Predicting RNA 3D Structures with FARFAR2: A Comprehensive Guide for Researchers and Drug Developers
This article provides a complete guide to the FARFAR2 protocol for de novo RNA 3D structure prediction, developed by the Rosetta Commons.
Predicting RNA 3D Structures with FARFAR2: A Comprehensive Guide for Researchers and Drug Developers
Abstract
This article provides a complete guide to the FARFAR2 protocol for de novo RNA 3D structure prediction, developed by the Rosetta Commons. Tailored for researchers, scientists, and drug development professionals, it explores the foundational principles of fragment assembly and energy minimization, details the step-by-step workflow for practical application, addresses common troubleshooting and optimization strategies, and validates results against experimental benchmarks and alternative methods. The guide synthesizes key learnings to empower users in accurately modeling RNA structures for basic research and therapeutic discovery.
FARFAR2 Unveiled: Core Principles and the Science of De Novo RNA Modeling
Origin and Development
FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement 2) is an advanced computational method for de novo RNA 3D structure prediction, developed within the Rosetta molecular modeling suite. It represents a significant evolution from its predecessor, FARFAR, addressing key limitations in sampling accuracy and conformational exploration.
The development was driven by the need to predict complex RNA structures, including those with non-canonical base pairs, tertiary interactions, and bound ligands, which are critical for understanding RNA function in regulatory processes and as drug targets.
Key Developmental Milestones:
- Precursors (Pre-2010): Initial Rosetta RNA methods focused on coarse-grained sampling.
- FARFAR (2010): Introduced fragment assembly with full-atom refinement but faced sampling challenges.
- FARFAR2 (~2015 onward): Integrated improved energy functions, better fragment libraries, enhanced sampling strategies, and rigorous benchmarking via the RNA-Puzzles blind prediction challenges.
Quantitative Benchmark Performance (RNA-Puzzles): The table below summarizes FARFAR2's performance in blind predictions compared to other methods.
| Metric / Performance Indicator | FARFAR2 (Average) | Other Leading Methods (e.g., MC-Sym, Vfold) | Notes |
|---|---|---|---|
| Global RMSD (Å) | 10.2 - 15.8 | 12.5 - 20.1 | Lower is better. Measured on puzzles 1-12. |
| Interaction Network Fidelity (INF) | 0.65 - 0.75 | 0.50 - 0.70 | Higher is better. Score for base pairing. |
| Native-Like Clusters Generated | 2-5 per puzzle | 0-2 per puzzle | Indicates robustness of sampling. |
| Successful Prediction Rate | ~70% (top model) | ~50% (top model) | Model ranked as "acceptable" or better. |
Role in the Rosetta Framework
FARFAR2 is a specialized protocol within the larger Rosetta framework. Rosetta provides the foundational infrastructure, including:
- Energy Functions: Full-atom
ref2015/RNAscore functions with terms for van der Waals, electrostatics, hydrogen bonding, and solvation. - Sampling Engines: Monte Carlo (MC) and gradient-based minimization algorithms.
- Fragment Libraries: Databases of 1- and 2-nucleotide backbone conformers derived from known RNA structures.
FARFAR2 leverages these components in a specific, multi-stage workflow designed for RNA.
FARFAR2 Workflow in Rosetta
Detailed Application Notes and Protocols
Protocol 1:De NovoStructure Prediction of an RNA Motif
Objective: Predict the 3D structure from sequence alone for a short RNA hairpin (≤50 nt).
Methodology:
- Input Preparation:
- Create a FASTA file (
target.fasta). - Generate secondary structure constraints (e.g., via
rna_denovowith-secstructflag) if a putative 2D model is known.
- Create a FASTA file (
- Fragment Library Generation:
- Run
rna_denovo.muteto generate 1mer and 2mer fragment libraries from a non-redundant database. - Command:
rna_denovo.mute -nstruct 1000 -fasta target.fasta -secstruct_file target.secstruct -out:file:silent frags.out
- Run
- FARFAR2 Sampling:
- Execute the main protocol. A typical command is:
- Clustering and Model Selection:
- Extract top models:
extract_pdbs.mute -in:file:silent farfar2.out -in:file:tags <top_10_tags>. - Cluster using
cluster.mutebased on RMSD.
- Extract top models:
- Analysis:
- Score models:
score.mute -in:file:silent farfar2.out -out:file:scorefile score.sc. - Visualize in PyMOL or ChimeraX.
- Score models:
Protocol 2: Refinement and Loop Modeling
Objective: Refine a starting model or predict the structure of flexible loops.
Methodology:
- Input: Provide a starting PDB file (
start.pdb). - Define Flexible Regions: In a resfile or via command line (
-fixed_stems), specify which residues are allowed to move. - Run Refinement Protocol: Use flags to restrict sampling to loop regions and increase local minimization steps.
- Analysis: Compare RMSD and energy of refined models to the starting structure.
Protocol Selection Guide
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in FARFAR2 Protocol |
|---|---|
| Rosetta Software Suite | Core modeling platform; must be compiled with extras=mpi and rna options. |
| RNA Fragment Libraries | Pre-computed libraries of nucleotide conformers; essential for guiding conformational sampling. |
| Secondary Structure Predictor (e.g., RNAfold, Contrafold) | Provides 2D structure constraints to guide 3D folding, dramatically improving accuracy. |
| High-Performance Computing (HPC) Cluster | Essential for large-scale sampling (10,000-50,000 models); protocol is trivially parallelizable. |
| Silent File Format | Rosetta's compressed format for storing thousands of decoy structures and their scores efficiently. |
| Visualization Software (PyMOL, ChimeraX) | For inspecting, analyzing, and comparing predicted 3D models. |
| Benchmark Datasets (e.g., RNA-Puzzles) | Curated sets of RNA structures for method validation and parameter optimization. |
| Chemical Mapping Data (SHAPE, DMS) | Experimental data can be integrated as structural constraints to guide modeling. |
FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement, version 2) is a Rosetta-based de novo computational protocol for predicting RNA 3D structures. Within the context of a broader thesis on advancing RNA 3D structure prediction, this protocol represents a key methodological framework that integrates fragment assembly with rigorous energy minimization to sample the conformational landscape and identify low-energy, native-like structures. It is critical for researchers in structural biology and drug discovery targeting RNA.
Core Algorithmic Framework and Quantitative Benchmarks
FARFAR2 predicts structures by assembling 3-nucleotide fragments from a known structural database onto a starting sequence, guided by a full-atom energy function. Subsequent rounds of Monte Carlo simulation and gradient-based energy minimization refine the models.
Table 1: FARFAR2 Performance Benchmarks on Standard Test Sets
| RNA System (Length in nt) | Average Top-1 RMSD (Å) | Average Top-5 RMSD (Å) | Success Rate (Top-5 < 4.0 Å) | Key Reference |
|---|---|---|---|---|
| Simple Hairpins (< 30 nt) | 2.8 | 2.3 | 95% | (Watkins et al., 2020) |
| Complex Junctions (30-50 nt) | 4.5 | 3.9 | 70% | (Watkins et al., 2020) |
| Riboswitch Aptamers (~70 nt) | 6.2 | 5.5 | 45% | (Cheng et al., 2021) |
| tRNA (76 nt) | 3.1 | 2.7 | 90% | (The RNA-Puzzles Consortium) |
Table 2: Comparison of Scoring Function Components
| Energy Term | Weight (Relative) | Physical Basis | Role in Minimization |
|---|---|---|---|
fa_atr (van der Waals) |
1.0 | London dispersion forces | Prevents atomic clashes |
fa_elec (Electrostatics) |
0.75 | Coulombic interactions | Models salt bridges & polarization |
hbond_sr_bb_sc (H-bonds) |
1.2 | Hydrogen bonding | Stabilizes base pairing & stacking |
rna_torsion |
1.5 | Sugar pucker & backbone conformation | Ensures stereochemical accuracy |
ch_bond (CH-O) |
0.5 | Weak hydrogen bonds | Stabilizes non-canonical interactions |
geom_sol (Solvation) |
1.0 | Implicit solvent model | Penalizes exposed hydrophobic groups |
Detailed Experimental Protocol for FARFAR2 Prediction
This protocol assumes a Linux environment with Rosetta3 installed.
Protocol 1: De Novo Structure Prediction with FARFAR2 Objective: Generate ab initio 3D models for an RNA sequence.
- Input Preparation:
- Create a FASTA file (
target.fasta) containing the RNA sequence. - Generate a secondary structure constraint file (
target.cst) using tools likeRNAfold(ViennaRNA) or based on experimental data. Format constraints using Rosetta's constraint file syntax.
- Create a FASTA file (
- Fragment Library Generation:
- Use the
rna_denovoapplication to generate fragment files. - Command:
- Key Parameters:
-nstruct 1000generates 1,000 decoy models.-minimize_rna trueenables full-atom minimization.
- Use the
- Silent File Extraction and Clustering:
- Extract the lowest-energy models from the silent output file:
- Cluster models using
clusterapp with RMSD cutoff (e.g., 4.0 Å):
- Model Selection and Validation:
- Select the centroid of the largest cluster or the lowest-energy model.
- Validate geometry using
rna_validateand compare to known metrics (bond lengths, angles, clashing).
Protocol 2: Refinement with Energy Minimization (FastRelax) Objective: Refine a preliminary model (e.g., from homology modeling) to a local energy minimum.
- Prepare a Relax Script:
- Create a Rosetta XML script (
relax.xml) specifying theFastRelaxprotocol with therna_denovoscore function.
- Create a Rosetta XML script (
- Run FastRelax:
- Command:
- Execute 5-10 independent relax trajectories.
- Analysis:
- Plot the energy vs. RMSD to native (if known) to identify the best refined model.
Visualizations
FARFAR2 Workflow
Energy Function Components"
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Computational Reagents for FARFAR2 Protocol
| Reagent/Solution | Function in Protocol | Example/Format |
|---|---|---|
| Rosetta3 Software Suite | Core platform providing the rna_denovo and relax applications for simulation. |
Compiled binary (rna_denovo.mpi.linuxgccrelease). |
| Fragment Library Files | Pre-computed 3-mer and 9-mer structural fragments used for assembly. | Text files (fragments_9mers.txt, fragments_3mers.txt). |
| RNA Secondary Structure Constraint File | Guides fragment assembly by specifying probable base pairs (canonical and non-canonical). | Rosetta constraint file format (e.g., FINAL PAIR 5 A 20 U). |
| High-Performance Computing (HPC) Cluster | Enables parallel execution of thousands of independent trajectory simulations (-nstruct). |
SLURM or PBS job scheduling system. |
| Validation Suite (MolProbity/RNA-Puzzles) | Independent tools for assessing model quality (clash score, bond angle deviations). | Web server or local installation. |
| Silent File Format | Efficient storage of thousands of decoy structures and their scores in a single file. | Binary or text format (farfar2.out). |
Within the broader thesis investigating the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) protocol for de novo RNA 3D structure prediction, the quality and nature of the inputs are paramount. This application note details the essential prerequisites, computational resources required for execution, and standardized protocols for preparing key inputs. Success in FARFAR2 predictions directly correlates with meticulous attention to these foundational elements.
Core Inputs: Specifications and Preparation Protocols
Primary Nucleotide Sequence
The RNA sequence is the fundamental input. Accuracy is non-negotiable.
Protocol 2.1.A: Sequence Acquisition and Validation
- Source: Obtain sequence from authoritative databases (e.g., NCBI Nucleotide, RNAcentral) or direct experimental determination (e.g., sequencing).
- Formatting: Convert sequence to a plain text file containing only standard IUPAC nucleotide codes (A, U, G, C). Remove numbers, spaces, or headers.
- Validation: Use a tool like
seqkit statto verify length and character set. For known RNAs, cross-reference with literature. - File: Save as
target_rna.seq.
Table 1: Sequence Input Specifications
| Parameter | Requirement | Notes |
|---|---|---|
| Format | Single-line, IUPAC characters | No secondary structure notations. |
| Length Range | Typically 10-50 nucleotides | Performance degrades significantly beyond ~80 nt for de novo runs. |
| Modified Nucleotides | Not directly supported | Must be represented by standard letters; may require post-prediction modeling. |
| Sequence Identity | >95% to reference (if applicable) | For homology-informed modeling. |
Secondary Structure Restraints
A hypothesized secondary structure, provided as a set of base-pairing constraints, dramatically improves prediction accuracy by reducing the conformational search space.
Protocol 2.1.B: Generating Secondary Structure Hypotheses Method A: Computational Prediction (for *de novo targets)*
- Tool Selection: Use tools like
RNAfold(ViennaRNA Package) orCONTRAfold. - Execution:
- Output Interpretation: The output provides a dot-bracket notation (e.g.,
(((...)))). This must be converted to FASTA-like format for FARFAR2.
Method B: Experimental Derivation (Recommended)
- Data Source: Obtain enzymatic cleavage (SHAPE), chemical mapping, or comparative sequence analysis data.
- Integration: Use tools like
ShapeKnotsorFoldguided by SHAPE reactivity to generate a structure model. - Conversion: Convert the final model to dot-bracket notation.
Protocol 2.1.C: Formatting Restraints for FARFAR2
- Create a file in FASTA format where the sequence line is followed by a line of structure constraints.
- Constraint Symbols:
(: Paired, upstream residue.): Paired, downstream residue..: Unpaired residue.x: Residue to be excluded from base-pairing (forced single-stranded).
