectable RNA [74]. These findings inform the next generation of LNP design.
Protocol 4.2.1: Formulation of Ionizable Lipid-Containing LNPs
Objective: To prepare and characterize LNPs with optimized endosomal escape efficiency through novel ionizable lipid components.
Materials:
- Ionizable lipid (e.g., MC3, novel proprietary lipids)
- Helper lipids (DSPC, DOPE)
- Cholesterol
- PEG-lipid (DMG-PEG2000)
- siRNA (unmodified or chemically modified)
- Microfluidic mixing device (NanoAssemblr, etc.)
- Dialysis membranes (MWCO 100kDa)
- Dynamic light scattering (DLS) instrument
- Cryo-electron microscopy
Procedure:
- Lipid Stock Preparation: Dissolve individual lipid components in ethanol at predetermined molar ratios (typical: ionizable lipid 50%, cholesterol 38.5%, DSPC 10%, PEG-lipid 1.5%).
- Aqueous Phase Preparation: Dilute siRNA in citrate buffer (pH 4.0) at a concentration of 0.2 mg/mL.
- Nanoparticle Formation:
- Set total flow rate to 12 mL/min with aqueous:ethanol ratio of 3:1.
- Use microfluidic device to rapidly mix lipid and aqueous streams.
- Collect resulting LNP suspension.
- Buffer Exchange: Dialyze against PBS (pH 7.4) for 24h at 4°C to remove ethanol and establish neutral pH.
- Characterization:
- Measure particle size, PDI, and zeta potential by DLS.
- Determine siRNA encapsulation efficiency using Ribogreen assay.
- Visualize morphology by cryo-EM.
- Functional Testing: Evaluate gene silencing efficiency in vitro and endosomal escape kinetics using galectin-9 recruitment assays.
Optimization Tips:
- Screen multiple ionizable lipids with pKa values between 6.0-6.5 for optimal endosomal disruption.
- Incorporate helper lipids like DOPE that promote hexagonal phase transition for enhanced membrane fusion.
- Consider incorporating fluorophores into both lipid and RNA components to track co-localization during intracellular trafficking.
Analytical Methods for Quantifying Endosomal Escape
Live-Cell Imaging of Galectin Recruitment
The discovery that galectins, particularly galectin-9, rapidly relocate to damaged vesicles provides a powerful tool for visualizing and quantifying endosomal escape events in real-time [74] [75].
Diagram Title: Experimental Workflow for Imaging Endosomal Escape
Protocol 5.1.1: Galectin-9 Recruitment Assay for Endosomal Damage
Objective: To visualize and quantify endosomal membrane disruption in live cells using galectin-9 as a sensitive biosensor.
Materials:
- Cell line stably expressing YFP-galectin-9 (or transiently transfected)
- siRNA delivery system (LNPs, polymer nanoparticles, or ligand-conjugated siRNA)
- Confocal live-cell imaging system with environmental control
- Image analysis software (ImageJ, Volocity, or similar)
Procedure:
- Cell Preparation: Seed YFP-galectin-9 expressing cells in glass-bottom imaging dishes at 50-60% confluence 24h before experiment.
- Treatment: Add siRNA delivery system at optimized concentration in pre-warmed imaging medium.
- Image Acquisition:
- Maintain cells at 37°C with 5% CO₂ throughout imaging.
- Acquire images every 30-60s for 2-24h depending on delivery kinetics.
- Include brightfield and YFP channels.
- Image Analysis:
- Identify de novo formation of galectin-9 foci as bright punctate structures.
- Track individual foci over time to distinguish from static background.
- Quantify number of galectin-9 positive vesicles per cell over time.
- Co-localization Studies (Optional): Use siRNA labeled with far-red fluorophore (Cy5, AlexaFluor 647) to assess coincidence of galectin-9 recruitment and siRNA presence.
Key Interpretation:
- Galectin-9 recruitment indicates membrane damage but does not guarantee productive RNA release.
- Recent studies show only 67-74% of galectin-9+ vesicles contain detectable siRNA for siRNA-LNPs, and only ~20% for mRNA-LNPs [74].
- Correlation with functional knockdown is essential to establish biological relevance.
Flow Cytometry-Based Escape Quantification
While imaging provides spatial information, flow cytometry offers higher throughput quantification of cytosolic delivery.
Protocol 5.2.1: Split-Luciferase Complementation Assay
Objective: To quantitatively measure cytosolic siRNA delivery using a split-luciferase complementation system.
Materials:
- Cells expressing N-terminal half of Gaussian luciferase (GLuc) fused to a localization signal
- siRNA complementary to GLuc mRNA and conjugated to C-terminal half of GLuc
- Luciferase substrate (coelenterazine)
- Luminometer or flow cytometer with luminescence detection
Procedure:
- Engineer Reporter Cell Line: Stably transfect cells with plasmid expressing N-terminal GLuc (aa 1-93) fused to a cytosolic protein.
- Treatment: Incubate cells with siRNA conjugated to C-terminal GLuc (aa 94-185).
- Measurement: After 24-48h, lyse cells and measure luminescence after addition of coelenterazine.
- Data Analysis: Normalize luminescence to protein concentration and compare to controls.
Advantages: This method directly measures functional cytosolic delivery rather than just endosomal damage, providing a more biologically relevant assessment of escape efficiency.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for siRNA Delivery Studies
| Reagent Category | Specific Products/Tools | Primary Function | Key Considerations |
|---|---|---|---|
| Delivery Systems | LNP formulations, Lipofectamine RNAiMAX, PEI-based transfection reagents | Facilitate cellular siRNA uptake | Varying efficiencies across cell types; potential cytotoxicity |
| Endosomal Escape Enhancers | Chloroquine, Siramesine, EndoPorter | Disrupt endosomal membranes to promote siRNA release | Require optimization of concentration and timing |
| Chemical Modification Reagents | 2'-fluoro-dUTP, 2'-O-methyl RNA, Phosphorothioate modifiers | Enhance siRNA stability and reduce immunogenicity | Modification patterns affect RISC loading and activity |
| Detection & Imaging Tools | Galectin-9 fluorescent constructs, pH-sensitive dyes (LysoTracker), Fluorophore-labeled siRNA | Visualize intracellular trafficking and endosomal escape | Galectin-9 most sensitive sensor for membrane damage [75] |
| Functional Assay Systems | Split-luciferase systems, qRT-PCR reagents, Western blot kits | Quantify gene silencing efficiency | Multiple validation methods recommended |
| Control siRNAs | Non-targeting scrambled siRNA, GAPDH or cyclophilin B targeting siRNA | Experimental controls for specificity | Essential for distinguishing sequence-specific effects |
The field of siRNA therapeutics has made remarkable progress since the discovery of RNAi, with multiple FDA-approved medicines now available. However, efficient cytosolic delivery remains the critical limiting factor for expanding their therapeutic applications. The strategies outlined in this Application Note—including advanced nanocarrier design, strategic chemical modifications, and small molecule enhancement—provide researchers with practical approaches to overcome the central challenges of cellular uptake and endosomal escape.
Future directions will likely focus on developing more sophisticated delivery systems with improved tissue specificity and endosomal escape efficiency. Recent research revealing the segregation of ionizable lipid and RNA payload during endosomal sorting [74] highlights the need for next-generation LNPs with improved co-trafficking characteristics. Additionally, the discovery that only a fraction of damaged endosomes release their siRNA cargo suggests opportunities for optimizing the timing and location of endosomal disruption.
As delivery technologies continue to evolve, siRNA therapeutics will expand beyond current primarily hepatotropic applications to target additional tissues, potentially enabling treatment of neurological disorders, solid tumors, and genetic diseases affecting extrahepatic tissues. The methodologies and principles described herein provide a foundation for these future advances, bringing us closer to fully realizing the therapeutic potential of RNA interference.
Optimizing Thermodynamic Properties for Efficient RISC Loading and Strand Selection
Within the framework of small interfering RNA (siRNA) research for targeted gene knockdown, the precise selection of the guide strand for incorporation into the RNA-induced silencing complex (RISC) is a critical determinant of experimental success and therapeutic efficacy [79]. This guide strand directs RISC to complementary messenger RNA (mRNA) for sequence-specific degradation, thereby silencing gene expression [5]. The process of RISC loading and strand selection is not random; it is profoundly influenced by the thermodynamic profile of the siRNA duplex [79] [80]. This application note details the underlying principles and provides validated protocols for designing and testing siRNAs with optimized thermodynamic properties to ensure efficient RISC loading and accurate strand selection, thereby maximizing on-target gene silencing while minimizing off-target effects.
The Critical Role of Thermodynamics in Strand Selection
The two strands of an siRNA duplex are not incorporated into RISC with equal probability. Cellular machinery, including the enzyme Dicer, functions as a gatekeeper that senses the relative thermodynamic stability at the two ends of the duplex [80]. The strand whose 5' end is less thermodynamically stable (typically denoted by a lower G/C content) is preferentially loaded into RISC as the guide strand [79]. This asymmetry is crucial because it ensures that the correct, target-complementary strand is used for silencing.
The biological rationale for this is rooted in the mechanism of RISC loading. An RNA helicase responsible for unwinding the siRNA duplex encounters less resistance at the end with lower stability, facilitating the ejection of the passenger strand and the retention of the guide strand [79]. When siRNAs are designed with this thermodynamic asymmetry, the antisense strand is efficiently selected, leading to higher efficacy and specificity by preventing the sense strand from guiding the degradation of non-target mRNAs [79].
Table 1: Key Thermodynamic Parameters for siRNA Design
| Parameter | Optimal Characteristic | Functional Impact |
|---|---|---|
| 5' Antisense Stability | Low thermodynamic stability (A/U rich) | Promotes preferential loading of the antisense strand into RISC [79]. |
| 3' Antisense Stability | High thermodynamic stability (G/C rich) | Stabilizes the bound siRNA within the RISC machinery [79]. |
| Overall GC Content | 30-50% | Prevents overly stable duplexes that resist RISC unwinding; avoids off-target effects [30]. |
| Terminal Base Pairs | Differential stability at ends | Recognized by Dicer for pre-RISC selection of highly functional siRNAs [80]. |
| Seed Region Stability | Moderate stability | Reduces miRNA-like off-target effects by discouraging prolonged interactions with non-target transcripts [30] [5]. |
Figure 1: Thermodynamic asymmetry dictates strand selection during RISC loading. The strand with the less stable 5' end is preferentially selected as the guide.
Protocol: Experimentally Validating siRNA Efficacy and Strand Selection
The following protocol provides a methodology for empirically testing the silencing efficiency of siRNA candidates designed with thermodynamic principles in mind, using a dual-luciferase reporter assay system.
Materials and Equipment
- Synthetic siRNAs: Designed with varying 5' terminal stabilities (e.g., low vs. high GC content at the 5' end of the antisense strand) [80].
- Reporter Plasmids: psiCHECK-2 vector (or similar) where the target sequence is cloned in the 3'-UTR of the Renilla luciferase gene [80].
- Cell Line: Adherent mammalian cell line suitable for transfection (e.g., HEK293, Hela) [79] [80].
- Transfection Reagent: Lipofectamine 2000 or equivalent [80].
- Dual-Luciferase Reporter Assay System: For sequential measurement of Renilla and Firefly luciferase activities [80].
- Real-Time PCR System: For quantifying endogenous mRNA levels (optional, for native context validation) [4] [79].
- Cell Culture Equipment: Laminar flow hood, CO₂ incubator, multi-well plates.