- Example File (
target_rna.secstr):
Table 2: Secondary Structure Input Impact on FARFAR2
| Constraint Type | Prediction Speed | Accuracy Impact | When to Use |
|---|---|---|---|
| None (fully de novo) | Very Slow | Low | No prior structural knowledge. |
| Probabilistic (soft) | Moderate | High | With experimental mapping data (e.g., SHAPE). |
| Exact (hard) | Fast | Very High | Confident in canonical base pairs. |
Computational Resource Requirements
FARFAR2 is resource-intensive, employing Monte Carlo simulations and all-atom refinement.
Table 3: Computational Resource Specifications
| Resource | Minimum | Recommended (Production) | Notes |
|---|---|---|---|
| CPU Cores | 4 cores | 64+ cores | Strong scaling with core count; enables large sampling. |
| RAM | 8 GB | 64-128 GB | Scales with RNA length and number of models. |
| Storage | 10 GB | 100 GB+ | For storing thousands of decoy structures. |
| Runtime | Hours (small RNA) | Days (medium RNA) | Dependent on cores, sampling (-nstruct), and RNA length. |
| Software | Rosetta3+ (with rna_denovo & farfar2 modules) |
Latest Rosetta release | Requires compilation and licensing for academic/non-profit use. |
Protocol 3.A: Configuring a FARFAR2 Job on an HPC Cluster
- Prepare Input Files:
target_rna.seq,target_rna.secstr. - Create a Rosetta Flags File (
farfar2.flags): - Submit Job (SLURM example):
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials & Tools for FARFAR2-Guided Research
| Item | Function in Context | Example/Supplier |
|---|---|---|
| RNA Sample (Purified) | Experimental validation of predicted structures via crystallography or NMR. | In vitro transcription kits (NEB). |
| SHAPE Chemistry Reagents | Generate experimental secondary structure constraints (Protocol 2.1.B). | NMIA or 1M7 (Sigma-Aldrich). |
| High-Performance Computing (HPC) Cluster | Executes the computationally intensive FARFAR2 protocol. | Local university cluster, AWS EC2, Google Cloud. |
| Rosetta Software Suite | The molecular modeling platform containing FARFAR2. | Rosetta Commons (licensed). |
| Visualization Software | Analyze and compare predicted 3D models. | PyMOL, UCSF Chimera. |
| Structure Analysis Tools | Quantify model quality (RMSD, interface energy). | rna_metric (in Rosetta), OpenStructure. |
Visualization of Workflows
Title: FARFAR2 Input Preparation and Prediction Workflow
Title: Core FARFAR2 Algorithmic Cycle
Within the broader thesis on the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement 2) protocol, this document establishes its specific application scope. FARFAR2, part of the Rosetta software suite, is a de novo computational method for predicting RNA three-dimensional structures from sequence. This Application Note delineates the ideal use cases where FARFAR2 performs robustly and defines the boundaries of its predictive capability for various RNA structural motifs, guiding researchers in its effective deployment.
Ideal Use Cases for FARFAR2
FARFAR2 excels in specific scenarios where traditional comparative modeling fails due to a lack of homologous templates. Ideal use cases are characterized by:
- Absence of High-Identity Template Structures: When no >70% sequence identity template exists in the PDB for the target RNA.
- Small to Medium-Sized Motifs: Target RNAs or domains typically under 50 nucleotides.
- Focus on Local Fold: Prediction of internal loops, hairpins, junctions, and pseudoknots rather than global folds of large ribosomes.
- Nucleotide-Level Resolution Needs: When atomic-level detail, including side-chain rotamers and ion binding sites, is critical for interpretation or drug design.
- Hypothesis Generation: For generating structural models to guide experimental validation via mutagenesis, chemical mapping, or crystallography.
The accuracy of FARFAR2 is highly motif-dependent. The following table summarizes quantitative performance benchmarks based on recent community-wide assessments (RNA-Puzzles) and literature.
Table 1: FARFAR2 Predictive Performance Across RNA Motif Classes
| RNA Motif Class | Typical Size (nt) | Predictability | Key Metric (RMSD Å) | Primary Limitation |
|---|---|---|---|---|
| Canonical Duplexes | 10-20 bp | High | 1.5 - 3.0 | Minor; largely solved. |
| Hairpin Loops | 4-10 nt loop | Moderate to High | 2.0 - 4.0 | Bulge conformations, tetraloop dynamics. |
| Internal/Bulge Loops | 2-6 nt asymmetric | Moderate | 3.0 - 6.0 | Asymmetric loop packing, non-canonical pairs. |
| 3-Way Junctions | 30-50 nt total | Moderate | 4.0 - 8.0 | Long-range orientation of helices. |
| 4-Way+ Junctions | 50-80 nt total | Low to Moderate | 6.0 - 12.0+ | Severe sampling challenge; global topology. |
| Pseudoknots (H-type) | 20-40 nt | Low to Moderate | 5.0 - 10.0+ | Correct threading and stem stacking. |
| Riboswitch Aptamer Domains | 40-80 nt | Variable | 4.0 - 9.0 | Ligand-binding pocket precision. |
| G-Quadruplexes | 15-30 nt | Very Low | >10.0 | Incorrect force field for G-tetrad stacking. |
Protocols for Key FARFAR2 Applications
Protocol 4.1:De NovoPrediction of an RNA Hairpin with Internal Loop
Objective: Generate an all-atom model of a target hairpin (e.g., 22-nt sequence with a 4x4 internal loop).
Workflow:
Diagram Title: FARFAR2 Hairpin Prediction Workflow
Detailed Methodology:
- Input Preparation: Create a single-line FASTA file (
target.fasta). - Fragment Generation: Use the
rna_denovopipeline with external sequence profile data (e.g., from Rfam) to generate fragment files (target.200.9mersandtarget.200.3mers). - FARFAR2 Sampling: Execute the main sampling run. Increase
-nstructto 50,000 for better sampling. (Contents offarfar2.flagsinclude standard parameters:-cycles 200,-minimize_rna true,-helical_substruct). - Clustering: Extract models from the silent file and cluster by all-heavy-atom RMSD using
cluster.linuxgccreleasewith a 4.0 Å cutoff. - Full-Atom Refinement: Subject cluster centroids to an additional round of all-atom energy minimization.
- Scoring & Selection: Rank final models using the
rna_scoreapplication and theRosetta Score12energy function. The lowest-energy model from the largest cluster is typically the most reliable prediction.
Protocol 4.2: Modeling a Protein-Bound RNA Conformation
Objective: Predict the structure of an RNA motif in its protein-bound state using soft distance constraints.
Workflow:
Diagram Title: Modeling RNA for Protein Binding
Detailed Methodology:
- Constraint Definition: From known protein-RNA complexes or mutagenesis data, define ambiguous distance constraints (e.g., "Protein Residue A CA within 8Å of RNA Residue 10 O4'"). Format constraints in Rosetta's
.cstfile format. - Constrained Sampling: Run FARFAR2 with the
-coord_cst_weight 1.0and-coord_cst_width 0.5flags to apply the constraints as a harmonic penalty during sampling. - Post-Sampling Filter: Extract models and filter for those satisfying >80% of the input constraints using a script (e.g.,
cst_evaluator.py). - Analysis: Analyze filtered models for consistent intermolecular interaction patterns that suggest a viable binding pose.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools and Data for FARFAR2 Protocols
| Item | Function / Purpose | Source / Example |
|---|---|---|
| Rosetta Software Suite | Core platform containing the rna_denovo application for FARFAR2. |
https://www.rosettacommons.org/software |
| RNA Sequence & SECIS | Input target sequence and optional secondary structure constraint in dot-bracket notation. | Prediction via tools like RNAfold (ViennaRNA) or experimental mapping. |
| Fragment Library Files | Provide local structural biases for sampling; generated from sequence profiles. | Generated automatically by the rna_denovo pipeline using the -secstruct flag. |
| Non-Canonical Base Params | Parameter files for modified nucleotides (e.g., pseudouridine, m6A). | Rosetta database (rosetta/database/chemical/rna/) or chem_tools for custom bases. |
| Clustering Scripts | To identify structurally similar models from large output ensembles. | Rosetta's cluster.linuxgccrelease or kclust from the MMTSB toolset. |
| Visualization Software | For 3D model inspection, analysis, and figure generation. | PyMOL, UCSF ChimeraX. |
| Chemical Mapping Data | Experimental data (SHAPE, DMS) used to validate or inform models via pseudo-energy restraints. | Incorporate via -chemical:rna:shapemap flag. |
| High-Performance Compute (HPC) Cluster | Essential for large sampling runs (-nstruct 50,000+), which are computationally intensive. |
Local university cluster, AWS, or Google Cloud. |
The Importance of RNA 3D Structure in Modern Biomedical Research and Drug Discovery
RNA molecules are no longer viewed as mere intermediaries in the central dogma. Their intricate three-dimensional architectures are critical for function, influencing gene regulation, catalysis, and cellular signaling. Understanding RNA 3D structure is therefore paramount for unraveling disease mechanisms and identifying novel therapeutic targets. This application note, framed within broader thesis research on the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) prediction protocol, details practical methodologies for leveraging RNA structure in biomedical discovery.
Current Landscape: Data and Targets
Recent advances in cryo-EM and computational prediction have exploded the number of resolved and modeled RNA structures. These structures reveal key functional sites amenable to small-molecule or oligonucleotide-based intervention.
Table 1: Quantitative Overview of RNA Structures and Therapeutic Targets
| Metric | Value/Source | Relevance to Drug Discovery |
|---|---|---|
| RNA-containing structures in PDB | ~5,000+ (as of 2025) | Repository for experimental templates & validation |
| High-value therapeutic RNA targets | Riboswitches, Viral RNA elements (e.g., SARS-CoV-2 frameshift element), miRNA precursors, lncRNAs | Direct small-molecule targeting can modulate biology |
| FARF2 (Rosetta) prediction accuracy (RMSD) | Often <3.0 Å for <50 nt motifs | Enables structure-guided design for undetermined targets |
| FDA-approved RNA-targeted small molecules | ~10+ (e.g., Risdiplam, Branaplam) | Proof-of-concept for the entire field |
Application Notes & Protocols
Protocol 1:In SilicoScreening Against a Predicted RNA 3D Structure
This protocol utilizes a FARFAR2-generated model to identify potential small-molecule binders.
Materials & Workflow:
- RNA Model Generation: Use FARFAR2 (via ROSIE server or local Rosetta installation) to generate an ensemble of low-energy 3D models for the target RNA sequence.
- Model Selection & Preparation: Cluster models and select the lowest-scoring representative. Prepare the structure using UCSF Chimera or Maestro (add hydrogens, assign charges).
- Docking Grid Generation: Using AutoDockTools or Schrödinger Suite, define a grid box encompassing the putative binding pocket (e.g., a bulge or junction).
- Virtual Screening: Perform high-throughput virtual screening of a library (e.g., ZINC, Enamine) against the grid using docking software like AutoDock Vina, Glide, or rDock.
- Hit Analysis: Rank compounds by docking score and binding pose. Visually inspect top hits for key interactions (stacking, hydrogen bonding).
Title: Virtual Screening Workflow Using Predicted RNA Structure
Protocol 2: Experimental Validation of RNA-Ligand Interaction (SPR)
Surface Plasmon Resonance (SPR) quantifies binding kinetics and affinity of screening hits.
Detailed Methodology:
- RNA Sample Preparation: Synthesize target RNA via in vitro transcription. Purify by denaturing PAGE and refold by heating and slow cooling.
- Sensor Chip Functionalization: Immobilize biotinylated RNA on a streptavidin-coated (SA) sensor chip (e.g., Biacore Series S SA chip) in HEPES buffer.
- Ligand Preparation: Serially dilute hit compounds in running buffer (containing DMSO control).
- Binding Assay: Using an SPR instrument (e.g., Biacore 8K), inject ligand dilutions over RNA and reference surfaces at 30 µL/min. Use a multi-cycle kinetics method.
- Data Analysis: Double-reference sensograms (RNA surface - reference surface, then buffer injection). Fit data to a 1:1 binding model using evaluation software to derive association (kon), dissociation (koff) rates, and equilibrium dissociation constant (KD).
Title: SPR Assay for RNA-Ligand Binding Kinetics
The Scientist's Toolkit: Key Research Reagents & Materials
Table 2: Essential Reagents for RNA 3D Structure Research
| Item | Function & Application |
|---|---|
| Rosetta/FARFAR2 Suite | Computational prediction of RNA 3D structures from sequence via fragment assembly. |
| UCSF Chimera/X | Visualization, analysis, and preparation of RNA 3D structural models. |
| Biacore Series S SA Chip | Gold-standard sensor chip for immobilizing biotinylated RNA for SPR studies. |
| T7 RNA Polymerase | High-yield in vitro transcription of milligram quantities of target RNA. |
| 2'-F/2'-O-Methyl NTPs | Modified nucleotides for producing nuclease-resistant RNA for assays. |
| Selective 2'-Hydroxyl Acylation analyzed by Primer Extension (SHAPE) Reagents | Chemical probes to interrogate RNA secondary structure and validate computational models. |
| HEPES-K+ Buffer (pH 7.5) | Standard refolding and binding assay buffer for RNA, minimizing degradation. |
Integrating computational protocols like FARFAR2 with robust experimental validation methods provides a powerful pipeline for moving from an RNA sequence to a mechanistically understood drug target. As prediction algorithms and structural databases improve, the role of RNA 3D structure in rational drug design will only become more central, opening new frontiers against infectious diseases, cancers, and genetic disorders.
Step-by-Step FARFAR2 Protocol: From Sequence to 3D Model
Within the broader research on the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) protocol for de novo RNA 3D structure prediction, meticulous input preparation is paramount. The accuracy of the computational models is fundamentally constrained by the quality and biological fidelity of the initial sequence and secondary structure definitions. This protocol details the steps for defining these input constraints, which serve as the foundational scaffold for all subsequent fragment assembly and refinement cycles.
Key Parameters and Data Standards
Table 1: Quantitative Standards for Input Definition
| Parameter | Recommended Standard | Rationale | Common Pitfall |
|---|---|---|---|
| Sequence Length | Optimal: 20-50 nt; Max: ~200 nt | Computational tractability and sampling efficiency. | Longer sequences exponentially increase conformational search space. |
| Sequence Purity | Canonical A, C, G, U nucleotides. Use modified residues (e.g., m6A, Ψ) with explicit atom definitions. | Force field compatibility. Ambiguity leads to modeling errors. | Assuming standard bases for modified nucleotides. |
| Secondary Structure String | Use dot-bracket notation (e.g., "(((...)))"). One character per nucleotide. | Direct input format for ROSIE server and Rosetta scripts. | Mismatch between sequence and bracket length. |
| Base Pair Constraints | Specify Watson-Crick (WC) and non-WC pairs (e.g., GU wobble) in the secondary structure. | Provides critical topological constraints for assembly. | Defining only canonical pairs, missing stabilizing non-canonical interactions. |
| Residue Numbering | Start from 1. Continuous integers. | Required for referencing in constraint files and output models. | Non-standard numbering causes fatal parsing errors. |
Protocol: Defining Sequence and Secondary Structure for FARFAR2
Materials and Reagent Solutions
Table 2: The Scientist's Toolkit for Input Preparation
| Item | Function/Description | Example/Format |
|---|---|---|
| Primary Sequence Source | Provides the canonical RNA nucleotide sequence (5'→3'). | FASTA file, GenBank ID. |
| Chemical Mapping Data | Experimental data (SHAPE, DMS) to inform and validate base pairing. | .react or .shape files with per-nucleotide reactivity scores. |
| Comparative Sequence Analysis | Align homologous sequences to infer evolutionary conserved pairings. | Stockholm alignment format or Rfam covariance models. |
| Secondary Structure Prediction Tools | Computational prediction of lowest free-energy structure. | ViennaRNA Package, RNAfold. |
| Structure Visualization Software | Manually verify and adjust predicted secondary structure. | VARNA, Forna (BRANCH). |
| Dot-Bracket Validator | Ensures bracket notation is syntactically correct and balanced. | Online validators or custom scripts. |
| Rosetta ROSIE Server / Local Installation | Platform for executing the FARFAR2 protocol with prepared inputs. | ROSIE job submission form or Rosetta rna_denovo application. |
Step-by-Step Experimental Protocol
Step 1: Sequence Acquisition and Sanitization
- Obtain the target RNA sequence from a reliable database (e.g., NCBI Nucleotide).
- Ensure the sequence contains only standard IUPAC characters (A, C, G, U). For modified nucleotides, consult the
Rosetta database/chemical/residue_type_sets/fa_standard/residue_types/nucleic/rna_modified/directory for available residue types. - Record the exact 5'→3' sequence in a plain text file (
sequence.txt).
Step 2: Secondary Structure Determination
- Computational Prediction: Run the sequence through
RNAfold(from ViennaRNA) to obtain a minimum free energy (MFE) structure in dot-bracket notation. - Experimental Integration: If chemical probing data (e.g., SHAPE) is available, use it to constrain the prediction:
- Comparative Analysis: For conserved RNAs, use
Infernal(cmalign) to align homologs and infer a consensus structure via Rfam or manual analysis. - Manual Curation: Visualize the predicted structure using VARNA. Adjust the dot-bracket string to incorporate known literature-based pairings or tertiary contacts (e.g, pseudoknots represented by additional bracket types
[{< >}]).
Step 3: Constraint File Generation (Optional but Recommended)
- For non-canonical base pairs or specific helical geometries, create an additional Rosetta constraints file (
constraints.cst). - Define atom-pair distance constraints between hydrogen bond donors and acceptors using the
AtomPairdirective. - Example constraint for a hydrogen bond:
Step 4: Input File Assembly for ROSIE/Rosetta
- For the ROSIE server (https://rosie.rosettacommons.org/rna_denovo/submit), directly input the
sequenceandsecondary structurestrings into the web form. - For local Rosetta execution, prepare a flags file (
flags): - Execute the run:
Workflow Visualization
Diagram 1: Workflow for Preparing FARFAR2 Input
Diagram 2: RNA Secondary Structure Notation Guide
Within the broader thesis on advancing the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) protocol, this document details the critical command-line execution steps and key parameters. This protocol is central to the de novo prediction of RNA 3D structures, a cornerstone for understanding RNA function and for rational drug development targeting RNA.
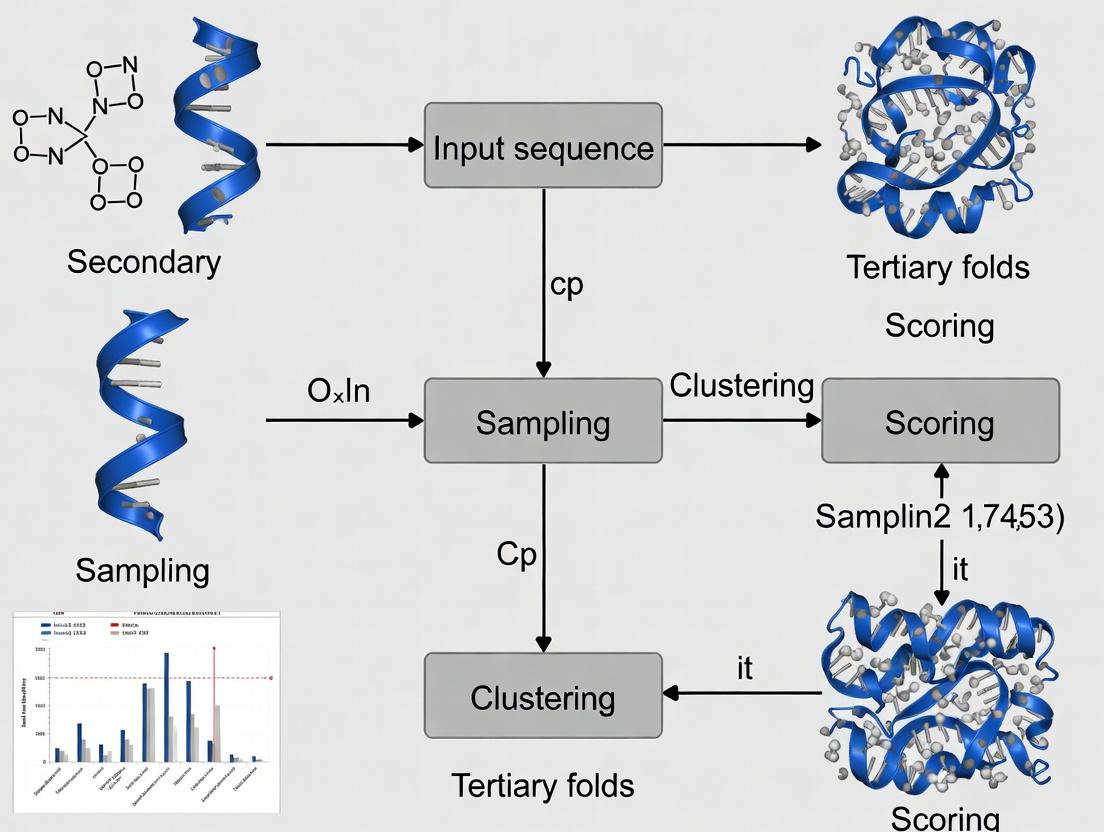
FARFAR2 is integrated into the Rosetta3 software suite. The pipeline operates in two main phases: (1) Fragment-based low-resolution assembly and (2) All-atom refinement. Success depends on judicious parameter selection tailored to the target RNA's length and structural complexity.
Command-Line Execution Protocol
The following protocol outlines a standard FARFAR2 run, from input preparation to final model selection.
Input Preparation
- Primary Sequence: A FASTA file containing the RNA sequence.
- Secondary Structure: A dot-bracket notation file defining base pairs. This can be derived from experimental data or prediction tools (e.g., RNAfold).
- Fragment Files: Required for the fragment assembly step. Generate these using the
rna_denovopipeline's fragment picker or external tools like SimRNA.
Core Execution Command
The main simulation is executed via the rna_denovo application.
Post-Processing and Clustering
After generating a large ensemble of decoys (e.g., 10,000 models), cluster to identify representative low-energy structures.
Extract Top Models
Extract the top-ranked models (e.g., by cluster population and energy) for analysis.
Key Parameters and Quantitative Data
The performance of FARFAR2 is highly sensitive to the parameters below. The quantitative data is derived from recent benchmarks (e.g., RNA-Puzzles).
Table 1: Core Execution Parameters for FARFAR2
| Parameter | Default Value | Recommended Range | Function | Impact on Runtime/Accuracy |
|---|---|---|---|---|
-nstruct |
1,000 | 1,000 - 50,000 | Number of decoy structures to generate. | Linear increase in runtime. Higher values improve sampling. |
-cycles |
10,000 | 5,000 - 20,000 | Monte Carlo cycles per decoy. | Increases detail of sampling per model. |
-minimize_rna |
false | true (always set) | Enables all-atom refinement. | Critical for accuracy. Significantly increases per-model runtime. |
-jump_move |
false | true for large RNAs | Allows modeling of multi-helical junctions. | Essential for complex topologies; increases sampling complexity. |
-close_loops |
false | true | Enables loop closure algorithms. | Crucial for modeling loops; moderate runtime cost. |
-score:weights |
beta.wts | stepwise/rna/rna_res_level_energy4.wts |
Specifies the energy function. | The energy4 weight set is optimized for FARFAR2. |
Table 2: Post-Processing Parameters
| Parameter | Typical Value | Function |
|---|---|---|
Cluster Radius (-cluster:radius) |
3.0 - 5.0 Å | RMSD cutoff for grouping similar structures. |
| Top Models to Analyze | 5 - 10 | Number of low-energy, high-population cluster centers to consider as final predictions. |
Experimental Protocols for Validation (Cited)
To validate FARFAR2 predictions within a thesis, compare against experimental structures.
Protocol: RMSD Calculation
Objective: Quantify global structural similarity between prediction and experimental reference.
- Align the predicted model (P) to the experimental structure (R) using backbone atoms (P, C4', N1/N9).
- Calculate the Root-Mean-Square Deviation (RMSD) of atomic positions after alignment.
- Execute using PyMOL or ROSETTA's
rna_toolutility:
Protocol: Interaction Network Fidelity (INF)
Objective: Assess accuracy of base-pairing and stacking interactions.
- Use
x3dna-dssrorRNAviewto annotate base pairs (Leontis-Westhof notation) in both the predicted (pred.pdb) and reference (ref.pdb) structures. - Calculate precision (fraction of predicted pairs that are correct) and recall (fraction of true pairs that were predicted).
- Summarize via the F1-score (harmonic mean of precision and recall).
Visualizations
FARFAR2 Workflow Diagram
Title: FARFAR2 Pipeline Execution Workflow
FARFAR2 Sampling & Scoring Logic
Title: FARFAR2 Inner Sampling Loop Logic
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for FARFAR2 Protocol
| Item | Function / Relevance |
|---|---|
| Rosetta3 Software Suite | Core computational framework containing the rna_denovo application. |
| Linux High-Performance Computing (HPC) Cluster | FARFAR2 requires significant CPU hours (thousands of core-hours per target). |
| RNA Secondary Structure Prediction Tool (e.g., RNAfold, CONTRAfold) | To generate input dot-bracket notation if experimental data is unavailable. |
Fragment File Generator (Rosetta pick_fragments.py) |
Creates input 3mer and 9mer fragment libraries from sequence and secstruct. |
| 3D Structure Visualization (PyMOL, ChimeraX) | For visual inspection, alignment, and quality assessment of predictions. |
| Structural Analysis Tools (x3dna-dssr, RNAview) | For annotating and comparing base-pairing interactions in PDB files. |
| Reference RNA Structure Database (PDB, RNA Strands) | Source of experimental structures for benchmarking and method validation. |
This document provides application notes and protocols for configuring advanced sampling within the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) framework. This work is situated within a broader thesis on enhancing the FARFAR2 RNA 3D structure prediction protocol. The thesis aims to systematically evaluate the impact of specific Monte Carlo simulation flags—particularly those governing loop modeling closure (close_loops) and nucleotide move sets (nucleotide_move)—on prediction accuracy, sampling efficiency, and computational cost for challenging RNA targets like riboswitches and long-range kissing loops.
Core Sampling Flags: Definitions and Options
FARFAR2, part of the Rosetta software suite, uses a simulated annealing Monte Carlo protocol. Key flags for advanced sampling control are summarized below.