Procedure
- Cell Seeding: Seed appropriate cells (e.g., HEK293) in a 24-well plate at 60-80% confluency in normal growth medium without antibiotics [80].
- Transfection Mixture Preparation:
- For each well, dilute 100 ng of the reporter plasmid DNA in a sterile buffer.
- Dilute siRNA to the desired concentration (e.g., 25-200 pM for highly active siRNAs) in the same buffer [79].
- Combine the diluted DNA and siRNA.
- Add 0.5 µL of Lipofectamine 2000 to the mixture, incubate for 15-20 minutes at room temperature to form complexes.
- Transfection: Add the entire transfection complex mixture to the cells in the well. Include controls: a non-targeting siRNA (negative control) and an untreated control [5].
- Incubation: Incubate cells for 24-48 hours at 37°C and 5% CO₂.
- Cell Lysis and Assay: Following incubation, lyse the cells and perform the dual-luciferase assay according to the manufacturer's instructions.
- Data Analysis: Calculate the normalized Renilla luciferase activity (Renilla/Firefly luciferase ratio). The percentage of silencing is determined by comparing this ratio in siRNA-treated cells to that in negative control siRNA-treated cells [80].
Table 2: Key Reagent Solutions for siRNA Validation
| Research Reagent | Function/Application |
|---|---|
| Pre-designed siRNAs (e.g., ON-TARGETplus) | Chemically modified siRNAs with reduced off-target effects; ideal for benchmarking and positive controls [5]. |
| Silencer Validated siRNAs | Algorithm-designed siRNAs with high success rates for endogenous gene silencing [79]. |
| Dual-Luciferase Reporter Assay System | Quantifies siRNA efficacy by measuring knockdown of a luciferase reporter gene [80]. |
| Transfection Reagents (Lipid-based) | Enables efficient delivery of synthetic siRNA duplexes into cultured cells [5]. |
| Accell siRNA | Modified siRNAs for delivery in difficult-to-transfect cells without the need for transfection reagents [5]. |
Protocol: Computational Design and In Silico Validation of siRNAs
Computational tools are indispensable for the initial screening and rational design of siRNA sequences, allowing for the incorporation of thermodynamic rules before costly synthesis.
Procedure
- Sequence Retrieval: Obtain the full mRNA coding sequence (CDS) of the target gene in FASTA format from a reliable database like NCBI Nucleotide [34] [19].
- siRNA Candidate Generation: Use design tools (e.g., siDirect, i-Score Designer) that implement established rules (Ui-Tei, Amarzguioui, Reynolds) to generate a list of potential siRNA candidate sequences targeting various regions of the mRNA [34].
- Thermodynamic Filtering:
- Calculate the GC content for each candidate and filter out sequences with >50-60% GC [30] [34].
- Use tools like RNAfold to predict the free energy (ΔG) for the 5' end of both strands. Select candidates where the antisense strand has a significantly less stable 5' end (more negative ΔG) than the passenger strand [30] [79].
- Specificity and Off-target Check: Perform BLAST or other homology searches to ensure minimal sequence identity to other genes. Filter out sequences with high complementarity in the "seed" region (nucleotides 2-8 of the guide strand) to avoid miRNA-like off-target effects [30] [79] [5].
- Molecular Docking (Advanced): For lead candidates, perform molecular docking against the Argonaute-2 (Ago2) protein structure to evaluate binding affinity and conformational fit, which can further predict silencing efficacy [34] [19].
Figure 2: A layered computational workflow for refining siRNA candidates based on thermodynamic and specificity rules.
The strategic optimization of siRNA thermodynamic properties is not merely a theoretical exercise but a practical necessity for achieving predictable and potent gene silencing. By deliberately designing siRNA duplexes with asymmetric terminal stability, researchers can exploit the cell's natural RISC loading machinery to favor the incorporation of the intended guide strand. The integration of computational design with empirical validation using reporter and native assays, as outlined in these protocols, provides a robust framework for developing high-quality siRNA reagents. Adhering to these principles is fundamental to advancing basic research and therapeutic development in the field of RNA interference.
Small interfering RNA (siRNA) represents a powerful therapeutic modality for achieving sequence-specific silencing of disease-related genes. However, the clinical application of conventional linear siRNA is often constrained by intrinsic limitations, including inadequate stability in biological environments and inefficient delivery to extrahepatic tissues. Overcoming these challenges is crucial for expanding the therapeutic potential of RNA interference (RNAi). This Application Note explores the development of prodrug-type circular siRNAs (circRNAs), a novel nucleic acid format that addresses these limitations through its unique covalently closed structure. We detail the design, in vitro and in vivo performance, and provide standardized protocols for researchers aiming to implement this technology in preclinical drug development.
Background and Mechanism of Action
Prodrug-type circular siRNA is a chemically synthesized, covalently closed molecule engineered to undergo a structural transformation in the intracellular environment. Its "prodrug" nature derives from a cleavable linker that maintains the circular configuration during systemic circulation, providing exceptional stability against nucleases. Upon cellular internalization, this linker is cleaved, converting the circular siRNA into its biologically active linear form, which then loads into the RNA-induced silencing complex (RISC) to mediate target messenger RNA (mRNA) knockdown [81].
The following diagram illustrates the core mechanism of this structural transformation from a stable circular prodrug to an active linear siRNA.
Key Experimental Data and Performance Metrics
In Vitro and In Vivo Performance of Circular siRNA
Extensive studies have characterized the advantageous properties of circular siRNA compared to traditional linear siRNA. The data summarized in the table below demonstrate its enhanced stability, cellular uptake, and knockdown efficacy.
Table 1: Comparative Performance of Circular siRNA versus Linear siRNA
| Parameter | Circular siRNA | Linear siRNA | Experimental Context |
|---|---|---|---|
| Serum Stability | High | Low | Incubation in mouse serum [81] |
| Exonuclease Resistance | High | Low | Challenge with exonucleases [81] |
| Cellular Uptake | Increased | Lower | In vitro cell culture without transfection reagent [81] |
| In Vitro Knockdown Activity | Stronger | Weaker | In vitro cell culture without transfection reagent [81] |
| Systemic Circulation Half-life | Prolonged | Short | Pharmacokinetics in mice after systemic administration [81] |
| In Vivo Knockdown Efficacy | Improved in liver, kidney, and muscle | Less effective | Target mRNA reduction in mouse tissues [81] |
| Safety Profile | No adverse effects reported (study limits) | N/A | Mouse model after systemic administration [81] |
Pharmacokinetics and Biodistribution
A key advantage of circular siRNA is its altered pharmacokinetic profile. Following systemic administration in mice, circular siRNA demonstrated prolonged circulation in the bloodstream compared to its linear counterpart. This enhanced stability contributes to improved tissue accumulation and knockdown activity not only in the liver but also in extrahepatic tissues such as the kidney and muscle, which are traditionally difficult to target with linear siRNA formats [81].
Alternative delivery strategies, such as enteral delivery of α-tocopherol (Toc)-conjugated siRNA formulated in lipid nanoparticles (LNPs), have also shown promise for hepatic targeting. In one study, rectal administration of Toc-siRNA LNPs resulted in liver accumulation levels approximately ten times lower than intravenous injection. However, the ratio of liver-to-serum concentration was significantly higher with the enteral route, suggesting a more efficient and selective delivery pathway to hepatocytes, likely mediated by the chylomicron-lymphatic transport system [82].
Experimental Protocols
Protocol: In Vitro Evaluation of Serum Stability
This protocol assesses the intrinsic stability of circular siRNA against nucleases present in biological fluids.
- Objective: To determine the degradation profile of circular siRNA compared to linear siRNA in serum.
Materials:
- Prodrug-type circular siRNA and traditional linear siRNA (positive control).
- Fetal Bovine Serum (FBS) or mouse serum.
- Incubation buffer (e.g., PBS or Tris-EDTA buffer).
- Proteinase K solution.
- Phenol-chloroform or suitable RNA extraction kit.
- Denaturing Polyacrylamide Gel Electrophoresis (PAGE) apparatus.
- Gel staining dye (e.g., SYBR Gold).
Procedure:
- Reaction Setup: Mix 2 µg of each siRNA with 90% FBS in a final volume of 50 µL. Incubate at 37°C.
- Time-point Sampling: Withdraw 5 µL aliquots at predetermined time points (e.g., 0, 1, 2, 4, 8, 24 hours).
- Reaction Termination: Immediately mix each aliquot with 5 µL of Proteinase K solution (e.g., 1 mg/mL) and incubate at 37°C for 15 minutes to digest serum proteins.
- RNA Extraction: Recover the siRNA using phenol-chloroform extraction or a commercial RNA cleanup kit. Elute in a small volume of nuclease-free water.
- Analysis: Load equal amounts of each sample onto a denaturing PAGE gel. Run the gel, stain, and visualize intact siRNA bands. The intensity of the full-length band over time quantifies stability [81].
Protocol: In Vivo Knockdown Efficacy in Mouse Models
This protocol outlines the procedure for evaluating the gene silencing activity of circular siRNA in vivo.
- Objective: To assess target mRNA knockdown in multiple tissues of mice after systemic administration of circular siRNA.
Materials:
- Purified prodrug-type circular siRNA and a scrambled sequence control.
- Experimental mice (e.g., C57BL/6).
- Appropriate injection supplies (sterile syringes, needles).
- RNAlater or similar RNA stabilization solution.
- Tissue homogenizer.
- RNA extraction kit, cDNA synthesis kit, and qPCR reagents.
Procedure:
- Animal Dosing: Systemically administer (e.g., via intravenous or intraperitoneal injection) a single dose of circular siRNA (e.g., 1-5 mg/kg) or control to mice. Include a group treated with linear siRNA for comparison.
- Tissue Collection: At the desired endpoint (e.g., 24-48 hours post-injection), euthanize the animals and harvest target tissues (liver, kidney, skeletal muscle). Snap-freeze tissue samples in liquid nitrogen and store at -80°C.
- RNA Isolation: Homogenize tissue samples and extract total RNA according to the manufacturer's instructions. Ensure RNA integrity and purity.
- Quantitative PCR (qPCR): Synthesize cDNA from equal amounts of total RNA. Perform qPCR using primers specific for the target mRNA and a housekeeping gene (e.g., GAPDH, β-actin).
- Data Analysis: Calculate the relative expression of the target mRNA using the 2^(-ΔΔCt) method. Compare the mRNA levels in the circular siRNA-treated group to the control group to determine the percentage of knockdown [81].
The following workflow maps the key stages of the in vivo efficacy study, from preparation to data analysis.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of circular siRNA technology relies on a suite of specialized reagents and materials. The following table lists essential components for research in this field.
Table 2: Essential Reagents and Materials for Circular siRNA Research
| Reagent/Material | Function/Description | Example Application |
|---|---|---|
| Chemically Synthesized Circular siRNA | The core active molecule with a cleavable linker, designed for optimal stability and intracellular activation. | In vitro and in vivo knockdown studies [81]. |
| N-acetylgalactosamine (GalNAc) Conjugate | A targeting ligand that binds specifically to the asialoglycoprotein receptor (ASGPR) on hepatocytes, facilitating liver-specific delivery. | Can be conjugated to siRNA (linear or circular) for enhanced hepatic uptake [83]. |
| Lipid Nanoparticles (LNPs) | A delivery system that encapsulates siRNA to protect it from degradation and improve cellular uptake. | Formulation for enteral delivery of Toc-siRNA [82] or systemic delivery [50]. |
| Alpha-Tocopherol (Vitamin E) Conjugate | A hydrophobic molecule conjugated to siRNA to facilitate association with lipoproteins (e.g., chylomicrons) for liver-targeted delivery via the lymphatic system. | Enteral (rectal) delivery of siRNA [82]. |
| Fully Chemically Modified siRNA | Linear siRNA with comprehensive backbone and sugar modifications to drastically improve metabolic stability and tissue retention. | Serves as a high-performance benchmark for comparing novel formats like circular siRNA [84]. |
| Group II Intron Plasmid System | A biological system (e.g., in E. coli) for the in vivo production of long circular RNAs, which can be adapted for sciRNA/siRNA production. | Scalable production of circular RNA [85]. |
Discussion and Future Perspectives
Prodrug-type circular siRNA represents a significant innovation in the RNAi therapeutic landscape. Its primary advantages lie in its enhanced nuclease resistance and prolonged systemic exposure, which collectively enable robust gene silencing in extrahepatic tissues—a major hurdle for current siRNA therapeutics. The synthetic feasibility of these molecules further supports their potential for broad application.
Future work will likely focus on expanding the tissue targeting repertoire of circular siRNAs by combining the circular scaffold with novel targeting ligands (e.g., peptides or antibodies) directed against receptors expressed in specific extrahepatic tissues. Furthermore, optimizing the cleavable linker chemistry for tissue-specific or stimulus-responsive activation could provide an additional layer of control over siRNA activity, enhancing both efficacy and safety. As production methodologies, such as those using group II intron systems in E. coli, continue to mature and scale, the accessibility and adoption of circular RNA formats are expected to increase [85].
In conclusion, prodrug-type circular siRNA provides a versatile and powerful platform that complements existing GalNAc-based liver delivery technologies. It offers a promising path forward for developing RNAi therapeutics for a wider range of diseases affecting organs beyond the liver.
Confirming Knockdown Efficacy: Robust Validation Systems and Technology Comparisons
Within the framework of small interfering RNA (siRNA) research for targeted gene knockdown, reporter-based validation systems are indispensable tools for quantifying silencing efficacy and understanding gene function. This application note details the integrated use of Enhanced Green Fluorescent Protein (EGFP) and Firefly Luciferase (FLuc) as sensitive, high-throughput-compatible reporters. We provide detailed protocols for employing these systems in siRNA screening workflows, supported by optimized experimental parameters and robust quantitative data. The methodologies outlined herein are designed to equip researchers and drug development professionals with reliable techniques to accelerate the discovery and validation of novel siRNA therapeutics.
Reporter Gene Assays (RGAs) are powerful techniques that investigate gene expression regulation and cellular signal transduction through easily detectable reporter genes [86]. In the context of siRNA-mediated gene knockdown, RGAs provide a direct, quantifiable readout of the silencing effect on a target gene of interest. The design of these assays is highly dependent on the drug mechanism, offering high accuracy and precision, which is critical for both basic research and quality control in biopharmaceutical development [86]. By creating transgenic cell lines where the expression of a reporter gene (e.g., EGFP or luciferase) is under the control of a specific pathway or promoter targeted by the siRNA, researchers can effectively simulate the mechanism of action and obtain reliable, high-throughput data. The combination of EGFP, which allows for visual tracking and fluorescence-based quantification, and luciferase, which offers high sensitivity and a broad dynamic range through bioluminescence, creates a versatile validation system ideal for screening and confirmatory experiments.
Key Reporter Systems: EGFP and Luciferase
Firefly Luciferase (FLuc) as a Highly Sensitive Reporter
Firefly luciferase is a quintessential reporter enzyme due to its exceptional sensitivity, low background, and wide dynamic range. Its application is well-established in high-throughput screening (HTS) formats. For instance, in antimalarial drug discovery, a stable transgenic Plasmodium falciparum line expressing high levels of firefly luciferase was used to develop a robust and reliable HTS assay. This assay was successfully miniaturized to a 384-well format, demonstrating an average Z′-factor of >0.7 and a coefficient of variation (CV) below 10%, which are key indicators of an excellent assay for large-scale screening [87]. The signal-to-background (S/B) ratio for this luciferase-based assay can be as high as 71, underscoring its exceptional sensitivity [87]. The principle involves measuring the light output (Relative Luminescent Units, RLU) produced when luciferase reacts with its substrate, providing a direct correlation to the level of gene expression or the efficacy of siRNA-mediated knockdown.
EGFP for Visualization and Quantification
Enhanced Green Fluorescent Protein (EGFP) serves as a complementary reporter that enables both spatial localization within cells and quantitative analysis via fluorescence microscopy or plate readers. A key application is found in dual-reporter systems, such as the pLTR-Luc2P-EGFP construct, where the expression of a single fusion protein, Luc2P-EGFP, is controlled by a specific promoter [88]. This system allows for initial visualization of successful transfection and reporter expression under a fluorescence microscope, followed by precise, high-throughput quantification of promoter activity via luciferase activity assays [88]. This dual approach combines the visual confirmation provided by EGFP with the superior quantitative accuracy of luciferase.
Combined and Specialized Reporter Applications
Engineering more sophisticated reporter systems can yield powerful tools for specific research questions. For example, researchers have engineered firefly luciferase to detect subtle abnormalities in protein biogenesis within the endoplasmic reticulum (ER) by introducing multiple cysteine substitutions and targeting the enzyme to the ER [89]. This engineered reporter exhibited outstanding sensitivity, reproducibility, and convenience in detecting defects in protein localization or disulfide bond formation [89]. Furthermore, the pLTR-Luc2P-EGFP system has been successfully used to quantify DNA methylation levels. When the CpG sites in the promoter are methylated, the expression of the Luc2P-EGFP reporter is silenced, and the degree of silencing, as measured by luciferase activity, can be used to generate standard curves for accurate DNA methylation quantification [88].
Quantitative Performance of Biological Assays
The selection of an appropriate assay is guided by its performance metrics. The table below summarizes the key performance characteristics of various biological activity methods, including reporter gene assays.
Table 1: Performance Comparison of Biological Activity Assay Methods [86]
| Classification | Detection Method | Limit of Detection (LOD) | Dynamic Range | Intra-batch CV (%) | Inter-batch CV (%) |
|---|---|---|---|---|---|
| Transgenic cell-based methods | Reporter Gene Assay (RGA) | ~ 10–12 M | 102–106 relative light units | Below 10% | Below 15% |
| Cell-based activity methods | Cell Proliferation Inhibition | ~ 10–9–10–12 M | Varies with cell ratio | Below 10% | Below 15% |
| Cytotoxicity Assay | ~ 100 cells per test well | 10–90% cell death | Below 10% | Below 15% | |
| ELISA | ~ 10–9–10–12 M | Wide, typically 102–105 | ~ 2–10 | ~ 5–15 | |
| New technology-based methods | Surface Plasmon Resonance (SPR) | ~ 10–9 M | Wide, typically 104—106 | ~ 1–5 | ~ 5–10 |
| Homogeneous Time-Resolved Fluorescence (HTRF) | ~ 10–12 M | Moderate, typically 102–104 | ~ 2–8 | ~ 5–12 | |
| Alpha Technology | ~ 10–11 M | Moderate, typically 102–104 | ~ 3–10 | ~ 6–15 |
As evidenced in the table, Reporter Gene Assays offer an excellent combination of sensitivity (low LOD), a wide dynamic range, and high reproducibility (low CV), making them particularly suitable for quantifying the effects of siRNA knockdown.
Experimental Protocols
Protocol 1: Validating siRNA Knockdown Efficacy Using a Dual Luciferase/EGFP Reporter System
This protocol outlines the steps for utilizing a dual-reporter system, such as pLTR-Luc2P-EGFP, to validate siRNA functionality [88].
Workflow Overview:
Materials & Reagents:
- Cells: Adherent mammalian cell line (e.g., HEK 293T, medaka OLHNI-2 cells [90]).
- Reporter Plasmid: pLTR-Luc2P-EGFP or similar construct [88].
- siRNA: Validated siRNA targeting your gene of interest and non-targeting negative control siRNA [91].
- Transfection Reagent: Suitable for siRNA/protein DNA co-transfection (e.g., X-tremeGENE siRNA Transfection Reagent) [90].
- Luciferase Assay Reagent: Commercially available one-step reagent (e.g., ONE-Glo Luciferase Assay System) [87].
- Equipment: Luminometer, fluorescence microscope, cell culture incubator, multi-well plates.
Step-by-Step Procedure:
- Cell Seeding: Seed appropriate cells in a 96-well or 384-well plate at an optimized density (e.g., 70-80% confluency at time of transfection) [91]. Use antibiotic-free medium for optimal health.
- Transfection Complex Preparation:
- Transfection: Add the transfection complex dropwise to the cells. Include controls: non-targeting siRNA, mock transfection (reagent only), and untreated cells [91].
- Incubation: Incubate the cells for 24-72 hours at 37°C with 5% CO₂. The optimal duration should be determined empirically and depends on protein turnover.
- EGFP Visualization (Qualitative Assessment): After 24-48 hours, observe the cells under a fluorescence microscope. A visible reduction in EGFP signal in the test siRNA group compared to the negative control indicates successful knockdown.
- Luciferase Assay (Quantitative Assessment):
- Equilibrate the ONE-Glo Luciferase Assay Reagent to room temperature.
- Add an equal volume of the reagent directly to the culture medium in each well. Mix gently.
- Incubate for 5-10 minutes to stabilize the signal.
- Measure the Relative Luminescent Unit (RLU) using a luminometer [87].
- Data Analysis:
- Normalize the RLU values of the test siRNA group to the negative control siRNA group.
- Calculate the percentage of remaining gene expression and percentage of knockdown using the following formulas, which are based on the ΔΔCT method adapted for reporter data [92]:
- % Remaining Gene Expression = 2^(-ΔΔCT) * 100% (Where ΔΔCT = (CTsiRNAtest - CTEndogenousControl) - (CTsiRNAcontrol - CTEndogenousControl))
- % Knockdown = 100% - % Remaining Gene Expression.
Protocol 2: High-Throughput siRNA Screening Using a Stable Luciferase Reporter Cell Line
This protocol describes an HTS-compatible assay using a stable cell line constitutively expressing firefly luciferase, ideal for large-scale siRNA library screening.
Workflow Overview:
Materials & Reagents:
- Stable Cell Line: A cell line (e.g., transgenic P. falciparum 3D7-Luc [87] or a mammalian cell line generated via CRISPR/Cas9-mediated targeted integration [86]) stably expressing firefly luciferase.
- siRNA Library: The defined set of siRNAs for screening.
- Transfection Reagent: Optimized for the stable cell line and HTS format.
- Luciferase Assay Reagent: ONE-Glo Luciferase Assay System for a simple "add-and-measure" procedure [87].
- Equipment: Automated liquid dispenser, luminescence microplate reader capable of reading 384-well plates, tissue culture facilities.
Step-by-Step Procedure:
- Cell Line Development: Generate a stable luciferase-expressing cell line. CRISPR/Cas9 technology is recommended for rapid and precise insertion of the luciferase gene into a specific genomic locus, ensuring consistent and reliable expression [86].
- Assay Miniaturization and Optimization:
- Optimize critical parameters such as cell seeding density (e.g., 1.6 x 10^5 cells/well for a 24-well plate), hematocrit (if using blood cells), and parasitemia in smaller plates before scaling up to 384-well format [87] [90].
- Determine the optimal siRNA concentration (e.g., 10-80 nM) to use in the screen to maximize knockdown while minimizing off-target effects [90] [91].