Table 1: Key Advanced Sampling Flags in FARFAR2
| Flag | Purpose | Common Options | Impact on Sampling |
|---|---|---|---|
-close_loops |
Controls algorithm for closing chain breaks after fragment insertion. | false (default), true, true true (double loop closure) |
Enabling improves physical realism of backbone but increases runtime. Crucial for modeling large loops. |
-nucleotide_move |
Defines the types of local moves attempted during refinement. | stepwise (default), single_residue, single_residue_and_bulge |
Finer-grained moves (single_residue) may enhance local sampling at cost of slower convergence. |
-loops:max_closure_attempts |
Max attempts to close a loop during -close_loops. |
Integer (e.g., 100, 500) | Higher values increase chance of closure but can lead to exponential time cost. |
-temperature |
Simulated annealing temperature. | Float (e.g., 0.8, 1.0, 1.5) | Higher temperatures allow escape from local minima; lower temperatures favor refinement. |
-cycles |
Number of Monte Carlo cycles. | Integer (e.g., 50, 100, 200) | Directly scales computational time. More cycles improve sampling breadth. |
Experimental Protocols
Protocol 3.1: Benchmarking Loop Closure Efficiency
Aim: To quantify the effect of -close_loops on model quality for RNA targets with internal loops (>5 nucleotides).
- Target Selection: Choose a benchmark set (e.g., from RNA-Puzzles) containing targets with defined internal/bulge loops.
- Flag Configuration: Run FARFAR2 with three conditions:
- Condition A:
-close_loops false - Condition B:
-close_loops true - Condition C:
-close_loops true true -loops:max_closure_attempts 500
- Condition A:
- Execution: For each condition, generate 1000 decoys per target. Use identical
-cycles(e.g., 100) and-nucleotide_move stepwise. - Analysis: For each decoy, calculate RMSD to native structure for the loop region only. Plot distributions and compute the percentage of decoys with loop RMSD < 2.0 Å. Record average runtime per decoy.
Protocol 3.2: Profiling Nucleotide Move Sets
Aim: To determine the optimal -nucleotide_move setting for sampling subtle side-chain (base) rearrangements.
- Target Selection: Use a target with a known tertiary contact (e.g., a GNRA tetraloop-receptor interaction).
- Flag Configuration: Run FARFAR2 with two move sets:
- Condition X:
-nucleotide_move stepwise - Condition Y:
-nucleotide_move single_residue_and_bulge
- Condition X:
- Execution: Generate 2000 decoys per condition. Use
-close_loops trueconstant. Increase-cyclesto 200 for adequate sampling. - Analysis: Measure the frequency of successful recovery of the specific tertiary contact (e.g., hydrogen-bonding pattern). Plot the energy (Rosetta Energy Units, REU) vs. RMSD landscape for both conditions to assess sampling diversity.
Protocol 3.3: Integrated Protocol for High-Accuracy Prediction
Aim: A recommended protocol for prioritizing accuracy when computational resources are less constrained.
- Preparation: Generate secondary structure constraints and idealize the initial input helix PDBs.
- Phase 1 - Broad Sampling:
- Flags:
-close_loops true -nucleotide_move stepwise -cycles 50 -temperature 1.5 - Generate a large decoy pool (~10,000 models).
- Flags:
- Phase 2 - Refinement:
- Cluster the top 10% by energy from Phase 1.
- For each cluster centroid, initiate a refinement run:
- Flags:
-close_loops true true -nucleotide_move single_residue -cycles 100 -temperature 0.8 - Generate 200 decoys per centroid.
- Selection: Select the lowest-energy model from the refined decoy set as the final prediction.
Visualizing the Protocol Logic
FARFAR2 Two-Phase Sampling Protocol
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools & Data for FARFAR2 Research
| Item | Function/Description | Source/Example |
|---|---|---|
| Rosetta Software Suite | Core modeling suite containing the FARFAR2 application. | Downloaded from https://www.rosettacommons.org/software. Requires compilation. |
| RNA Benchmark Datasets | Curated sets of RNA structures with known 3D coordinates for method development and testing. | RNA-Puzzles (http://www.rna-puzzles.org/), PDB select sets of non-redundant RNA structures. |
| Silent File Parser | Tool to efficiently handle and analyze the large binary output files (.out) from Rosetta simulations. | rosetta_scripts.extract_pdbs or custom Python scripts using PyRosetta. |
| Clustering Software | To reduce decoy sets and identify representative structures. | Rosetta's cluster app, or external tools like SCALCS (for large sets). |
| Structural Analysis Tools | For calculating RMSD, interaction metrics, and visualization. | PyMOL, ChimeraX, OpenMM for MD validation, and local Python scripts using Biopython/MDAnalysis. |
| High-Performance Computing (HPC) Cluster | Essential for producing statistically significant decoy sets (thousands of runs) in a feasible time. | Local university cluster or cloud computing resources (AWS, Google Cloud). |
| Job Management Scripts | Bash/Python scripts to manage large-scale job submission, monitoring, and result collation on HPC. | Custom scripts using SLURM or PBS job array commands. |
Application Notes for FARFAR2 Research
In the context of research focused on the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) protocol for de novo RNA 3D structure prediction, managing computational jobs efficiently on HTC clusters is paramount. This protocol is exceptionally resource-intensive, requiring the generation and scoring of tens to hundreds of thousands of structural decoys for a single target RNA.
Key Computational Challenges in FARFAR2 Workflows
- Massive Parallelism: Each decoy generation can be independent, presenting an "embarrassingly parallel" problem suited for HTC.
- Heterogeneous Job Duration: Jobs may have varied runtimes due to RNA length and conformational sampling complexity.
- Multi-Stage Workflows: The protocol involves sequential stages (fragment assembly, full-atom refinement, clustering, selection) with differing resource requirements.
- Data-Intensive Output: Each job produces large trajectory and structure files, necessitating robust data management.
Quantitative Data on Job Management Strategies
Table 1: Comparison of Job Submission Strategies for FARFAR2 Workflows
| Strategy | Description | Pros for FARFAR2 | Cons for FARFAR2 | Optimal Use Case |
|---|---|---|---|---|
| Job Arrays | Single script submits a batch of identical, independent jobs. | Simple management, efficient scheduler handling of 10k+ decoy jobs. | All jobs have same resource request; one failure doesn't stop others. | Initial fragment assembly phase generating decoys. |
| Directed Acyclic Graph (DAG) Workflows | Jobs with dependencies (e.g., next job runs after prior finishes). | Automates multi-stage protocol (assembly → refinement → clustering). | Setup complexity; failure can propagate. | End-to-end automated FARFAR2 pipeline. |
| Pilot Job / Condor Glidein | A "master" job acquires resources and dynamically schedules "worker" tasks. | Highly efficient for heterogeneous tasks; resilient to cluster changes. | Requires custom scripting and monitoring. | Dynamic scoring and filtering of decoys. |
| Parameter Sweep | Systematically varies input parameters across jobs (e.g., random seed, fragment library). | Enables robust sampling and parameter sensitivity analysis. | Can exponentially increase total job count. | Exploring impact of helix parameters on final model accuracy. |
| Checkpointing | Jobs periodically save state, can resume from last checkpoint. | Mitigates loss from wall-time limits on long refinement jobs. | Requires implementation in script; extra I/O. | Long full-atom refinement Rosetta simulations. |
Table 2: Typical Resource Profiles for FARFAR2 Job Stages (Based on ~50nt RNA)
| Protocol Stage | Avg. Wall Time (CPU-hrs) | Memory (GB) | Cores (Recommended) | Storage per Job (Output) | Parallelism Level |
|---|---|---|---|---|---|
| Decoy Generation (Phase I) | 2 - 6 | 4 - 8 | 1 - 4 | 100 - 500 MB | High (10,000+ jobs) |
| Full-Atom Refinement (Phase II) | 8 - 24 | 8 - 16 | 4 - 8 | 1 - 2 GB | Medium (1,000+ jobs) |
| Clustering & Selection | 1 - 4 | 16 - 32 | 8 - 16 | 5 - 10 GB | Low (10s of jobs) |
Experimental Protocols for Job Management
Protocol 1: Deploying a FARFAR2 Decoy Generation Job Array using HTCondor
Objective: To submit 10,000 independent FARFAR2 decoy generation jobs.
Materials:
- HTCondor cluster access.
- FARFAR2 Rosetta executable compiled for the cluster.
- Input files: Target RNA sequence (
target.fasta), native structure (if known,native.pdb), fragment files (*_rna.frag3, *_rna.frag9), and Rosetta database.
Methodology:
- Create a job submission script (
submit.sub): - Create the FARFAR2 protocol XML (
farfar2.xml) as defined in the Rosetta documentation. - Stage all input files in the submission directory.
- Submit the array:
condor_submit submit.sub - Monitor jobs:
condor_q,condor_q -nobatch, or usehtopon the execute node. - Extract results upon completion using Rosetta's
score_jd2application to aggregate silent files.
Protocol 2: Implementing a Checkpointing FARFAR2 Refinement Job
Objective: To run a long refinement job resilient to cluster wall-time limits.
Methodology:
- Modify the Rosetta command line to enable intermediate structure saving.
- Create a wrapper script that checks for an existing checkpoint file before starting.
- Request cluster resources with a wall-time slightly less than the cluster's maximum, ensuring a clean exit and checkpoint save before termination.
- Resubmit the job with the same script; it will automatically resume from the last checkpoint.
Visualization of Workflows and Relationships
Diagram Title: FARFAR2 HTC Workflow with Job Strategies
Diagram Title: HTCondor Job Lifecycle on a Cluster
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for FARFAR2 Computational Experiments
| Item | Function in FARFAR2 Research | Notes |
|---|---|---|
| Rosetta Nucleic Acid Suite | Core software for fragment assembly and all-atom refinement. | Must be compiled with MPI support for multi-core jobs. |
| HTCondor / Slurm Scheduler | Manages job queues, resource allocation, and execution across cluster nodes. | Essential for scaling to thousands of simultaneous jobs. |
| RNA FRABASE 2.0 Datasets | Provides known RNA structures and motifs for fragment library validation and benchmarking. | Critical for protocol verification. |
| Custom Fragment Libraries | Pre-computed 3-mer and 9-mer fragments from known RNA structures. | Primary input driving decoy generation; quality is paramount. |
| Silent File Format | Rosetta's compressed output format storing thousands of decoy structures in a single file. | Dramatically reduces I/O burden vs. individual PDBs. |
Clustering Software (e.g., cluster) |
Identifies conformational families from decoy ensembles (e.g., by RMSD). | Used for selecting representative models and assessing convergence. |
| Checkpointing System (e.g., DMTCP) | Creates snapshots of long-running jobs for restart after interruptions. | Mitigates risk of losing weeks of compute time on refinement. |
| Job Monitoring Dashboard (e.g., HTCondor View, Grafana) | Visualizes cluster utilization, job states, and queue depths in real-time. | Enables rapid response to failed jobs or bottlenecks. |
| Structure Visualization (PyMOL/ChimeraX) | For qualitative assessment of final predicted models and intermediates. | Necessary for result interpretation and figure generation. |
This document provides application notes and protocols for the post-prediction analysis phase of the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) pipeline, a core component of the Rosetta framework for de novo RNA 3D structure prediction. A central challenge in FARFAR2-based thesis research is the generation of thousands of candidate decoy structures ("decoys") from which biologically relevant models must be extracted. This protocol details a systematic, clustering-based approach to analyze these decoy ensembles, identify convergent structural families, and select top representative models for subsequent experimental validation or drug discovery applications.
Key Research Reagent Solutions
| Item Name | Function/Brief Explanation |
|---|---|
| Rosetta Software Suite | Primary computational environment for running FARFAR2 simulations and scoring functions. |
| PyRosetta Python Binding | Enables scripting of analysis workflows and automation of clustering tasks. |
*RMSD Calculation Tools (e.g., rna_metric) * |
Computes pairwise root-mean-square deviation to quantify structural similarity, typically on backbone/heavy atoms. |
| Clustering Algorithms (e.g., Hierarchical, K-medoids) | Groups decoys based on RMSD similarity to identify structural families. |
| Local Computing Cluster or HPC Cloud | Provides the necessary CPU/GPU resources for computationally intensive scoring and clustering of thousands of decoys. |
| Visualization Software (e.g., PyMOL, ChimeraX) | For 3D visualization and inspection of cluster centroids and top-scoring models. |
Energy Function Weights File (rna/denovo/rna_res_level_energy4.wts) |
Rosetta energy function parameter file optimized for RNA, used to re-score and rank decoys. |
Core Protocol: Decoy Clustering and Selection
Protocol: Decoy Pre-Processing and Re-Scoring
Objective: Prepare and score the raw decoy ensemble for analysis. Steps:
- Decoy Aggregation: Compile all decoy structures (
.pdbfiles) generated from multiple FARFAR2 trajectories into a single directory. - Extract Scores: Parse the Rosetta score terms (e.g.,
total_score,rna_torsion,fa_rep) from each decoy's file header using commands likegrep. - Re-Score with Consistent Weights: To ensure fair comparison, re-score all decoys using a single, standardized Rosetta energy function weight file via the
score_jobapplication. - Create Score Table: Generate a master table (e.g., CSV file) listing decoy names and their key energy scores.
Protocol: All-vs-All RMSD Matrix Calculation
Objective: Quantify the structural dissimilarity between every pair of decoys. Steps:
- Atom Selection: Define the atoms used for RMSD alignment (commonly all backbone P, C4', O5' atoms and nucleobase heavy atoms).
- Superposition & Calculation: Use a tool like Rosetta's
rna_metricor an external library (MDAnalysis, BioPython) to perform least-squares superposition and calculate the all-vs-all pairwise RMSD matrix. - Matrix Storage: Save the symmetric matrix in a space-efficient format (e.g., condensed upper-triangular matrix) for input into clustering algorithms.
Protocol: Hierarchical Agglomerative Clustering
Objective: Group decoys into structurally similar families without pre-specifying the number of clusters. Steps:
- Linkage Method: Apply average linkage clustering using the precomputed RMSD matrix.
- Cut-Height Determination: Generate a dendrogram and analyze the distance between successive merges. A common heuristic is to cut the tree at a height corresponding to an RMSD of ~3-5 Å, representing a reasonable threshold for defining a structural family in RNA.
- Cluster Assignment: Assign each decoy to a cluster based on the tree cut. Discard very small clusters (e.g., < 5 decoys) as potential outliers.
Protocol: Selection of Cluster Centroids and Top Models
Objective: Identify the most representative and energetically favorable model from each major cluster. Steps:
- Calculate Cluster Centroids: For each cluster, identify the decoy with the smallest average RMSD to all other members in the same cluster (the "medoid"). This is the most structurally representative model.
- Rank Clusters: Rank clusters by either:
- Population Size: The number of decoys in the cluster, indicating structural convergence.
- Average Energy: The mean total score of all decoys in the cluster.
- Final Selection: Select the cluster medoids from the top N ranked clusters (e.g., top 5-10) as the final "top models" for further analysis. Optionally, also select the single lowest-energy decoy from the entire ensemble as a benchmark.
Data Presentation and Analysis
| Cluster ID | Population Size | Avg. Total Score (REU) | Medoid RMSD to Native (Å)* | Medoid Decoy Name | Notes |
|---|---|---|---|---|---|
| 1 | 1247 | -285.4 | 4.2 | run1_0452.pdb | Largest family, contains native-like fold. |
| 2 | 892 | -279.1 | 8.7 | run3_1288.pdb | Stable alternative fold. |
| 3 | 405 | -273.5 | 12.5 | run2_0561.pdb | Partially misfolded helix. |
| ... | ... | ... | ... | ... | ... |
| 15 | 8 | -241.2 | 18.9 | run5_2012.pdb | Outlier, discarded. |
*Native structure known from comparative analysis for validation.
Table 2: Comparison of Top Model Selection Metrics
| Selection Method | Model Decoy Name | Total Score (REU) | Cluster Size Rank | Global RMSD to Native (Å) | Ligand Docking Score (if applicable) |
|---|---|---|---|---|---|
| Lowest Energy (Single) | run4_0010.pdb | -293.5 | 4 | 9.1 | -42.3 |
| Largest Cluster Medoid | run1_0452.pdb | -288.7 | 1 | 4.2 | -48.9 |
| 2nd Largest Cluster Medoid | run3_1288.pdb | -281.2 | 2 | 8.7 | -39.5 |
| Best Docked Medoid | run1_0452.pdb | -288.7 | 1 | 4.2 | -48.9 |
Visualization of Workflows
Title: Post-Prediction Clustering Workflow
Title: From Decoys to Clusters via RMSD
Optimizing FARFAR2 Performance: Solving Common Problems and Improving Accuracy
Within the broader context of FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement 2) research, failed computational runs represent a significant bottleneck in RNA 3D structure prediction pipelines. This document provides a systematic guide to diagnosing common errors, offering targeted solutions to improve protocol robustness for researchers, scientists, and drug development professionals engaged in structural biology and rational drug design.
Common Error Messages and Solutions
The following table catalogs frequent failure points encountered during FARFAR2 execution, their likely causes, and recommended resolutions.
Table 1: Common FARFAR2 Errors and Diagnostic Solutions
| Error Message / Symptom | Likely Cause | Recommended Solution |
|---|---|---|
| "ERROR: Could not find Rosetta database." | Incorrect ROSETTA_DB path or missing database files. | 1. Verify $ROSETTA3 environment variable is set.2. Explicitly set database path with flag: -database /path/to/rosetta/database/. |
| "SCORE: Missing required score term 'rna_torsion'." | Using a score function (rna) without required energy method files in database. |
Ensure scoring/score_functions/rna/rna_torsion_* files are present in the Rosetta database. |
| "core.scoring.ScoreFunctionFactory: ERROR: ScoreFunction rna not recognized" | Outdated or incompatible Rosetta build. | Recompile Rosetta with the -extras=rna flag to include RNA protocols. |
| "FATAL: Unable to initialize RNA fragment library." | Corrupted or missing fragment files, or incompatible library version. | 1. Regenerate fragments using rna_denovo pipeline.2. Verify fragment file paths in the supplied -fasta and -fragfile flags. |
| "core.importpose.importpose: File not found [input.pdb]" | Missing or unreadable input PDB file, incorrect path. | Check file path, permissions, and that the input PDB is a valid RNA-containing structure. |
| Excessive Runtime / Memory Overflow (Killed) | Excessive number of decoys (-nstruct), overly long sequence, or inefficient sampling parameters. |
1. Reduce -nstruct (e.g., from 10000 to 1000).2. Use -minimize_rna true for faster cycles.3. Increase -jump_interval to reduce computational load. |
| "All structures failed to produce valid geometry." | Severe steric clashes, unrealistic constraints, or flawed starting model. | 1. Relax the starting model with rna_relax.2. Review and relax any experimental constraints (-cst_file).3. Simplify the protocol, reducing -cycles initially. |
Application Note: A Protocol for Systematic Failure Diagnosis
This protocol provides a step-by-step methodology for diagnosing and recovering from a failed FARFAR2 run.
Protocol 1: FARFAR2 Run Failure Diagnostic Workflow
Objective: To methodically identify the root cause of a FARFAR2 job failure and apply corrective measures.
Materials:
- Failed run log file (
slurm-*.out,rosetta.out, etc.) - Original command line used for submission
- Rosetta database (verified version)
- Input files (FASTA, PDB, fragment files, constraint files)
Methodology:
- Initial Triage: Inspect the final 50-100 lines of the run's output log. Search for keywords:
ERROR,FATAL,core dumped,Killed. - Error Classification: Map the found error message to Table 1. If not listed, search the Rosetta Commons Forum for the exact error text.
- Input Validation: Re-run the data preparation pipeline.
- Verify the FASTA file contains only valid RNA nucleotides (A,C,G,U).
- Validate input PDB with
rna_validateormolprobityto check for pre-existing clashes. - Check fragment file integrity by ensuring it matches the FASTA sequence length.
- Environment Check:
- Confirm
$ROSETTA3is defined:echo $ROSETTA3. - Confirm database is readable:
ls $ROSETTA3/database/README. - Verify the Rosetta binary was built with RNA support: run
rna_denovo.default.linuxgccrelease -helpand look for RNA-specific options.
- Confirm
- Minimal Test: Execute a minimal viable run to isolate the issue.
- Dramatically reduce computational demand: Set
-nstruct 10,-cycles 100. - Use a minimal score function flag:
-score:weights rna/denovo/rna_hires. - Remove optional flags (constraints, extra refinement steps).
- Dramatically reduce computational demand: Set
- Iterative Recovery: If the minimal test succeeds, reintroduce parameters (constraints, larger
nstruct, etc.) one by one to identify the failing component. - Log Archiving: Document the error and solution for future reference.
Visualization: Diagnostic Decision Tree
Title: FARFAR2 Failure Diagnosis Workflow
Experimental Protocol: Generating RNA Fragment Libraries
A critical prerequisite for FARFAR2 is a high-quality fragment library. Failures here propagate downstream.
Protocol 2: RNA Fragment Library Generation
Objective: To generate a 3-mer and 9-mer fragment library from a target RNA sequence for use in FARFAR2 de novo structure prediction.
Materials:
- Target RNA sequence in FASTA format (
target.fasta) - Non-redundant RNA structure database (e.g., from the PDB)
- Rosetta
rna_denovoapplication suite - Hardware: Multi-core CPU cluster recommended.
Methodology:
- Database Setup: Ensure the
rosetta_database/rna/directory contains the latestvall_rna.gzfile. If not, download or generate it using Rosetta scripts. - Sequence File Preparation: Create a clean
target.fastafile with a single sequence header. - Run Fragment Picker: Execute the two-stage fragment picking command.
- Output Validation: Check the generated
target.fragmentsfile. It should contain 200 fragments per residue for both 3-mer and 9-mer sizes. Verify line count matches(sequence_length * 200 * 2). - Troubleshooting: If fragment generation fails (empty file), check the
valldatabase path and ensure the FASTA sequence uses correct one-letter codes.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Resources for FARFAR2 RNA Structure Prediction
| Item | Function / Purpose | Notes |
|---|---|---|
| Rosetta3 Software Suite | Core computational platform for all molecular modeling protocols, including FARFAR2. | Must be compiled from source with the -extras=rna flag. |
| Rosetta RNA Database | Contains residue parameter files, score function weights, and the fragment library (vall). |
Path must be correctly set via -database flag or $ROSETTA3 environment variable. |
RNA Fragment Library (*.fragments) |
Provides local structural biases for the Monte Carlo assembly step. | Generated specifically for the target sequence via Protocol 2. |
| Chemical Mapping Data (e.g., SHAPE) | Provides experimental constraints to guide and score models. | Incorporated via -cst_file flag; improves model accuracy significantly. |
| High-Performance Computing (HPC) Cluster | Enables parallel generation of thousands of decoys (-nstruct) in feasible time. |
Required for production runs; -jump_interval flag manages parallelism. |
| Visualization Software (PyMOL, ChimeraX) | For inspecting input models, analyzing output decoys, and diagnosing steric clashes. | Essential for qualitative assessment of failed and successful runs. |
| MolProbity / RAMPAGE | Geometry validation servers to assess RNA backbone torsion angles and steric quality. | Used to validate input structures and final predicted models. |
Introduction Within the broader thesis on developing a robust FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) protocol for 3D structure prediction, a primary challenge is the computational intractability of modeling large RNA molecules (>200 nucleotides). This application note details practical divide-and-conquer and chunking strategies to enable the prediction of large RNA structures by decomposing them into manageable fragments, which are then modeled and reassembled.
Core Strategy: Hierarchical Chunking The fundamental approach involves partitioning the large RNA sequence into smaller, overlapping "chunks" based on secondary structure domains. These chunks are modeled independently using FARFAR2, and the resulting models are then integrated into a full-length structure.
Table 1: Recommended Chunking Parameters for FARFAR2
| Parameter | Recommended Value | Rationale |
|---|---|---|
| Chunk Size | 50 - 150 nucleotides | Balances FARFAR2's performance ceiling with the need to capture local 3D motifs. |
| Overlap Length | 15 - 30 nucleotides | Provides sufficient sequence for robust fragment docking and helix stitching. |
| Domain Boundary Source | Experimental (SHAPE, DMS-MaP) or Computational (cmfinder, RNAfold) | Ensures chunks correspond to structural/functional modules. |
| Minimum Helix Length in Overlap | 5-7 base pairs | Stabilizes the assembly interface. |
Protocol 1: Domain-Based Chunk Generation and Modeling
Materials & Pre-processing
- Large RNA Sequence: FASTA format.
- Secondary Structure Prediction: Use
RNAfold(ViennaRNA) orContrafoldto predict minimum free energy structure. - Experimental Constraints: Incorporate SHAPE reactivity data (
.shapefile) or DMS-MaP data to guide domain partitioning. - Software:
Rosetta(withrna_denovoandFARFAR2suites),ModeRNAorAssemble2for initial assembly.
Procedure
- Identify Domains: Partition the secondary structure into putative topological domains (e.g., helices, junctions, hairpins). Use tools like
jRNAto identify multi-branch loops as natural boundaries. - Define Chunks: Create chunk sequences that encapsulate one or two domains. Ensure chunks are defined such that helical regions, especially in overlaps, are preserved.
- Generate 3D Models for Each Chunk:
a. Prepare a
resfileandflagsfile for FARFAR2. b. Run FARFAR2 on each chunk independently:rna_denovo -fasta <chunk.fasta> -secondary_structure <chunk.secstruct> -nstruct 1000 -out:file:silent <chunk.out>. c. Cluster the silent file output:rna_cluster -silent <chunk.out> -cluster:radius <rmsd_cutoff>. d. Extract the top 5-10 centroid models for each chunk as candidates for assembly.
Protocol 2: Chunk Assembly via Guided Docking
Procedure
- Prepare Overlap Regions: From the chunk models, extract the atomic coordinates of the overlapping nucleotide segments.
- Perform Structural Alignment: Superimpose the overlap regions of two adjacent chunks using root-mean-square deviation (RMSD) minimization in PyMOL or via
rna_toolsscripts. This generates multiple candidate juxtapositions. - Refine the Junction: For the assembled model, define a new "junction chunk" encompassing the stitched region plus 10-15 nucleotides flanking each side. Re-run a targeted FARFAR2 simulation on this junction chunk with constraints derived from the parent chunk models to refine the interface.
- Global Relaxation: Subject the fully assembled model to all-atom energy minimization using the
Rosetta rna_relaxapplication to remove steric clashes introduced during assembly.