- HTS Execution:
- Using an automated dispenser, seed the stable cells into white 384-well assay plates.
- Transfert the siRNA library into the cells. Include controls on each plate: a negative control (non-targeting siRNA) and a positive control (e.g., a known effective siRNA or a control for background signal like media-only).
- Incubate the plates for 72 hours to allow for sufficient gene knockdown and depletion of the luciferase protein.
- Signal Detection:
- Add a pre-optimized volume of ONE-Glo reagent directly to each well without removing the culture medium.
- Incubate the plates at room temperature for 5 minutes to lyse the cells and stabilize the luminescent signal.
- Read the RLU on a luminescence microplate reader [87].
- Data Analysis and Hit Selection:
- Calculate the percentage of inhibition for each well:
% Inhibition = [1 - (RLU_sample / RLU_negative_control)] * 100%. - Assess the quality of the HTS screen by calculating the Z′-factor for each plate:
Z' = 1 - [3*(StdDev_positive_control + StdDev_negative_control) / |Mean_positive_control - Mean_negative_control|]. A Z′-factor > 0.5 indicates an excellent assay [87]. - Define "hit" siRNAs as those showing greater than the average inhibition plus three times the standard deviation of all samples [87].
- Calculate the percentage of inhibition for each well:
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of reporter-based siRNA validation requires a suite of reliable reagents. The following table lists key materials and their functions.
Table 2: Essential Research Reagents for Reporter-Based siRNA Screening
| Reagent / Material | Function / Application | Examples / Specifications |
|---|---|---|
| Firefly Luciferase | Sensitive bioluminescent reporter for quantitative HTS. | Engineered variants for stability and high signal [89]. |
| EGFP | Fluorescent reporter for visualization and quantification. | Used in dual-reporter systems like Luc2P-EGFP [88]. |
| Reporter Plasmids | Vectors carrying reporter genes under specific promoters. | pLTR-Luc2P-EGFP for methylation studies [88]. |
| Stable Cell Lines | Consistent reporter expression for HTS; generated via genetic engineering. | CRISPR/Cas9-mediated targeted integration [86]. |
| siRNAs | Effector molecules for targeted gene knockdown. | 21-23 nt, 30-50% G/C content, Stealth modified for stability [90] [91]. |
| Transfection Reagents | Facilitate delivery of siRNA and/or plasmid DNA into cells. | X-tremeGENE siRNA, Lipofectamine 2000, INTERFERin [90]. |
| Luciferase Assay Kits | Provide optimized substrates for light emission. | ONE-Glo, Steady-Glo Assay Systems [87]. |
| Control siRNAs | Critical for data interpretation and assay validation. | Non-targeting (negative), fluorescently-labeled (transfection efficiency), and known functional (positive) [91]. |
The integration of EGFP and luciferase reporter systems provides a robust, sensitive, and high-throughput-capable framework for validating siRNA efficacy in targeted gene knockdown research. The detailed protocols and performance data outlined in this application note demonstrate that these methods meet the rigorous demands of modern drug discovery and functional genomics. By following the optimized workflows and utilizing the essential reagents described, scientists can confidently generate high-quality, reproducible data to drive their research and therapeutic development programs forward.
In the field of functional genomics and drug discovery, small interfering RNA (siRNA) technology serves as a cornerstone for targeted gene knockdown research, enabling researchers to elucidate gene function and validate therapeutic targets [93]. The core principle of RNA interference (RNAi) involves introducing double-stranded RNA into cells, where it is processed by the Dicer enzyme into siRNAs approximately 21-23 nucleotides in length [94]. These siRNAs subsequently associate with the RNA-induced silencing complex (RISC), which guides the cleavage of complementary mRNA sequences, thereby preventing translation of the target gene [94]. Assessing the efficiency of this gene silencing process is critical for experimental validity, particularly in pharmaceutical development where accurate target validation can significantly impact downstream research trajectories. This application note provides detailed methodologies for evaluating siRNA-mediated knockdown efficiency using three complementary techniques: quantitative RT-PCR (qRT-PCR), Western blot (WB), and flow cytometry, framed within the context of a comprehensive siRNA research workflow.
The siRNA Knockdown Workflow and Assessment Strategy
A typical siRNA experiment follows a sequential workflow from design to validation. The process initiates with careful siRNA design and selection, considering factors such as GC content, seed region composition, and avoidance of secondary structures [94]. Following the selection of target sequences, siRNA molecules are introduced into cells via various delivery methods, including synthetic siRNA transfection, viral vector delivery, or newer formats such as paperclip RNA (pcRNA) [94]. After a suitable incubation period to allow for mRNA degradation and protein turnover, knockdown efficiency must be validated using multiple assessment methods to confirm both molecular and functional effects.
The diagram below illustrates the integrated experimental workflow for siRNA-mediated gene knockdown and efficiency assessment:
Key Assessment Methods: Principles and Applications
Each method for assessing knockdown efficiency provides unique insights into different stages of gene expression. The table below summarizes the primary applications and key technical considerations for the three core techniques discussed in this protocol:
Table 1: Overview of Knockdown Assessment Methods
| Method | Target Molecule | Key Applications | Technical Considerations |
|---|---|---|---|
| qRT-PCR | mRNA | - Rapid initial validation- Quantitative measurement- High sensitivity | - Does not confirm protein reduction- Requires proper normalization- RNA quality critical |
| Western Blot | Protein | - Confirms functional knockdown- Detects post-translational modifications- Provides molecular weight confirmation | - Protein stability affects results- Antibody specificity crucial- Semi-quantitative without standardization |
| Flow Cytometry | Cell surface proteins | - Single-cell analysis- Multiparameter detection- Live cell applications | - Limited to accessible epitopes- Requires cell suspension- Fluorescence compensation needed |
Quantitative RT-PCR (qRT-PCR) for Transcript-Level Assessment
qRT-PCR represents the most rapid and sensitive method for initial assessment of knockdown efficiency at the transcriptional level. This technique quantifies mRNA expression levels through reverse transcription followed by real-time PCR amplification.
Experimental Protocol:
- Total RNA Extraction: Isolate total RNA using TRIzol reagent or commercial kits 48-72 hours post-transfection [95]. Treat samples with DNase I to eliminate genomic DNA contamination.
- cDNA Synthesis: Synthesize cDNA using reverse transcriptase with oligo(dT) primers or random hexamers. Use 0.5-1μg total RNA in a 20μL reaction volume [96].
- qPCR Amplification: Prepare reactions with SYBR Green Master Mix, gene-specific primers (0.2-0.5μM each), and cDNA template. Utilize the following cycling conditions: initial denaturation at 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute [95] [97].
- Data Analysis: Calculate relative expression using the 2^(-ΔΔCt) method with normalization to appropriate housekeeping genes (e.g., GAPDH, β-actin) [95].
Troubleshooting Guide:
- Inconsistent results: Validate primer efficiency (90-110%) and specificity using standard curves and melt curve analysis [98].
- High variability between replicates: Ensure consistent RNA quality and avoid repeated freeze-thaw cycles of samples.
- Unstable reference genes: Validate housekeeping gene stability under experimental conditions; consider using multiple reference genes [98].
Western Blot for Protein-Level Confirmation
Western blot analysis provides essential confirmation that mRNA knockdown translates to reduced target protein expression. This method separates proteins by molecular weight, transfers them to a membrane, and detects specific proteins using antibodies.
Experimental Protocol:
- Protein Extraction: Lyse cells in RIPA buffer supplemented with protease and phosphatase inhibitors 72-96 hours post-transfection to allow for protein turnover [95].
- Protein Quantification: Determine protein concentration using BCA assay with bovine serum albumin standards [95].
- Gel Electrophoresis: Load 15-30μg total protein per lane on 8-12% SDS-polyacrylamide gels. Separate proteins at 80-120V for 1-2 hours [95].
- Membrane Transfer: Transfer proteins to PVDF membranes at constant current (400mA) for 2 hours using wet or semi-dry transfer systems [95].
- Immunoblotting: Block membranes with 5% non-fat milk for 1 hour, incubate with primary antibody overnight at 4°C, followed by species-matched HRP-conjugated secondary antibody for 1 hour at room temperature [95].
- Detection: Develop blots using enhanced chemiluminescence substrate and image with chemiluminescence detection systems [95].
Technical Considerations:
- Include loading controls (e.g., GAPDH, β-actin) to normalize for protein loading variations [98].
- Optimize antibody concentrations through preliminary titration experiments.
- Consider protein half-life when determining harvest timepoints; some proteins may require longer periods for noticeable reduction.
Flow Cytometry for Surface Protein Analysis
Flow cytometry offers unique advantages for detecting cell surface proteins and performing multi-parameter analysis at the single-cell level, making it invaluable for immunology research and membrane protein studies.
Experimental Protocol:
- Cell Harvesting: Collect cells 72-96 hours post-transfection and wash with PBS containing 1% FBS [97].
- Antibody Staining: Resuspend 0.5-1×10^6 cells in 100μL staining buffer containing fluorochrome-conjugated antibodies. Incubate for 30 minutes at 4°C protected from light.
- Washing and Fixation: Wash cells twice to remove unbound antibody, then fix with 1-4% paraformaldehyde if required for downstream analysis [97].
- Data Acquisition: Analyze samples using flow cytometer, collecting a minimum of 10,000 events per sample. Use unstained and isotype controls to establish background fluorescence and gating parameters.
- Data Analysis: Calculate geometric mean fluorescence intensity (MFI) or percentage of positive cells compared to control samples.
Optimization Tips:
- Perform antibody titration experiments to determine optimal signal-to-noise ratios.
- Implement fluorescence-minus-one (FMO) controls for accurate gating in multicolor panels.
- Use viability dyes to exclude dead cells from analysis, which may non-specifically bind antibodies.
Method Integration and Data Interpretation
Effective knockdown assessment requires integration of multiple methods to build a comprehensive understanding of siRNA efficacy. The complementary nature of these techniques addresses different aspects of gene silencing, from initial transcript reduction to ultimate functional protein depletion. The diagram below outlines a logical framework for interpreting concordant and discordant results across assessment methods:
Addressing Discrepancies Between Methods
Discrepancies between qRT-PCR and Western blot results are common in knockdown experiments and often reveal important biological insights or technical issues. The table below outlines frequent scenarios and their potential biological and technical explanations:
Table 2: Troubleshooting Discordant Knockdown Results
| qRT-PCR Result | Western Blot Result | Potential Biological Causes | Technical Considerations |
|---|---|---|---|
| Significant reduction | Minimal reduction | - Long protein half-life- Translational compensation- Protein stabilization | - Insufficient incubation time- Antibody specificity issues- Inefficient protein transfer |
| Minimal reduction | Significant reduction | - Altered protein degradation- Feedback mechanisms- miRNA regulation | - RNA degradation during isolation- Primer efficiency problems- Reference gene instability |
| Significant reduction | Significant reduction | Successful knockdown at both transcript and protein levels | Optimal experimental conditions |
Recent research demonstrates effective application of integrated assessment methods. In B-cell non-Hodgkin lymphoma studies, B7-H7 knockdown efficiency was validated through both qRT-PCR and Western blot, confirming reduced mRNA levels and corresponding protein reduction, which translated to inhibited tumor growth and increased drug sensitivity [95]. Similarly, in hepatocellular carcinoma research, FANCI knockdown was confirmed through multiple methods, with functional assays demonstrating suppressed cell proliferation and colony formation [99].
Essential Research Reagent Solutions
Successful knockdown experiments require high-quality reagents and optimized systems. The following table outlines essential research tools for implementing the protocols described in this application note:
Table 3: Essential Research Reagents for siRNA Knockdown Studies
| Reagent Category | Specific Examples | Function | Selection Considerations |
|---|---|---|---|
| siRNA Design Tools | siDirect, NUPACK | Design specific siRNA sequences with minimal off-target effects | Algorithm specificity, species compatibility, validation data [94] |
| Delivery Vehicles | Lipofectamine, lentiviral particles (e.g., Santa Cruz sc-78498-V) | Introduce siRNA into cells | Cell type compatibility, efficiency, cytotoxicity [95] |
| RNA Isolation Kits | TRIzol reagent, column-based kits | Extract high-quality RNA for qRT-PCR | Yield, purity, processing time, DNase treatment option [95] [97] |
| qPCR Reagents | SYBR Green Master Mix, reverse transcriptase | Convert RNA to cDNA and amplify target sequences | Efficiency, specificity, compatibility with detection systems [95] [96] |
| Antibodies | Primary (e.g., Abcam ab214327), HRP-conjugated secondary | Detect specific proteins in Western blot | Specificity, species reactivity, validation in applications [95] |
| Flow Cytometry Reagents | Fluorochrome-conjugated antibodies, viability dyes | Label and detect surface proteins on live cells | Brightness, compatibility with instrument lasers, minimal spillover [97] |
Robust assessment of siRNA-mediated knockdown requires a multi-faceted approach that evaluates both transcriptional and translational suppression. While qRT-PCR offers rapid and sensitive confirmation of mRNA reduction, Western blot provides essential protein-level validation, and flow cytometry enables single-cell analysis of surface proteins. The integrated application of these methods, with careful attention to experimental timing, appropriate controls, and reagent quality, ensures accurate interpretation of knockdown efficiency. Furthermore, understanding the potential biological and technical reasons for discrepant results between methods enhances experimental troubleshooting and strengthens conclusions drawn from siRNA-based experiments. As RNAi technology continues to evolve toward more sophisticated applications, including conditional systems like ORIENTR [100] and therapeutic development, rigorous efficiency assessment remains fundamental to generating reliable, reproducible research outcomes.
RNA interference (RNAi) is a fundamental biological process that enables sequence-specific silencing of gene expression. For research and therapeutic development, two primary synthetic tools harness this pathway: small interfering RNA (siRNA) and short hairpin RNA (shRNA) [101]. While both aim to degrade target messenger RNA (mRNA), their molecular structures, mechanisms of delivery, and temporal applications differ significantly. The choice between siRNA and shRNA is critical and hinges on the experimental needs—specifically, whether the goal is a rapid, transient knockdown or a long-term, stable gene silencing [102]. This article provides a detailed comparison and protocols to guide researchers and drug development professionals in selecting and implementing the appropriate RNAi tool.
Key Differences Between siRNA and shRNA
Understanding the distinct characteristics of siRNA and shRNA is the first step in selection. The table below summarizes their core properties for direct comparison.
Table 1: Fundamental Characteristics of siRNA and shRNA
| Characteristic | siRNA | shRNA |
|---|---|---|
| Structure | Short, ~21-25 nucleotide double-stranded RNA with 2-nucleotide 3' overhangs [101] | ~57-58 nucleotide single-stranded RNA that folds into a stem-loop (hairpin) structure [101] |
| Delivery Method | Transfection (e.g., liposomes, lipid nanoparticles) or electroporation [101] [103] | Viral vector transduction (e.g., lentivirus, adenovirus) [101] [102] |
| Cellular Processing | Pre-formed duplex is directly loaded into the RNA-induced silencing complex (RISC) in the cytoplasm [101] | DNA vector is transcribed in the nucleus, processed by Drosha and Exportin-5, then exported to cytoplasm where Dicer cleaves it into functional siRNA [101] |
| Duration of Effect | Transient (typically 3-7 days) [101] [102] | Stable (weeks to months); can be propagated in dividing cells due to genomic integration [101] [102] |
| Primary Application | Rapid, high-throughput, and transient knockdown studies; easily titratable [101] [102] | Long-term knockdown, studies in hard-to-transfect cells (e.g., primary cells), and in vivo models [101] [102] |
The decision flowchart below synthesizes the key selection criteria to guide researchers in choosing the right tool for their experimental goals.
Experimental Protocols for RNAi
Protocol for Transient Gene Silencing Using siRNA
This protocol is optimized for reverse transfection in cultured cells, which saves time and can improve efficiency for certain cell lines [104].
Workflow Overview:
Step-by-Step Methodology:
Cell Plating and Transfection Complex Formation:
- Plate cells at 40-80% confluency in appropriate growth medium without antibiotics. Using cells at a low passage number (e.g., <50) is critical for consistency [104].
- For a 24-well plate, dilute 5-20 nM siRNA in a sterile, serum-free medium (e.g., 50 µL per well) [104].
- Dilute the transfection reagent (e.g., RNAiMax) in an equal volume of serum-free medium (50 µL per well) and incubate for 5 minutes at room temperature.
- Combine the diluted siRNA and diluted transfection reagent. Mix gently and incubate for 15-20 minutes at room temperature to allow complex formation.
- Add the siRNA-transfection reagent complexes dropwise onto the plated cells. Gently swirl the plate to ensure even distribution.
Incubation and Media Change:
- Incubate cells at 37°C for 24-48 hours. If cytotoxicity is observed, replace the transfection complex-containing media with normal growth media after 8-24 hours to maintain cell health [104].
Knockdown Validation:
- mRNA Analysis: Quantify target mRNA levels using qRT-PCR 24-48 hours post-transfection to directly assess silencing efficiency [104].
- Protein Analysis: Assess protein knockdown by Western blot, immunofluorescence, or flow cytometry 48-72 hours post-transfection. The timing depends on the half-life of the target protein [104].
Protocol for Stable Gene Silencing Using shRNA
This protocol utilizes lentiviral transduction for stable integration of shRNA into the host genome, enabling long-term silencing.
Workflow Overview:
Step-by-Step Methodology:
Viral Particle Production:
- Co-transfect a packaging cell line (e.g., HEK293T) with the shRNA expression plasmid and viral packaging plasmids using a standard transfection reagent.
- Harvest the virus-containing supernatant 48-72 hours post-transfection. Concentrate the supernatant if necessary.
Cell Transduction:
- Plate target cells at a density that will be 30-50% confluent at the time of transduction.
- Infect cells with the viral supernatant in the presence of a transduction enhancer (e.g., polybrene). A low viral titer is recommended to increase the probability of single shRNA integration per cell, minimizing combinatorial effects [101].
- Centrifuge the plate (spinoculation) to enhance infection efficiency.
Selection of Stable Cells:
- Begin antibiotic selection (e.g., Puromycin) 48 hours post-transduction. The optimal antibiotic concentration should be determined by a kill curve beforehand.
- Maintain selection pressure for at least 3-7 days until all non-transduced control cells are dead. For pooled populations, continue culture under selection to establish a stable polyclonal line.
Knockdown Validation:
- Validate mRNA and protein knockdown as described in the siRNA protocol. Monitor knockdown efficiency over multiple passages, as it can decline if cells with lower shRNA expression are over-represented [101].
Research Reagent Solutions
Successful RNAi experiments depend on high-quality reagents. The table below lists essential materials and their functions.
Table 2: Essential Reagents for RNAi Experiments
| Reagent / Material | Function & Importance |
|---|---|
| Validated siRNA/siPOOLs | Defined pools of siRNAs targeting a single gene; reduce off-target effects compared to single siRNAs or small pools [101]. |
| shRNA Expression Vector | Plasmid or viral vector containing the shRNA sequence, often driven by a U6 or H1 promoter, and frequently including a reporter gene (e.g., GFP) or antibiotic resistance marker for selection [101] [102]. |
| Lipid-Based Transfection Reagents | Form complexes with negatively charged siRNA, facilitating cellular uptake through endocytosis; suitable for standard cell lines (e.g., HeLa, HEK293) [104]. |
| Lipid Nanoparticles (LNPs) | Advanced delivery system that encapsulates siRNA, offering superior protection from degradation and enhanced cellular uptake with low toxicity; available as ready-to-use formulations [103]. |
| Viral Packaging System | Plasmids providing viral structural proteins (gag, pol) and envelope protein (vs.v.g.) required to produce replication-incompetent viral particles for shRNA delivery [101]. |
| Selection Antibiotics | (e.g., Puromycin, Hygromycin): Allow for the elimination of non-transduced cells, ensuring a pure population of stably expressing shRNA cells [101]. |
Troubleshooting and Optimization Guidelines
- Optimizing siRNA Delivery: For difficult-to-transfect cells (e.g., primary cells, neurons), try different transfection reagents or use electroporation [104]. Always include a positive control siRNA (targeting an essential or easily detectable gene) and a negative control (scrambled or non-targeting sequence) to monitor efficiency and specificity [104].
- Minimizing Off-Target Effects: Both siRNA and shRNA can cause off-target effects. shRNAs are subject to additional variables, as high expression from strong promoters can saturate RNAi machinery, and heterogeneous Dicer processing can generate multiple siRNA sequences [101]. Using defined siRNA pools (siPOOLs) and inducible shRNA systems can mitigate these issues [101].
- Validation is Critical: Due to documented poor overlap between hits from siRNA and shRNA screens, always validate key findings with an alternative tool, such as CRISPR-Cas9 gene knockout or pharmacological inhibitors [101].
The choice between siRNA and shRNA is strategic, defined by the need for either transient (siRNA) or stable (shRNA) gene silencing. siRNA offers rapid, titratable knockdown ideal for initial functional screening, while shRNA enables long-term studies and is applicable to a wider range of cell types via viral delivery.
In drug development, this distinction is paramount. siRNA therapeutics have seen clinical success, with six FDA-approved drugs for metabolic and genetic disorders as of 2024 [26] [105]. These leverage siRNA's transient nature for treatments requiring periodic administration. The oncology field, however, presents greater challenges like targeted delivery, though over 260 siRNA candidates are in preclinical or clinical development [3] [26]. Innovations in delivery systems, particularly lipid nanoparticles (LNPs) and GalNAc conjugates, along with sophisticated chemical modifications, are enhancing stability, specificity, and efficacy, positioning siRNA and shRNA technologies as cornerstones of next-generation precision therapeutics [106] [3] [105].
In the field of targeted genetic research, three powerful technologies have emerged as cornerstone methodologies: small interfering RNA (siRNA), CRISPR/Cas9 gene editing, and antisense oligonucleotides (ASOs). Each offers a distinct mechanism for modulating gene expression, with unique applications, advantages, and limitations. For researchers focused on gene knockdown, understanding the comparative landscape of these tools is fundamental to experimental design and therapeutic development. siRNA, a component of the RNA interference (RNAi) pathway, enables transient gene silencing at the post-transcriptional level through mRNA degradation [107] [2]. In contrast, CRISPR/Cas9 provides a permanent DNA-level modification by introducing double-strand breaks in the genome, enabling gene knockout, knock-in, or correction [108] [109]. ASOs, single-stranded DNA or RNA molecules, employ a versatile modulation strategy, capable of inducing mRNA degradation or sterically blocking translation and splicing events [106] [110]. This application note provides a comparative analysis framed within siRNA-centric research, detailing protocols and strategic considerations for employing these technologies.
The following table provides a high-level quantitative comparison of the core characteristics of siRNA, CRISPR/Cas9, and ASOs.