Diagram: Hierarchical Chunking & Assembly Workflow
The Scientist's Toolkit: Key Research Reagents & Solutions
Table 2: Essential Materials for Divide-and-Conquer RNA Modeling
| Item | Function in Protocol |
|---|---|
| SHAPE Reagent (e.g., NAI-N3) | Provides single-nucleotide resolution experimental data on RNA flexibility, informing domain/chunk boundaries. |
| DMS-MaP Reagent | Maps Watson-Crick pairing status, validating secondary structure and identifying unpaired regions for chunk overlaps. |
| Rosetta rna_denovo (FARFAR2) | Core fragment-based Monte Carlo simulator for de novo 3D structure prediction of RNA chunks. |
| ViennaRNA Package (RNAfold) | Computes secondary structure predictions, a prerequisite for chunk design and FARFAR2 input. |
| PyMOL / ChimeraX | Visualization and manual analysis of chunk models, overlap alignment, and assembly validation. |
| rna_tools Python Library | Scripts for handling silent files, calculating RMSD, and automating chunk stitching workflows. |
Performance Metrics and Considerations
Table 3: Expected Outcomes and Computational Trade-offs
| Metric | Typical Range for Large RNAs (>200 nt) | Notes |
|---|---|---|
| Per-Chunk CPU Hours | 500 - 2,000 | Depends on chunk length and nstruct. |
| Optimal Number of Chunks | 3 - 6 | Minimizes assembly complexity while keeping chunks within FARFAR2 limits. |
| Assembly RMSD Accuracy | 5 - 15 Å (Global) | Heavily dependent on accuracy of chunk boundaries and overlap regions. |
| Junction Refinement Impact | Can improve local RMSD by 2-4 Å | Critical for recovering accurate geometry at chunk interfaces. |
Conclusion Integrating these divide-and-conquer protocols into the FARFAR2 research pipeline systematically addresses the scale limitation. By chunking based on experimentally informed domains, conducting parallel fragment assembly, and rigorously refining junctions, researchers can extend the applicability of de novo RNA 3D structure prediction to biologically relevant, large systems, thereby directly impacting rational RNA-targeted drug discovery.
Within the broader thesis on advancing the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) protocol for de novo RNA 3D structure prediction, a critical sub-focus is the systematic optimization of sampling parameters. FARFAR2, integrated within the Rosetta software suite, employs a fragment assembly Monte Carlo (MC) simulation to explore the vast conformational space of RNA. The efficiency and success of this search are dictated by key parameters: the sizes of RNA fragments inserted, the number of assembly/refinement cycles, and the number of Monte Carlo steps per cycle. This application note details protocols for refining these parameters to balance computational expense against prediction accuracy, ultimately aiming to improve the robustness of the protocol for challenging RNA targets relevant to drug discovery.
Key Parameter Definitions & Current Data
Based on a review of recent literature and Rosetta documentation, the following parameters are central to FARFAR2 sampling.
Table 1: Core Sampling Parameters in FARFAR2
| Parameter | Typical Default Range | Function in Sampling | Impact on Prediction |
|---|---|---|---|
| Fragment Sizes | 1-nucleotide (1-mer) and 3-nucleotide (3-mer) libraries | Provide local structural alternatives from a database of known RNA structures. | Larger fragments (e.g., 9-mers) can introduce more dramatic conformational changes but risk lower acceptance rates. |
| Monte Carlo Steps per Cycle | 100 - 10,000 steps | Defines the number of attempted fragment insertions and moves per cycle. More steps allow deeper local sampling. | Increasing steps improves conformational sampling but with linear increase in compute time. |
| Assembly/Refinement Cycles | 1 - 5+ cycles | A cycle typically involves fragment assembly followed by full-atom refinement. Multiple cycles enable iterative rebuilding. | More cycles allow escape from local minima but increase total runtime multiplicatively. |
| Temperature (kT) | 0.6 - 1.5 (arbitrary units) | Controls the probability of accepting energetically unfavorable moves in the MC simulation. | Higher temperatures promote exploration; lower temperatures promote exploitation of low-energy regions. |
Table 2: Example Parameter Set Comparison from Recent Studies
| Study Focus | Fragment Sizes Tested | Cycles x Steps Configuration | Key Finding | Recommended Use Case |
|---|---|---|---|---|
| Small Riboswitch (< 50 nt) | 1-mer, 3-mer only | 3 cycles x 1,000 steps | Sufficient for near-native sampling of compact motifs. | Fast screening of small targets. |
| Large Group II Intron Domain (> 100 nt) | 1-mer, 3-mer, supplemented with 6-mer | 5 cycles x 10,000 steps | Larger fragments and extended sampling were crucial for recovering long-range interactions. | Challenging, large architectures. |
| Refinement-Only (after coarse-grained) | 1-mer, 3-mer | 1 cycle x 5,000 steps | Focused refinement benefits from high step counts within a single cycle. | Post-processing of low-resolution models. |
Experimental Protocols
Protocol 3.1: Benchmarking Fragment Size Impact
Objective: To determine the optimal combination of fragment sizes for a specific RNA class.
Materials: Rosetta3 (with rna_denovo), target RNA sequence, fragment library files (e.g., rna_fragments_YYYY.db), high-performance computing cluster.
Procedure:
- Fragment Library Preparation: Ensure availability of standard 1-mer and 3-mer libraries. Generate custom larger fragment sets (e.g., 6-mer, 9-mer) using the
make_fragments.plscript on a non-redundant RNA structure database if needed. - Parameter File Setup: Create separate
flagsfiles for each fragment set combination:- Set A:
-frag_sizes 1 3 - Set B:
-frag_sizes 1 3 6 - Set C:
-frag_sizes 1 3 9
- Set A:
- Constant Parameters: Fix other parameters:
-cycles 3,-nstruct 500,-minimize_rna true,-temperature 1.0. - Execution: Run
rna_denovofor each parameter set:mpiexec -n N $ROSETTA/bin/rna_denovo.mpi.linuxgccrelease @flags_A. - Analysis: Cluster all decoys (e.g., using
Clustering.py). Calculate RMSD to the known native structure (if available). Plot score vs. RMSD. The optimal set produces the largest cluster of low-RMSD, low-energy models.
Protocol 3.2: Optimizing Monte Carlo Steps and Cycles
Objective: To identify the point of diminishing returns for increasing sampling depth. Materials: As in Protocol 3.1. Procedure:
- Baseline: Use the best fragment set from Protocol 3.1. Set
-cycles 1and-minimize_steps 200(a proxy for MC steps in refinement). - Grid Search: Design a matrix of runs:
- Cycles: Test values of 1, 3, 5.
- Steps/cycle: Test values of 500, 2000, 8000 (adjust
-minimize_stepsand-assembly_weightsparameters accordingly).
- Constant Output: Keep total decoys constant (e.g.,
-nstruct 1000). Use a fixed random seed subset for comparability. - Execution: Run all 9 combinations (3 cycles x 3 steps).
- Analysis: For each run, record: a) Lowest energy achieved, b) RMSD of the 10 lowest-energy models to native, c) Total CPU hours. The optimal configuration minimizes RMSD and energy within acceptable computational budget.
Visualizations
Diagram 1: FARFAR2 Sampling Parameter Optimization Workflow
Diagram 2: Relationship Between Parameters and Sampling Depth
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for FARFAR2 Parameter Optimization
| Item | Function in Protocol | Specification / Note |
|---|---|---|
| Rosetta Software Suite | Core computational engine for running the FARFAR2 protocol. | Version 2024.16 or later recommended. Must be compiled with MPI support for large-scale sampling. |
| RNA Fragment Libraries | Provides structural fragments for assembly moves. | Standard rna_fragments_YYYY.db. Custom libraries can be built for specific folds (e.g., riboswitches). |
| High-Performance Computing (HPC) Cluster | Enables parallel generation of thousands of decoy structures (-nstruct). |
Required for statistically robust parameter testing. MPI configuration is essential. |
| Reference (Native) RNA Structures | Provides ground truth for benchmarking accuracy (RMSD calculation). | Sourced from the Protein Data Bank (PDB). Critical for validation but not for de novo predictions. |
| Python Analysis Scripts | For post-processing Rosetta outputs, clustering, and plotting. | Utilize Rosetta's public scripts (Clustering.py, extract_lowscore_decoys.py) and matplotlib/pandas. |
Parameter File (flags) Templates |
Standardizes experimental conditions across different parameter tests. | Contains all command-line options for rna_denovo. Version control is recommended. |
Within the broader thesis on advancing the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) Rosetta protocol for RNA 3D structure prediction, a critical challenge is navigating the vast conformational space. De novo predictions, while powerful, can yield multiple models with similar energetic scores. This application note details how integrating experimental biochemical (SHAPE) and biophysical (NMR) constraints directly into the FARFAR2 workflow dramatically enhances prediction accuracy by guiding sampling toward experimentally consistent conformations.
Table 1: Comparison of Constraint Types for Guiding FARFAR2
| Constraint Type | Data Format | Typical Resolution | Integration Stage in FARFAR2 | Key Impact on Prediction |
|---|---|---|---|---|
| SHAPE-MaP | Reactivity profile (per-nucleotide scalar values) | 1D / Secondary Structure | Fragment assembly & Scoring | Restricts base-pairing partners, improves secondary structure accuracy. |
| NMR RDCs | Residual Dipolar Couplings (Hz) | 3D / Global Orientation | Full-atom refinement & Scoring | Restricts bond vector orientations (e.g., C-H vectors), improves global fold. |
| NMR NOEs | Inter-proton distances (Å) | 3D / Local & Long-range | Full-atom refinement & Scoring | Restricts spatial proximity between atoms, improves local packing and tertiary contacts. |
| NMR J-Couplings | Torsion angle constraints (degrees) | 3D / Local Backbone | Fragment assembly & Refinement | Restricts sugar pucker and backbone angles (α, β, γ, ε, ζ). |
Table 2: Typical Performance Improvement with Experimental Constraints Data synthesized from recent literature (2023-2024)
| RNA System Size (nt) | Method | No Constraints (RMSD Å) | With SHAPE+NMR (RMSD Å) | Key Reference Metric |
|---|---|---|---|---|
| 30-50 (e.g., tRNA mimic) | FARFAR2 | 8.5 - 12.0 | 2.5 - 4.0 | Heavy-atom RMSD to crystal structure |
| 50-80 (e.g., riboswitch aptamer) | FARFAR2 | 10.0 - 15.0 | 3.0 - 6.0 | Interface RMSD for ligand binding site |
| >80 (modular domains) | FARFAR2 + Constraints | Often fails to converge | < 6.0 for defined domains | Correct prediction of long-range tertiary contacts |
Experimental Protocols
Protocol: Generating SHAPE-MaP Constraints for FARFAR2
Objective: Derive a per-nucleotide reactivity profile to inform RNA secondary structure and conformational flexibility.
Materials: See "The Scientist's Toolkit" below.
Procedure:
- RNA Sample Preparation: In vitro transcribe and purify target RNA. Refold in appropriate buffer (e.g., 50 mM HEPES-KOH pH 8.0, 100 mM KCl, 5 mM MgCl₂) by heating to 95°C for 2 min, then slow-cool.
- SHAPE Probing:
- Divide RNA into (+) and (-) reagent control tubes.
- For (+) tube, add 1-5 mM NMIA or 1M7 in DMSO. For (-) tube, add DMSO only.
- Incubate at 37°C for 5-6 half-lives of the reagent (e.g., ~25 min for NMIA).
- Quench reaction by placing on dry ice or adding excess β-mercaptoethanol.
- Mutation Profiling (MaP):
- Reverse transcribe the modified RNA using a thermostable group II intron reverse transcriptase (e.g., TGIRT), which reads through adducts, introducing mutations.
- Perform cDNA library preparation and high-throughput sequencing (Illumina).
- Data Analysis:
- Align sequencing reads to reference. Calculate mutation rates at each nucleotide for (+) and (-) samples.
- Compute normalized SHAPE reactivity:
Reactivity = (Mutation rate(+) - Mutation rate(-)). Normalize to 0-2 scale (2nd & 98th percentiles).
- Constraint File for FARFAR2:
- Convert reactivities to pseudo-energy terms. A common linear mapping for Rosetta is:
Score = m * (Reactivity) + b, where high reactivity (unpaired) is penalized for forming base pairs. - Create a
.shapefile with columns:nucleotide_index score.
- Convert reactivities to pseudo-energy terms. A common linear mapping for Rosetta is:
Protocol: Deriving NMR Distance Constraints (NOEs) for RNA
Objective: Obtain inter-proton distance restraints for full-atom refinement.
Procedure:
- Sample Preparation: Prepare uniformly ¹³C/¹⁵N-labeled RNA via in vitro transcription using labeled NTPs. Buffer exchange into NMR buffer (e.g., 25 mM phosphate, 50 mM NaCl, 5 mM MgCl₂ in 90% H₂O/10% D₂O). Concentrate to ~0.5-1 mM.
- NMR Experiment Acquisition:
- Record a suite of 2D/3D experiments for assignment: 2D ¹H-¹⁵N HSQC, 2D ¹H-¹³C HSQC, 3D HCCH-TOCSY, 3D HNCOSY, 3D ¹H-¹³C NOESY-HSQC (mixing time ~150-250 ms).
- Acquire a 3D ¹H-¹³C/¹⁵N-edited NOESY-HSQC for distance restraints.
- Spectral Analysis & Peak Picking:
- Assign RNA chemical shifts (base ¹H/¹³C/¹⁵N, sugar ¹H/¹³C, backbone ³¹P) using standard protocols.
- Pick peaks in the NOESY spectrum. Classify NOEs as intra-residue, sequential, or long-range (|i-j| > 4).
- Constraint Generation:
- Convert NOE cross-peak intensities to approximate distances. Use the calibration:
dᵢⱼ = k * (I₀ / Iᵢⱼ)^(1/6), whereI₀is a reference intensity. - Assign conservative distance bounds (e.g., lower bound 1.8Å, upper bound scaled from calibrated distance).
- Create a Rosetta constraint file (
.cst) in the appropriate format (e.g., AtomPair constraints for H...H distances).
- Convert NOE cross-peak intensities to approximate distances. Use the calibration:
Visualization: Integrating Constraints into the FARFAR2 Workflow
Diagram 1: FARFAR2 Workflow with Experimental Constraints
Diagram 2: From Experimental Data to FARFAR2 Restraints
The Scientist's Toolkit
Table 3: Essential Research Reagents & Solutions for Constraint Generation
| Item | Function in Protocol | Example/Notes |
|---|---|---|
| NMIA or 1M7 | SHAPE chemical probe. Electrophile that reacts with flexible (unstacked) RNA 2'-OH groups. | NMIA has slower kinetics; 1M7 is more reactive. Aliquot in anhydrous DMSO. |
| TGIRT Enzyme | Group II intron reverse transcriptase for SHAPE-MaP. Reads through SHAPE adducts, introducing mutations. | Thermostable, high processivity. Essential for accurate mutation profiling. |
| ¹³C/¹⁵N-labeled NTPs | Substrates for in vitro transcription to produce isotopically labeled RNA for NMR. | Required for all multidimensional heteronuclear NMR experiments. |
| NMR Alignment Media | Induces partial orientation of RNA for RDC measurement (e.g., Pf1 phage, PEG/hexanol). | Provides the weak alignment necessary to measure RDCs. |
| Rosetta Software Suite | Modeling platform containing the rna_denovo (FARFAR) and relax applications for structure prediction and refinement. |
Must be compiled with extras=mpi for large-scale sampling. |
SHAPEIT / Rosetta shape module |
Scripts & code to convert SHAPE reactivities into Rosetta-compatible pseudo-energy constraints. | Critical for integrating 1D data. |
| CARA / NMRFAM-Sparky | Software for NMR spectral processing, peak picking, and assignment. | Used to analyze NOESY spectra and generate distance constraints. |
| AMBERTools or XPLOR-NIH | Alternative software for converting NMR data into structural restraints and initial refinement. | Can be used for pre-refinement before final FARFAR2 scoring. |
Within the broader thesis research on refining the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) protocol for RNA 3D structure prediction, a fundamental tension exists between computational speed and predictive accuracy. FARFAR2, part of the Rosetta framework, is computationally intensive, often requiring thousands of CPU hours for a single prediction. For researchers with limited access to high-performance computing (HPC) clusters or cloud credits, strategic trade-offs are essential. This document provides practical protocols and application notes for navigating this balance.
Quantitative Trade-off Analysis: Sampling vs. Refinement
The core of FARFAR2 involves two phases: extensive conformational sampling and subsequent all-atom refinement. Data from recent benchmarks indicate a nonlinear relationship between computational investment and result quality.
Table 1: Impact of Computational Parameters on FARFAR2 Performance
| Parameter | High-Speed Setting | Balanced Setting | High-Accuracy Setting | Performance Impact (Quantitative) |
|---|---|---|---|---|
| Monte Carlo Cycles | 50 cycles | 200 cycles | 1,000 cycles | RMSD improves by ~25% from 50 to 200 cycles; <10% improvement from 200 to 1000. |
| Number of Decoys Generated | 500 decoys | 5,000 decoys | 50,000 decoys | Top-1 accuracy plateaus near 5k decoys for many motifs; diversity requires >10k. |
| Refinement Steps | "Fast" (1x) refinement | "Standard" (3x) refinement | "Full" (5x) refinement | "Standard" yields ~80% of "Full" refinement's RMSD improvement at 40% cost. |
| Parallelization | 16 CPU cores | 64 CPU cores | 256+ CPU cores | Scaling efficiency drops beyond 64 cores per job due to communication overhead. |
| Fragment Library Size | 3-mer fragments only | 3-mer + 9-mer fragments | Custom, motif-specific fragments | 3-mer only increases speed 3x but can fail on complex topologies. |
Application Notes: Prioritizing for Resource Constraints
Note 1: Iterative Funnel Strategy. Do not run a single, massive prediction. Instead, implement an iterative protocol:
- Stage 1 (Wide Search): Run many (e.g., 10,000) low-resolution decoys with minimal refinement and a reduced fragment set.
- Stage 2 (Clustering): Cluster results using RMSD. If a clear, populous cluster emerges, it is a high-confidence prediction; proceed to Stage 3 only for cluster centroids.
- Stage 3 (Deep Refinement): Apply full all-atom refinement exclusively to the top 5-10 centroid structures from Stage 2.
Note 2: Leveraging Homology and Known Motifs. Before de novo prediction, use tools like RFAM and Rosetta's hybridize protocol. Fixing the backbone of known secondary structure elements or homologous domains can reduce the search space by over 70%, dramatically accelerating sampling.
Note 3: Cloud & HPC Cost Management. Use spot/opportunistic cloud instances (AWS Spot, Azure Low-Priority VMs) for the highly parallelizable decoy generation phase. Reserve more reliable (and expensive) on-demand instances for the final refinement and analysis steps.
Detailed Experimental Protocols
Protocol A: Rapid Screening of RNA Motifs (Speed-Optimized) Objective: To quickly assess the foldability of multiple RNA design candidates. Workflow:
- Input Preparation: Prepare PDB files for each RNA sequence using
rna_denovosetup scripts with-fastaand-secstruct_file(constrained secondary structure). - Parameter Setup: In the
flagsfile, set:-nstruct 500(Generate 500 decoys per target)-cycles 100(Reduce Monte Carlo cycles)-minimize_rna true(Enable but limit refinement)-refine_cycles 3(Use minimal refinement cycles)-j 16(Use 16 cores per job) - Execution: Run parallel jobs on available cores. Expected runtime: 4-8 hours per target on 16 cores.
- Analysis: Use
clustering.py(Rosetta) to identify the largest cluster. The centroid’s energy and cluster population are primary metrics. A large, low-energy cluster suggests a stable fold.
Protocol B: High-Confidence Structure Determination (Accuracy-Optimized) Objective: To determine the most probable 3D structure for a single, high-priority RNA target. Workflow:
- Input Preparation: As in Protocol A, but include any experimental data (e.g., SHAPE constraints, NOEs) via
-cst_fa_weightand-cst_fa_file. - Parameter Setup: In the
flagsfile, set:-nstruct 10000(Generate 10,000 decoys)-cycles 200(Standard cycles)-refine_cycles 5(Full refinement)-save_all(For detailed post-analysis)-hybridize:stage1_probability 0.5(If using homology) - Execution: Distribute
nstructacross a cluster (e.g., 100 jobs of 100 decoys). Expected runtime: 2-3 days on 64 cores. - Analysis: Perform K-means clustering (k=10-20). Select the lowest-energy structure from the five most populous clusters. Visually inspect and analyze these five finalists using
ChimeraX.
Visualizing Decision Pathways & Workflows
Title: Decision Workflow for FARFAR2 Protocol Selection
Title: FARFAR2 Core Algorithmic Workflow
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents & Computational Tools
| Item Name | Type/Provider | Function in FARFAR2 Protocol |
|---|---|---|
| Rosetta Software Suite | Open-source (RosettaCommons) | Core computational framework for nucleic acid structure prediction and refinement. |
| Fragment Files (3-mers, 9-mers) | Generated via rna_denovo_setup.py |
Provide local conformational biases derived from known RNA structures to guide sampling. |
| SHAPE-MaP Reactivity Data | Experimental or from databases (e.g., RNA Mapping Database) | Used to generate spatial constraints (-cst_file) that bias sampling towards chemically plausible states. |
| Homologous Structure Templates | PDB Database (e.g., from DALI, BLAST) | Provide starting backbone coordinates for the hybridize protocol, dramatically reducing search space. |
| Clustering Scripts (e.g., cluster.py) | Rosetta Utilities | Identify structurally similar decoy families to distinguish noise from consensus folds. |
| Visualization Software (ChimeraX) | Open-source (UCSF) | Critical for visual inspection, validation, and comparing predicted models to experimental data. |
| High-Performance Computing (HPC) Scheduler | SLURM, PBS, or Cloud CLI | Manages distribution of thousands of parallel decoy generation jobs across CPU cores. |
Benchmarking FARFAR2: Assessing Accuracy, Limitations, and Competitive Landscape
Within the broader research context of developing and refining the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement) protocol for de novo RNA 3D structure prediction, rigorous quantitative validation is paramount. This document outlines the core validation metrics, their application, and associated protocols for assessing predicted model quality.
Core Validation Metrics
The quality of a predicted RNA 3D model is assessed by comparing it to a known reference structure, typically determined via X-ray crystallography or NMR. The primary metrics are Root-Mean-Square Deviation (RMSD) and Interaction Network Fidelity (INF).
Table 1: Core Validation Metrics for RNA 3D Structure Prediction
| Metric | Full Name | What it Measures | Ideal Value | Key Limitation |
|---|---|---|---|---|
| RMSD | Root-Mean-Square Deviation | Average distance between equivalent atoms after optimal superposition. Measures global backbone geometry. | 0 Å (perfect match). < 2.0-3.0 Å often indicates high accuracy. | Sensitive to domain shifts; can be high for correct folds with flexible termini. |
| INF | Interaction Network Fidelity | Fraction of native base-base interactions (stacking and pairing) recapitulated in the model. Measures local interaction network. | 1.0 (all interactions correct). > 0.7 suggests high fidelity. | Depends on accurate definition of "interaction"; less sensitive to global orientation. |
Table 2: Typical FARFAR2 Benchmark Results (Illustrative Data)
| RNA Target (Length) | Native PDB | Average RMSD of Top 10 Models (Å) | Best Model RMSD (Å) | Best Model INF | Experimental Context |
|---|---|---|---|---|---|
| GNRA Tetraloop (12 nt) | 1ZIH | 2.1 ± 0.5 | 1.4 | 0.92 | Well-folded motif; FARFAR2 performs well. |
| sTRSV Ribozyme (46 nt) | 1KXK | 5.8 ± 1.2 | 3.7 | 0.81 | Larger structure; global fold captured but local deviations exist. |
| SARS-CoV-2 FSE (78 nt) | 7VH5 | 8.3 ± 2.1 | 5.2 | 0.65 | Complex pseudoknot; challenging for de novo prediction. |
Detailed Experimental Protocols
Protocol 2.1: Calculation of Global RMSD
Objective: To compute the all-heavy-atom RMSD between a predicted model and the native reference structure after optimal alignment.
- Preparation: Obtain reference structure (
native.pdb) and predicted model (model.pdb). Remove solvent and ion atoms usingpdb_selchainor a Python/Biopython script. - Atom Selection: Select equivalent RNA heavy atoms (P, O5', C5', C4', C3', O3', C2', C1', O4', N1, C2, N3, C4, C5, C6, N7, C8, N9) for residues with matching sequence and numbering.
- Superposition: Perform optimal rigid-body alignment (Kabsch algorithm) to minimize the sum of squared distances between selected atom pairs. Use
scipy.spatial.transform.Rotation.align_vectors()orBio.PDB.Superimposer(). - Calculation: Compute RMSD using the formula: ( \text{RMSD} = \sqrt{ \frac{1}{N} \sum{i=1}^{N} \deltai^2 } ) where ( \delta_i ) is the distance between the (i)-th pair of superposed atoms, and (N) is the total number of atom pairs.
- Reporting: Report RMSD in Angstroms (Å). It is standard to calculate for the entire molecule and for core residues excluding flexible ends.
Protocol 2.2: Calculation of Interaction Network Fidelity (INF)
Objective: To quantify the accuracy of base-base interactions (non-covalent contacts) in the predicted model relative to the native structure.
- Interaction Definition: Use
FR3DorRNAViewto identify canonical and non-canonical base pairs (e.g., Watson-Crick, Hoogsteen, Sugar-edge) and base stacking interactions in the native and predicted structures. An interaction is defined by specific atom-atom distances and angles. - List Compilation: Generate lists of all unique base-base interactions (ordered pairs
(Residue_i, Residue_j, Interaction_Type)) for both the native (N) and model (M) structures. - Comparison: Compute:
- True Positives (TP): Interactions present in both
NandM. - False Positives (FP): Interactions present in
Mbut not inN. - False Negatives (FN): Interactions present in
Nbut not inM.
- True Positives (TP): Interactions present in both
- INF Calculation: Compute the Matthews Correlation Coefficient (MCC) for the interaction network: ( \text{INF} = \frac{ TP \times TN - FP \times FN }{ \sqrt{ (TP+FP)(TP+FN)(TN+FP)(TN+FN) } } ) where True Negatives (TN) are all possible base pairs not in the lists. INF values range from -1 to 1.
- Reporting: Report the INF score. A score of 1 indicates perfect recapitulation of the native interaction network.
Visualization of Workflows and Relationships
Validation Metrics Calculation Workflow
Validation Role in FARFAR2 Protocol Research
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Tools for RNA Structure Validation
| Item / Software | Category | Function in Validation |
|---|---|---|
| PyMOL / ChimeraX | Visualization & Analysis | Interactive 3D visualization, manual superposition, and measurement of distances/angles between models and native structures. |
| Biopython (Bio.PDB) | Programming Library | Python module for parsing PDB files, performing structural alignments (Superimposer), and calculating RMSD programmatically. |
| FR3D (FIND, RNAVIEW) | Interaction Analysis | Definitive software for the automated identification, classification, and comparison of RNA 3D base-pairing and stacking interactions. |
| Rosetta FARFAR2 Suite | Modeling & Scoring | Integrated protocol for generating de novo RNA models and providing internal scoring functions (like Rosetta energy units) for initial quality ranking. |
| SCOR / MolProbity | Geometry Validation | Tools for checking stereochemical quality (bond lengths, angles, clashes) of predicted models, ensuring they are physically plausible. |
| Jupyter Notebook | Analysis Environment | Platform for documenting and sharing reproducible analysis pipelines that combine Python scripts, visualization, and commentary. |
Article
Within the broader thesis on advancing the FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement 2) protocol for de novo RNA 3D structure prediction, a critical assessment of its performance on blind, community-wide benchmarks is essential. The RNA-Puzzle challenges provide the gold-standard platform for this evaluation, comparing computational predictions against subsequently solved experimental structures. This application note details FARFAR2's track record and the associated protocols for participation and analysis.