Table 1: Core Technology Comparison
| Feature | siRNA | CRISPR/Cas9 | Antisense Oligonucleotides (ASOs) |
|---|---|---|---|
| Molecular Type | Double-stranded RNA (~20-25 bp) [5] | RNA-guided DNA endonuclease (Cas9 protein + gRNA) [111] | Single-stranded DNA/RNA (12-25 nucleotides) [106] [107] |
| Cellular Target | Cytoplasmic mRNA [2] | Nuclear DNA [109] | RNA (mRNA, pre-mRNA) [106] |
| Primary Mechanism | mRNA degradation via RISC [2] | DNA double-strand break via Cas9 [111] | RNase H1 degradation or steric blockade [106] |
| Genetic Effect | Reversible knockdown | Permanent editing | Transient to semi-permanent modulation |
| Key Application | Loss-of-function studies, therapeutic gene silencing [107] [2] | Gene knockout, knock-in, base editing, functional genomics [108] [109] | Exon skipping, translational inhibition, therapeutic splicing modulation [106] [107] |
| Typical Delivery | Lipid nanoparticles, electroporation, viral vectors [107] [5] | Viral vectors (AAV, LV), electroporation, nanoparticles [111] | Lipid conjugates (e.g., GalNAc), nanoparticles, free uptake [112] |
| Major Challenge | Off-target effects, transient effect, endosomal escape [5] [2] | Off-target editing, delivery efficiency, ethical concerns [108] [111] | Off-target effects, cellular uptake, stability [106] |
| Therapeutic Approval | Multiple approved (e.g., Patisiran, Givosiran) [107] [2] | Early clinical trials (e.g., for genetic diseases, cancer) [111] | Multiple approved (e.g., Nusinersen, Eteplirsen) [107] [113] |
Mechanism of Action and Workflow
The following diagrams illustrate the core mechanisms and experimental workflows for each technology, highlighting key differences in their approach to gene modulation.
siRNA Mechanism and Workflow
CRISPR-Cas9 Mechanism and Workflow
ASO Mechanism and Workflow
Key Research Reagent Solutions
Successful implementation of gene modulation technologies requires a suite of specialized reagents and tools. The following table catalogs essential solutions for research applications.
Table 2: Essential Research Reagents and Tools
| Reagent Category | Specific Examples & Formats | Primary Function | Key Considerations |
|---|---|---|---|
| siRNA Reagents | ON-TARGETplus siRNA [5], Accell siRNA [5], Silencer Select siRNA | Gene silencing with reduced off-targets; delivery in difficult cells | Chemical modifications (2'-OMe) enhance specificity [5]; pooling siRNAs reduces off-targets [5] |
| CRISPR-Cas9 Tools | SpCas9 expression plasmids, synthetic sgRNAs, Alt-R CRISPR-Cas9 systems [108] | Gene knockout, knock-in, base editing | gRNA design tools critical for specificity [111]; HDR efficiency requires optimization [108] |
| ASO Reagents | Gapmer ASOs [106], Steric-blocking ASOs [106], Morpholinos [106] | mRNA degradation or splicing modulation | Generation (1st-3rd) affects affinity/stability [106]; backbone modifications (PS) improve nuclease resistance [106] |
| Delivery Systems | Lipid Nanoparticles (LNPs) [107] [112], Electroporation systems [111], GalNAc conjugates [112] | Encapsulate and deliver nucleic acids to cells | LNPs effective for siRNA/mRNA [107] [112]; electroporation for hard-to-transfect cells [5] [111] |
| Validation Tools | qPCR assays, Western blot kits, NGS services (RNA-seq, WGS) | Confirm gene expression changes, protein knockdown, or on-target editing | Multi-level validation (RNA, protein, phenotype) is essential; NGS identifies off-target effects [111] |
Detailed Experimental Protocols
Protocol: siRNA-Mediated Gene Knockdown in Mammalian Cells
This protocol outlines a standard procedure for transient gene knockdown using synthetic siRNA in adherent mammalian cell lines, a foundational technique for functional genomics.
Materials & Reagents
- ON-TARGETplus SMARTpool siRNA or similar validated siRNA reagents [5]
- Appropriate cell culture medium and supplements
- Transfection reagent (e.g., Lipofectamine RNAiMAX)
- Opti-MEM or similar serum-free reduced-medium
- Validated positive control siRNA (e.g., targeting GAPDH or POLR2A)
- Non-targeting negative control siRNA [5]
Procedure
- Cell Seeding: One day prior to transfection, seed cells in an appropriate multi-well plate to achieve 30-50% confluence at the time of transfersion. Use standard growth medium without antibiotics.
- Transfection Complex Preparation:
- For a single well of a 24-well plate: Dilute 5 pmol of siRNA (e.g., 1 µL of a 5 µM stock) in 50 µL of Opti-MEM. In a separate tube, dilute 1-2 µL of transfection reagent in 50 µL of Opti-MEM. Incubate both for 5 minutes at room temperature.
- Combine the diluted siRNA with the diluted transfection reagent. Mix gently and incubate for 15-20 minutes at room temperature to allow lipid-siRNA complex formation.
- Transfection: Add the 100 µL of complex dropwise to the cells containing the complete growth medium. Gently swirl the plate to ensure even distribution.
- Incubation and Analysis:
- Incubate cells for 24-96 hours at 37°C and 5% CO₂.
- Timecourse Note: Maximal knockdown for most mRNAs is typically observed at 48-72 hours post-transfection. The effect is transient, generally lasting 2-4 days [5].
- Validation:
- Harvest cells and extract total RNA or protein.
- Quantify knockdown efficiency via qRT-PCR (mRNA level) and/or Western blot (protein level). Compare results to negative control siRNA-treated cells.
Technical Notes
- Controls: Always include a non-targeting negative control siRNA and a validated positive control siRNA to monitor transfection efficiency and assay performance [5].
- Optimization: For new cell lines, optimize the siRNA concentration (typically 1-50 nM) and the transfection reagent ratio in a dose-response experiment.
- Specificity: Use chemically modified siRNAs (e.g., ON-TARGETplus) and pool designs to minimize sequence-specific off-target effects [5].
Protocol: CRISPR-Cas9 Mediated Gene Knockout
This protocol describes the generation of constitutive gene knockout cell lines using a plasmid-based CRISPR-Cas9 system, enabling permanent loss-of-function studies.
Materials & Reagents
- Cas9 expression plasmid (e.g., pSpCas9(BB))
- sgRNA cloning vector or synthetic sgRNA
- Appropriate transfection reagent (e.g., Lipofectamine 3000) or electroporation kit
- Selection antibiotic (e.g., Puromycin) if using a plasmid with a resistance marker
- Cell culture medium and supplements
Procedure
- sgRNA Design and Preparation:
- Design sgRNAs targeting early exons of the target gene using established bioinformatic tools (e.g., from Broad Institute) to minimize off-target effects [111].
- Clone the designed sgRNA sequence into the CRISPR plasmid, or purchase synthetic sgRNA.
- Delivery:
- Transfect the Cas9-sgRNA plasmid (or ribonucleoprotein complex of Cas9 protein and synthetic sgRNA) into the target cells using a method appropriate for the cell type (e.g., lipofection, electroporation) [111].
- For plasmids, include a fluorescent or antibiotic selection marker to enrich for transfected cells.
- Selection and Expansion: 24-48 hours post-delivery, begin antibiotic selection (if applicable) for 3-7 days. Allow the population to recover and expand.
- Validation of Knockout:
- Screening: Perform a rapid genomic DNA extraction from the bulk population. Use a T7 Endonuclease I or Surveyor assay to detect indel mutations at the target site.
- Clonal Isolation: To isolate pure knockout clones, perform serial dilution or single-cell sorting to establish single-cell-derived colonies.
- Confirmation: Expand individual clones and validate the knockout by Sanger sequencing of the target locus and by Western blot to confirm the absence of the target protein.
Technical Notes
- Mosaicism: When working with embryos or primary cells, be aware that initial populations may be mosaic, containing a mix of edited and unedited cells [108].
- Off-target Analysis: For critical applications, employ next-generation sequencing to screen for potential off-target edits at predicted genomic sites [111].
- Phenotyping: Always use multiple, independently derived clones for phenotypic assays to control for clonal variation and potential off-target effects.
Protocol: Antisense Oligonucleotide (ASO) Treatment for Gene Silencing
This protocol covers the use of gapmer ASOs to induce RNase H1-mediated degradation of target mRNA in mammalian cells.
Materials & Reagents
- Chemically modified Gapmer ASOs (e.g., LNA/DNA mixmer) [106]
- Transfection reagent (e.g., Lipofectamine 3000) or free uptake medium for gymnotic delivery
- Opti-MEM or serum-free medium
- Cell culture medium
Procedure
- ASO Design: Design gapmer ASOs with a central DNA "gap" region (for RNase H1 recruitment) flanked by modified wings (e.g., 2'-MOE, LNA) to enhance binding affinity and nuclease resistance [106].
- Cell Treatment:
- Transfection-based Delivery: Follow steps similar to the siRNA protocol (Sections 5.1.2-5.1.3), using typically higher ASO concentrations (10-100 nM).
- Gymnotic (Free Uptake) Delivery: For ASOs designed for free uptake, simply add the ASO directly to the cell culture medium at a higher concentration (e.g., 1-5 µM) in the presence of serum. This method is less efficient but avoids transfection artifacts.
- Incubation: Incubate cells for 24-72 hours. Gapmer ASOs lead to mRNA degradation, with effects observable within hours and lasting several days.
- Analysis: Harvest cells and analyze knockdown efficacy by qRT-PCR and/or Western blot, as in Section 5.1.5.
Technical Notes
- Mechanism Selection: For applications other than degradation (e.g., splice modulation), use steric-blocking ASOs (e.g., Morpholinos, PMOs) which do not recruit RNase H1 [106].
- Stability: Chemical modifications (e.g., phosphorothioate backbone, 2'-MOE) are crucial to protect ASOs from nuclease degradation and improve pharmacokinetics [106].
- Specificity: As with siRNA, design controls and use bioinformatic tools to minimize off-targeting due to seed region homology or unintended RNA interactions.
The choice between siRNA, CRISPR/Cas9, and ASOs is dictated by experimental goals. siRNA is ideal for rapid, transient knockdown and high-throughput screens. CRISPR/Cas9 is unmatched for creating permanent, DNA-level changes and generating stable cell lines or animal models. ASOs offer unique capabilities in splicing modulation and can be effective in systems where transfection is challenging. For a thesis focused on siRNA, understanding its performance relative to these other technologies provides a comprehensive framework for critiquing results and planning future experiments. As these fields evolve, improvements in delivery, specificity, and chemical modification will further solidify their roles as indispensable tools in biological research and therapeutic development [106] [107] [112].
In the face of a significant reproducibility crisis in biomedical research, largely driven by poorly validated antibody reagents, the implementation of robust negative controls is paramount. This application note details industry best practices for employing short interfering RNA (siRNA)-mediated knockdown as a critical negative control to validate antibody specificity. We provide definitive experimental protocols for integrating genetic knockdown with Western blot analysis, enabling researchers and drug development professionals to confirm that their antibodies specifically recognize the intended target protein, thereby ensuring data integrity and accelerating therapeutic development.
Antibodies are fundamental tools in life science research, yet their improper validation poses a significant challenge to scientific reproducibility. It is estimated that the research community wastes approximately $800 million annually on low-quality antibodies, underscoring the scale of this issue [114]. A primary cause is the reliance on validation protocols that incorporate positive controls but lack critical negative controls [115]. Without such controls, nonspecific antibodies that bind to off-target proteins can go undetected, leading to irreproducible results and flawed scientific conclusions [115] [116].
The international scientific community has recognized this problem, with groups like the International Working Group for Antibody Validation (IWGAV) establishing guidelines to improve standards [116]. siRNA knockdown addresses this need directly by providing a powerful genetic negative control. This method verifies that an observed signal is indeed due to the specific antibody-target interaction, as a true specific signal will diminish when the target protein is genetically depleted [117].
siRNA Knockdown as a Gold-Standard Negative Control
Core Principle and Mechanism
siRNA-mediated knockdown utilizes the cell's native RNA interference (RNAi) pathway to selectively degrade target mRNA, thereby reducing the expression of the protein of interest [117]. The process can be summarized as follows:
- Introduction of siRNA: Double-stranded siRNA is introduced into cells via transfection.
- RISC Loading and Strand Selection: The siRNA is loaded into the RNA-induced silencing complex (RISC). The complex is asymmetric and favors the strand with the least thermodynamically stable 5' terminus [118].
- Target Cleavage: The antisense "guide" strand directs RISC to the complementary mRNA sequence, which is subsequently cleaved and degraded [115] [117].
- Protein Knockdown: The degradation of mRNA leads to a significant reduction in the synthesis of the target protein.
For antibody validation, this protein reduction is key. A specific antibody will show a markedly diminished signal (e.g., on a Western blot) in the knockdown sample compared to controls, confirming that the antibody is binding specifically to the target protein [115] [117].
Experimental Workflow for Knockdown Validation
The following diagram illustrates the integrated workflow for validating antibody specificity using siRNA knockdown:
Detailed Experimental Protocol
Establishing Experimental Conditions
A well-designed siRNA knockdown experiment for antibody validation requires three distinct conditions to be run in parallel [115]:
- Condition 1 (Test Knockdown): Cells transfected with siRNA targeting the gene of interest.
- Condition 2 (Scrambled Control): Cells transfected with a scrambled siRNA sequence with no significant homology to the genome. This controls for nonspecific effects of the transfection process and the presence of siRNA itself [115] [31].
- Condition 3 (Untransfected Control): Cells that are not subjected to transfection, providing a baseline for protein expression.
For optimal results, cell density should be around 70% confluence at the time of transfection, and culture conditions should be kept constant throughout the experiment [115]. Titration of the siRNA concentration may be necessary to optimize knockdown efficiency while minimizing off-target effects [115].
siRNA Design and Selection Guidelines
Effective siRNA design is critical for successful knockdown. While proprietary algorithms exist, the following general guidelines can be applied for designing or selecting siRNAs [31]:
Table 1: Guidelines for Effective siRNA Design
| Guideline | Description | Rationale |
|---|---|---|
| Sequence Selection | Start with 21 nt sequences that begin with an AA dinucleotide and record the 3' adjacent 19 nucleotides. | Promotes effective RISC binding and cleavage; compatible with U6 pol III promoter systems [31]. |
| GC Content | Select sequences with 30-50% GC content. | siRNAs with higher G/C content can be less active [31]. |
| Specificity Check | Compare potential target sequences to the organism's genome database using BLAST. Eliminate sequences with >16-17 contiguous base pairs of homology to other genes. | Minimizes off-target effects by ensuring specificity for the intended gene [31]. |
| Internal Repeats | Avoid stretches of >4 T's or A's in the target sequence. | Prevents premature transcription termination when using RNA pol III promoters [31]. |
| Target Position | Select 2-4 target sites at different positions along the gene sequence. | Reduces the chance of targeting an inaccessible region of the mRNA due to secondary structure or protein binding [31]. |
While several algorithms exist to predict siRNA efficacy, their performance can be variable, particularly for short hairpin RNAs (shRNAs) [118]. Therefore, empirical testing of multiple siRNAs per target is strongly recommended. Many suppliers offer pre-designed and validated siRNA pools that guarantee knockdown efficacy.
Validation and Analysis via Western Blotting
Following the knockdown period (typically 48-72 hours to allow for protein turnover), cells are harvested and lysed. The lysates are then analyzed by Western blotting:
- Gel Electrophoresis and Transfer: Load equal amounts of protein from each of the three conditions. Include a molecular weight marker.
- Immunodetection: Probe the membrane with the antibody being validated. A well-validated loading control antibody (e.g., against Actin or GAPDH) should also be used to ensure equal loading.
- Result Interpretation: A specific antibody will show a strong band in the scrambled and untransfected control lanes, and a significantly diminished or absent band in the target siRNA lane [115] [117]. The loading control should show consistent signal across all lanes.
Table 2: Expected Results for Antibody Specificity Validation by Western Blot
| Experimental Condition | Expected Result with a Specific Antibody | Expected Result with a Non-specific Antibody |
|---|---|---|
| Untransfected Cells | Strong band at expected molecular weight | Band(s) present |
| Scrambled siRNA Control | Strong band at expected molecular weight | Band(s) present |
| Target-specific siRNA | Significantly reduced or absent band | Band(s) remain unchanged |
The following diagram details the molecular mechanism of siRNA action that underlies this validation method:
Successful execution of this protocol requires careful selection of reagents. The following table lists key solutions and their critical functions.
Table 3: Research Reagent Solutions for Knockdown Validation
| Essential Reagent | Function & Importance | Selection Criteria |
|---|---|---|
| Validated Primary Antibodies | Binds specifically to the target protein for detection in Western blot. | Choose antibodies already validated for Western blot. Seek vendors that provide knockdown/knockout validation data [116] [117]. |
| siRNA (Target-specific) | Mediates sequence-specific degradation of the target mRNA. | Use pre-designed, validated pools for best results. Alternatively, design using guidelines in Table 1 [31]. |
| Scrambled siRNA | A negative control siRNA with no significant sequence homology to the genome. | Crucial for distinguishing specific knockdown from non-specific cellular responses to transfection or siRNA presence [115] [31]. |
| Transfection Reagent | Facilitates the delivery of siRNA into the cells. | Must be optimized for the specific cell line used. Efficiency can be monitored with fluorescently-labeled siRNA [115]. |
| Positive Control Lysates | Cell lines known to express the target protein provide a positive control. | Confirms the immunodetection protocol is working. Resources like Expression Atlas or the Human Protein Atlas can help identify suitable cell lines [116]. |
Integrating siRNA knockdown as a routine negative control is an industry best practice that is essential for demonstrating antibody specificity. This rigorous validation method directly addresses the root causes of the reproducibility crisis by weeding out non-specific antibodies, thereby ensuring that research data and conclusions are built on a solid foundation. By adhering to the detailed protocols and guidelines outlined in this application note, researchers and drug developers can significantly enhance the reliability of their experimental outcomes, save valuable time and resources, and accelerate the pace of scientific discovery and therapeutic innovation.
Conclusion
siRNA technology has matured from a powerful research tool into a validated therapeutic modality, as evidenced by multiple FDA-approved drugs. Success hinges on a integrated approach that combines sophisticated sequence design, strategic chemical modifications, and advanced delivery systems to overcome challenges related to stability, off-target effects, and tissue-specific targeting. Looking forward, the field is poised for significant growth by expanding delivery beyond the liver, improving the duration of silencing, and leveraging machine learning for more predictive siRNA design. These advancements will further solidify the role of siRNA in personalized medicine and unlock new treatments for a broader range of diseases.
References
- 1. RNAi-Based Therapeutics and Novel RNA Bioengineering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9827509/]
- 2. Small interfering RNA: From designing to therapeutic in cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC11999615/]
- 3. New breakthroughs in siRNA therapeutics expand the drug ... [https://www.cas.org/resources/cas-insights/sirna-therapeutics]
- 4. Systematic analysis of siRNA and mRNA features impacting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12205987/]
- 5. siRNA - Applications [https://horizondiscovery.com/en/applications/rnai/sirna-applications]
- 6. Life of RISC: Formation, action, and degradation of RNA ... [https://www.sciencedirect.com/science/article/pii/S1097276521010285]
- 7. Harnessing Argonaute 2 as an Innovative Delivery System ... [https://www.sciencedirect.com/science/article/abs/pii/S1773224725008627]
- 8. The guide-RNA sequence dictates the slicing kinetics and ... [https://www.sciencedirect.com/science/article/pii/S1097276524005331]
- 9. Small Interfering RNA (siRNA) Therapy - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK580472/]
- 10. Clinical Pharmacology Overview of Approved siRNA Drugs [https://www.bocsci.com/blog/summary-of-clinical-pharmacology-of-approved-sirna-drugs/?srsltid=AfmBOoq9SmlR9LL7u_zXLhV_q1TNdVwsr2NT5H0p67RTI_b3Zj464-CL]
- 11. A Comparison of Currently Approved Small Interfering RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11054365/]
- 12. INTRODUCTION TO C. elegans ANATOMY [https://www.wormatlas.org/hermaphrodite/introduction/mainframe.htm]
- 13. Caenorhabditis Elegans: Development from the Perspective of ... [https://www.ncbi.nlm.nih.gov/books/NBK26861/]
- 14. Caenorhabditis elegans [https://en.wikipedia.org/wiki/Caenorhabditis_elegans]
- 15. In vivo efficacy of NRL knockdown with cell-penetrating ... [https://www.nature.com/articles/s41598-025-07299-6]
- 16. Structural Diversity Repertoire of Gene Silencing Small ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3198623/]
- 17. siRNA Repositioning for Guide Strand Selection by Human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3143821/]
- 18. Structure of the Dicer-2–R2D2 heterodimer bound to a ... [https://www.nature.com/articles/s41586-022-04790-2]
- 19. Molecular dynamics simulations based siRNA design ... [https://www.nature.com/articles/s41598-025-16936-z]
- 20. Therapeutic siRNA: state of the art [https://www.nature.com/articles/s41392-020-0207-x]
- 21. Knocking down barriers: advances in siRNA delivery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7097568/]
- 22. Recent Update on siRNA Therapeutics [https://www.mdpi.com/1422-0067/26/8/3456]
- 23. Small interfering RNA (siRNA) Therapeutics | DelveInsight [https://www.delveinsight.com/blog/small-interfering-rna-sirna-therapeutics-competitive-landscape-technology-and-pipeline-insights-2015-a-delveinsights-report]
- 24. The new era of siRNA therapy: Advances in cancer treatment [https://pmc.ncbi.nlm.nih.gov/articles/PMC12148947/]
- 25. New molecular technology targets tumors and ... [https://unclineberger.org/news/new-molecular-technology-targets-tumors-and-simultaneously-silences-two-undruggable-cancer-genes/]
- 26. Frontiers | Clinical development prospects of siRNA drugs for tumor therapy: analysis of clinical trial registration data from 2004 to 2024 [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1637958/full]
- 27. Development of conditional-siRNA programmable riboswitch ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12375242/]
- 28. Capricor Therapeutics Presents New Data Demonstrating ... [https://www.barchart.com/story/news/36285430/capricor-therapeutics-presents-new-data-demonstrating-a-scalable-framework-for-loading-therapeutic-oligonucleotides-into-exosomes-at-aaev-2025]
- 29. Global siRNA screen identifies human host factors critical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12204624/]
- 30. Mastering siRNA Design: Steps to Achieve Precision Gene ... [https://www.genscript.com/synthetic-biology-news/mastering-sirna-design-precision-gene-silencing.html]
- 31. siRNA Design Guidelines | Technical Bulletin #506 [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rnai-sirna/general-articles/-sirna-design-guidelines.html]
- 32. How to design effective siRNA for gene knockdown ... [https://synapse.patsnap.com/article/how-to-design-effective-sirna-for-gene-knockdown-experiments]
- 33. Strategies for Improving siRNA-Induced Gene Silencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5788215/]
- 34. Designing & Screening of siRNA molecules for silencing ... [https://www.sciencedirect.com/science/article/abs/pii/S147692712500369X]
- 35. Top Ten Ways to Optimize siRNA Delivery in Cultured Cells [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rnai-sirna/general-articles/top-ten-ways-to-optimize-sirna-delivery-in-cultured-cells.html]
- 36. An artificial cationic oligosaccharide combined with ... [https://www.nature.com/articles/s41598-020-71896-w]
- 37. 2′-O-Methyl at 20-mer Guide Strand 3′ Termini May ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7327867/]
- 38. Systematic Evaluation of Position-Specific Tolerability ... [https://pubmed.ncbi.nlm.nih.gov/40418189/]
- 39. Chirality matters: stereo-defined phosphorothioate linkages ... [https://pubmed.ncbi.nlm.nih.gov/34268578/]
- 40. Minimal 2'-O-methyl phosphorothioate linkage modification ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0188593]
- 41. A guide to large-scale RNA sample preparation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5937877/]
- 42. Targeted delivery systems of siRNA based on ionizable ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975025000321]
- 43. Preclinical testing of antiviral siRNA therapeutics delivered in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12507954/]
- 44. GalNAc-siRNA Conjugates: Leading the Way for Delivery of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5994659/]
- 45. progress and challenges of targeted delivery of siRNA ... [https://pubmed.ncbi.nlm.nih.gov/25660205/]
- 46. Genomic medicine in hepatology: mechanisms and liver ... [https://molmed.biomedcentral.com/articles/10.1186/s10020-025-01358-4]
- 47. Ligand-tethered lipid nanoparticles for targeted RNA ... [https://www.nature.com/articles/s41467-022-35637-z]
- 48. GalNAc-Lipid nanoparticles enable non-LDLR dependent ... [https://www.nature.com/articles/s41467-023-37465-1]
- 49. Going beyond the liver: Progress and challenges of ... [https://www.sciencedirect.com/science/article/pii/S0168365915000930]
- 50. A Novel In Vivo siRNA Delivery System Specifically Targeting ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0044138]
- 51. siRNA Therapeutics for the Treatment of Hereditary ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12428872/]
- 52. The current and future landscape of RNA-based therapies ... [https://www.nature.com/articles/s43856-025-01166-1]
- 53. Optimizing siRNA Therapeutics: Addressing Nonclinical ... [https://novotech-cro.com/whitepapers/optimizing-sirna-therapeutics-addressing-nonclinical-and-manufacturing-complexities]
- 54. Oncology is back for siRNA, courtesy of AbbVie | ApexOnco [https://www.oncologypipeline.com/apexonco/oncology-back-sirna-courtesy-abbvie]
- 55. Development of siRNA therapeutics to combat microbial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12592196/]
- 56. Preclinical and clinical development of siRNA-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4504743/]
- 57. FDA Approves Patisiran, First-Ever RNA Interference… [https://www.asgct.org/news-publications/asgct-news/fda-approves-patisiran-first-ever-rna-interference-therapeutic-approved-for-clinical-use]
- 58. Alnylam Announces First-Ever FDA Approval of an RNAi ... [https://investors.alnylam.com/press-release?id=22946]
- 59. Patisiran - StatPearls - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK589768/]
- 60. Inclisiran: a new generation of lipid-lowering siRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10611522/]
- 61. Inclisiran: Uses, Interactions, Mechanism of Action [https://go.drugbank.com/drugs/DB14901]
- 62. Inclisiran for the treatment of hypercholesterolaemia [https://www.drugsincontext.com/inclisiran-for-the-treatment-of-hypercholesterolaemia/]
- 63. Novartis presents new long-term Leqvio® (inclisiran) data ... [https://www.novartis.com/news/media-releases/novartis-presents-new-long-term-leqvio-inclisiran-data-demonstrating-consistent-efficacy-and-safety-beyond-six-years]
- 64. siRNA Specificity: RNAi Mechanisms and Strategies to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7876455/]
- 65. Methods for reducing siRNA off-target binding [https://eclipsebio.com/eblogs/sirna-off-target-strategies/]
- 66. Minimizing off-target effects by using diced siRNAs for RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2737221/]
- 67. New chemical modification reduces off-target effects in ... [https://www.news-medical.net/news/20240909/New-chemical-modification-reduces-off-target-effects-in-siRNA-drugs.aspx]
- 68. siRMSD: A Structural Parameter to Reduce Sequence ... [https://pubmed.ncbi.nlm.nih.gov/40877521/]
- 69. siRNA and the immune system [https://www.biosyn.com/tew/siRNA-and-the-immune-system.aspx]
- 70. siRNA as a criterion in host immunity and cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12374835/]
- 71. An siRMSD parameter of structural distortion induced by ... [https://www.sciencedirect.com/science/article/pii/S2162253125002471]
- 72. RNA interference [https://en.wikipedia.org/wiki/RNA_interference]
- 73. RNA Interference: Biology, Mechanism, and Applications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC309050/]
- 74. Cellular and biophysical barriers to lipid nanoparticle ... [https://www.nature.com/articles/s41467-025-60959-z]
- 75. Imaging small molecule-induced endosomal escape of ... [https://www.nature.com/articles/s41467-020-15300-1]
- 76. Key Mechanistic Principles and Considerations ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2020.01237/full]
- 77. Dry powder inhalation of siRNA therapeutics [https://pubmed.ncbi.nlm.nih.gov/41274344/]
- 78. Endosomal escape kinetics of mesoporous silica-based ... [https://pubmed.ncbi.nlm.nih.gov/23524121/]
- 79. siRNA Design It's All in the Algorithm [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rnai-sirna/tech-notes/sirna-design-it-s-all-in-the-algorithm.html]
- 80. A role for human Dicer in pre-RISC loading of siRNAs - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3045585/]
- 81. Development of Prodrug Type Circular siRNA for In Vivo ... [https://pubmed.ncbi.nlm.nih.gov/33016851/]
- 82. Enteral siRNA delivery technique for therapeutic gene ... [https://www.nature.com/articles/srep17035]
- 83. Hepatocyte-specific delivery of siRNAs conjugated to novel ... [https://pubmed.ncbi.nlm.nih.gov/25786782/]
- 84. [PDF] Comparison of partially and fully chemically-modified ... [https://www.semanticscholar.org/paper/Comparison-of-partially-and-fully-siRNA-in-delivery-Hassler-Turanov/322d492262c2a8cc5854ac46e04242397b55a673]
- 85. A Simple and Efficient One-Step Synthesis System for ... [https://www.mdpi.com/2218-273X/14/11/1416]
- 86. Advancements in the application of reporter gene cell lines ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12460214/]
- 87. Validating a Firefly Luciferase-Based High-Throughput ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3277734/]
- 88. A Luciferase-EGFP Reporter System for the Evaluation of ... [https://link.springer.com/article/10.1134/S0026893321040099]
- 89. Development of luciferase-based highly sensitive reporters ... [https://www.sciencedirect.com/science/article/pii/S2589004224024143]
- 90. Establishing an effective gene knockdown system using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9171500/]
- 91. 10 tips on how to best optimize siRNA transfection [https://www.ptglab.com/news/blog/10-tips-on-how-to-best-optimize-sirna-transfection/?srsltid=AfmBOoqxLlLXdT8X_F3b0aSlHpZvu7ZjeN3WzD2qh1MDFwMCwLMqwwxv]
- 92. Understanding Calculations for siRNA Data [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rnai-sirna/tech-notes/understanding-calculations-for-sirna-data.html]
- 93. RNAi vs. CRISPR: Guide to Selecting the Best Gene ... [https://www.synthego.com/blog/rnai-vs-crispr-guide/]
- 94. Assessment of RNA interference effectiveness mediated by ... [https://www.sciencedirect.com/science/article/abs/pii/S0965174825001626]
- 95. B7-H7 knockdown suppresses the proliferation, metastasis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12571572/]
- 96. The oncogenic role of TIMM8A in cancer and ... [https://www.nature.com/articles/s41598-025-03331-x]
- 97. Loss of TACC2 impairs chemokine CCL3 and CCL4 ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02354-2]
- 98. Why qPCR and WB results are not consistent 2025- ... [https://www.antibodysystem.com/archive/380.html]
- 99. Knockdown of FANCI suppresses hepatocellular ... [https://www.sciencedirect.com/science/article/pii/S2405844025011120]
- 100. Conditional RNA interference in mammalian cells via ... [https://www.nature.com/articles/s41467-024-50600-w]
- 101. siRNA vs shRNA - applications and off-targeting [https://www.sitoolsbiotech.com/blog/sirna-vs-shrna-applications-and-off-targeting]
- 102. What criteria should one use in choosing between siRNA ... [https://www.qiagen.com/us/resources/faq/2771]
- 103. LNP- siRNA for Gene Silencing - Ready-to-use siRNA Delivery [https://ozbiosciences.com/blog/lnp-sirna-for-efficient-gene-silencing-n123]
- 104. Top Ten Ways to Optimize siRNA in Cultured Cells Delivery [https://www.thermofisher.com/tw/zt/home/references/ambion-tech-support/rnai-sirna/general-articles/top-ten-ways-to-optimize-sirna-delivery-in-cultured-cells.html]
- 105. Novotech Report Reveals Growth in siRNA Research [https://novotech-cro.com/news/novotech-report-reveals-growth-sirna-research-over-150-industry-sponsored-clinical-trials]
- 106. RNA-Targeting Techniques: A Comparative Analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12563179/]
- 107. siRNA-based delivery systems: Technologies, carriers ... [https://www.sciencedirect.com/science/article/abs/pii/S0014299925001955]
- 108. CRISPR Genome Engineering: Advantages and Limitations [https://www.taconic.com/resources/crispr-genome-engineering-advantages-limitations]
- 109. Recent applications, future perspectives, and limitations of ... [https://www.sciencedirect.com/science/article/pii/S216225312500188X]
- 110. Application of Antisense Oligonucleotides as an Alternative ... [https://pubmed.ncbi.nlm.nih.gov/41226561/]
- 111. Challenges and Opportunities in the Application of CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503735/]
- 112. The Oligonucleotide Market in 2025 and Beyond, Key ... [https://www.linkedin.com/pulse/oligonucleotide-market-2025-beyond-key-trends-luke-mclaughlin-sghuf]
- 113. Antisense Oligonucleotides Market Trends Report 2025-2035 [https://finance.yahoo.com/news/antisense-oligonucleotides-market-trends-report-080700834.html]
- 114. GEN: Validating Antibodies for Specificity [https://www.ptglab.com/news/company-news/gen-validating-antibodies-for-specificity/?srsltid=AfmBOoqmE5PBfmF8mg9E4N_TXMxuneSwk7NZuXNY6nciJ2oOs2yXg-Xx]
- 115. How siRNA Knockdown Antibody Validation Works [https://www.labmanager.com/how-sirna-knockdown-antibody-validation-works-3374]
- 116. Antibody validation for Western blot: By the user, for the user [https://pmc.ncbi.nlm.nih.gov/articles/PMC6983856/]
- 117. Knockout and Knockdown Antibody Validation [https://www.thermofisher.com/us/en/home/life-science/antibodies/invitrogen-antibody-validation/knockout-knockdown-antibody-validation.html]
- 118. Criteria for effective design, construction, and gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1409772/]