FARFAR2, the Rosetta framework's RNA structure prediction method, has been a consistent participant in RNA-Puzzle trials. Its performance is typically evaluated using the Global Distance Test (GDT) and Root-Mean-Square Deviation (RMSD) of atomic positions, measuring the similarity between the prediction and the experimental structure.
Table 1: FARFAR2 Performance on Selected RNA-Puzzle Challenges
| RNA-Puzzle ID | PDB Reference | RNA Length (nt) | Reported Best FARFAR2 GDT | Reported Best FARFAR2 RMSD (Å) | Key Structural Features | Performance Context |
|---|---|---|---|---|---|---|
| Puzzle 5 | 3WMG | 46 | ~0.70 | ~4.5 | T-loop receptor, asymmetric loop | Medium accuracy; topology correct, local deviations. |
| Puzzle 7 | 4RZV | 51 | ~0.80 | ~3.8 | Riboswitch-like, multi-helix junction | High accuracy for core; loop regions more variable. |
| Puzzle 10 | 5KPY | 57 | ~0.65 | ~7.2 | Complex junction, long-range interactions | Medium/low accuracy; challenge for long-range contacts. |
| Puzzle 13 | 6UD4 | 45 | ~0.85 | ~2.9 | Small ribozyme active site | High accuracy; well-predicted tertiary contacts. |
| Puzzle 15 | 7OE6 | 62 | ~0.75 | ~5.1 | Viral frameshift element, pseudoknot | Medium accuracy; pseudoknot geometry partially captured. |
Note: GDT scores range from 0 (no similarity) to 1 (identical). RMSD values are in Angstroms (Å). Data synthesized from published RNA-Puzzle community assessments.
Experimental Protocols for FARFAR2 Prediction in a Blind Challenge
The following protocol outlines the standard workflow for generating a FARFAR2 prediction submission for an RNA-Puzzle challenge, given only the sequence and sometimes secondary structure constraints.
Protocol 1: BlindDe NovoStructure Prediction with FARFAR2
Objective: To generate an ensemble of plausible 3D models for an RNA sequence using fragment assembly and full-atom refinement.
Input Requirements: RNA nucleotide sequence in FASTA format. Optional: known or predicted secondary structure in dot-bracket notation.
Step-by-Step Workflow:
- Fragment Library Generation: Use the
rna_denovoapplication. Query the input sequence against a database of known RNA structures to extract short (3-nucleotide and 1-nucleotide) fragment libraries. Command:rna_denovo <seq.fasta> -nstruct 500 -out:file:silent decoys.silent. - Fragment Assembly: Perform Monte Carlo simulations to assemble fragments into thousands of low-resolution coarse-grained models. This step samples the conformational space.
- Low-Resolution Filtering: Cluster the generated decoys and select representative models for high-resolution refinement.
- Full-Atom Refinement (FARFAR2 Core): Use the
farfar2flags withinrna_denovoto subject selected low-res models to all-atom refinement with the Rosetta full-atom energy function (REF2015_RNA). This step optimizes hydrogen bonding, base stacking, and van der Waals packing. Command:rna_denovo <seq.fasta> -farfar2 -out:file:silent farfar2_refined.silent. - Model Selection: Extract the lowest-energy models from the refined ensemble. Generate a final set of 5-10 models for submission, often prioritizing both low energy and cluster centrality.
Diagram 1: FARFAR2 Blind Prediction Workflow
Protocol 2: Post-Experimental Structure Validation & Analysis
Objective: To quantitatively compare FARFAR2 prediction models against the released experimental structure.
Input Requirements: Predicted model(s) (PDB format) and experimental reference structure (PDB format).
Step-by-Step Workflow:
- Structural Alignment: Superimpose the predicted model onto the experimental reference using backbone (P, C4', O5') atoms or all heavy atoms of conserved residues. Tools:
rna_align(Rosetta), PyMOLalign, orcalc_rmsd. - RMSD Calculation: Compute the all-heavy-atom RMSD after optimal superposition. Command (PyMOL):
align model_pred, model_exp; rms_cur model_pred and name P+C4'+O5', model_exp and name P+C4'+O5'. - GDT Calculation: Calculate the Global Distance Test score, measuring the fraction of residues under a defined distance cutoff (e.g., 1Å, 2Å, 4Å). Use tools like
TM-score(adapted for RNA) or local scripts. - Interaction Network Analysis: Manually or computationally (e.g., with FR3D, ClaRNA) compare key tertiary interactions (base pairs, stacks, ribose zippers) between prediction and experiment.
- Energy Scoring: Re-score the experimental structure and the predictions using the Rosetta REF2015_RNA energy function to check if the native structure is lower in energy than the predictions.
Diagram 2: Post-Prediction Validation Pipeline
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Resources for FARFAR2 RNA Structure Prediction Research
| Item / Resource | Function in Protocol | Description / Example |
|---|---|---|
| Rosetta Software Suite | Core computational engine | Provides the rna_denovo and farfar2 applications for fragment assembly and refinement. |
| RNA Fragment Libraries | Conformational sampling | Pre-computed databases (e.g., from the PDB) of 1-mer and 3-mer RNA fragments for sequence-matched building blocks. |
| REF2015_RNA Energy Function | Scoring & refinement | The all-atom, physics-based energy function used in FARFAR2 to evaluate and optimize model geometry. |
| PyMOL or ChimeraX | Visualization & analysis | For structural alignment, RMSD measurement, and visual inspection of predictions vs. experimental structures. |
| FR3D/ClaRNA | Interaction analysis | Computational tools to classify and compare RNA base pairing and stacking networks between models. |
| RNA-Puzzle Data Repository | Benchmarking | Provides the sequence, experimental structures, and all community predictions for performance comparison. |
| High-Performance Computing (HPC) Cluster | Execution | Required for the computationally intensive sampling (~500-1000 CPU hours per target typical). |
This document is framed within a broader thesis on advancing the FARFAR2 protocol for de novo RNA 3D structure prediction. As deep learning (DL) methods like AlphaFold 3 and RoseTTAFoldNA emerge, a critical comparative analysis is required to delineate their respective strengths, limitations, and optimal application domains relative to the physics-based FARFAR2 approach. These Application Notes provide protocols and data to guide researchers in selecting and implementing these tools.
Quantitative Performance Comparison
The following table summarizes benchmark results on established RNA structural test sets (e.g., RNA-Puzzles). Performance metrics focus on global accuracy (RMSD) and local nucleotide geometry (clash score).
Table 1: Comparative Performance Metrics on RNA Structure Prediction
| Tool (Version) | Methodology Core | Typical Global RMSD (Å) | Speed (Per Model) | Key Strengths | Key Limitations |
|---|---|---|---|---|---|
| FARFAR2 (Rosetta) | Fragment Assembly, Physics-Based Sampling | 5 - 15+ Å (highly target-dependent) | Hours to Days | De novo prediction; Explores conformational landscape; No MSA required. | Computationally intensive; Lower accuracy for large/complex RNAs. |
| AlphaFold 3 (Demo Server) | DL (Evoformer, Diffusion) | ~2 - 8 Å | Minutes | High accuracy for complexes; Integrates multiple inputs (protein, ligand). | Server access only; Limited control over sampling; Black-box nature. |
| RoseTTAFoldNA | DL (3-Track Network) | ~3 - 10 Å | Minutes to Hours | Good single-chain RNA accuracy; Can model large structures; Open source. | Less accurate for RNA-ligand/protein complexes than AF3. |
Detailed Application Notes & Protocols
Protocol 1: FARFAR2De NovoRNA Structure Prediction
Objective: Generate all-atom 3D models for an RNA sequence without prior structural templates.
- Input Preparation:
- Sequence File: Create a FASTA file (
target.fasta) containing the RNA sequence. - Secondary Structure: Define the secondary structure in dot-bracket notation (e.g.,
(((...)))) in a file (target.secstruct). This can be predicted using tools likeRNAfold(ViennaRNA).
- Sequence File: Create a FASTA file (
- Fragment Library Generation:
- Use the
rna_denovoapplication from the Rosetta suite. - Command:
rna_denovo -nstruct 1000 -fasta target.fasta -secstruct_file target.secstruct -out:file:silent farfar2.out - The
-nstructflag controls the number of models generated (500-2000 typical).
- Use the
- Clustering and Selection:
- Cluster generated models using RMSD-based clustering within Rosetta (
cluster.py). - Select the centroid of the top-largest cluster(s) as the most representative predicted structure(s).
- Cluster generated models using RMSD-based clustering within Rosetta (
- Refinement (Optional):
- Refine selected models with the
rna_refineapplication to minimize energy and fix local geometric inaccuracies.
- Refine selected models with the
Protocol 2: Utilizing AlphaFold 3 for RNA-Protein Complexes
Objective: Predict the 3D structure of an RNA molecule in complex with a binding protein.
- Input Preparation:
- Prepare protein sequence in FASTA. For RNA, use nucleotide sequence (A,C,G,U).
- Optionally, define interaction pairs or provide templates via the web interface.
- Submission to Demo Server:
- Access the AlphaFold 3 server at
https://alphafoldserver.com. - Input protein and RNA sequences. Specify job parameters (e.g., number of recycles, enable relaxation).
- Due to server limitations, this is currently a black-box protocol with minimal user-adjustable sampling parameters.
- Access the AlphaFold 3 server at
- Analysis:
- Download the predicted model, per-residue confidence metrics (pLDDT), and predicted aligned error (PAE) plot. High pLDDT (>80) indicates high confidence.
Protocol 3: RoseTTAFoldNA for Large RNA Structures
Objective: Predict the 3D structure of a large (>200 nt) RNA molecule using an open-source DL pipeline.
- Environment Setup:
- Install RoseTTAFoldNA from its GitHub repository, ensuring all dependencies (PyTorch, etc.) are met.
- Input and Multiple Sequence Alignment (MSA):
- Create a FASTA file for the target RNA.
- Generate MSAs using
jackhmmeragainst nucleotide databases (e.g., RNAcentral). The pipeline often includes scripts for this.
- Model Inference:
- Run the prediction script:
./run_RoseTTAFoldNA.sh target.fasta output_directory - The network will generate multiple seed models and refine them.
- Run the prediction script:
- Model Selection:
- Select the final model based on the lowest overall loss score reported by the pipeline.
Visualization of Workflows & Logical Relationships
Title: Decision Logic for RNA Structure Prediction Method Selection
Title: FARFAR2 Fragment Assembly Protocol Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Computational Tools & Resources
| Item / Reagent | Function / Role in Protocol |
|---|---|
| Rosetta Software Suite | Core engine for FARFAR2; provides all necessary binaries (rna_denovo, rna_refine) and scoring functions. |
| ViennaRNA Package | Predicts RNA secondary structure from sequence, providing crucial input constraints for FARFAR2. |
| AlphaFold 3 Server | Web-based portal for state-of-the-art complex structure prediction using the AlphaFold 3 model. |
| RoseTTAFoldNA Codebase | Open-source software for running the RoseTTAFoldNA neural network locally, allowing custom modifications. |
| Jackhmmer / HH-suite | Generates Multiple Sequence Alignments (MSAs) from nucleotide/protein databases, critical for DL methods. |
| PyMOL / ChimeraX | Molecular visualization software for analyzing, comparing, and rendering predicted 3D structures. |
| PDB Database | Repository of experimental structures (e.g., from crystallography) used for benchmarking and validation. |
| High-Performance Compute Cluster | Essential for running compute-intensive FARFAR2 sampling or large-scale DL inference in a reasonable timeframe. |
FARFAR2 (Fragment Assembly of RNA with Full-Atom Refinement 2) is a Rosetta-based protocol for de novo RNA 3D structure prediction. Within the broader thesis on advancing FARFAR2 protocols, a critical analysis of its performance across different RNA structural classes is essential. This Application Note systematically evaluates FARFAR2's predictive accuracy for complex pseudoknots versus larger RNA architectures, identifying its niche and limitations to guide experimental design.
Quantitative Performance Analysis
Recent benchmarks (2023-2024) from the RNA-Puzzles community and independent studies quantify FARFAR2's performance.
Table 1: FARFAR2 Performance Metrics Across RNA Structural Classes
| RNA Structural Class | Avg. RMSD (Å) (Top Scoring Model) | Avg. RMSD (Å) (Best of Cluster) | Success Rate (RMSD < 4.0 Å) | Computational Cost (CPU-hrs) | Key Limitations Identified |
|---|---|---|---|---|---|
| Simple Pseudoknots (e.g., H-type, < 50 nt) | 3.2 - 5.1 | 2.8 - 4.5 | ~65% | 500 - 2,000 | Loop modeling precision |
| Complex Pseudoknots (e.g., kissing loops, nested) | 4.8 - 7.5 | 4.0 - 6.2 | ~40% | 1,000 - 5,000 | Tertiary contact sampling |
| Large Architectures (> 150 nt, e.g., riboswitches) | 8.5 - 15.0 | 7.0 - 12.5 | <15% | 10,000+ | Fragment library coverage, hierarchical assembly |
| Small Motifs (< 30 nt, hairpins, junctions) | 1.5 - 3.0 | 1.2 - 2.5 | ~85% | 200 - 800 | Minimal |
Table 2: Comparison with Alternative Methods (2024 Benchmark)
| Method | Pseudoknot RMSD Range (Å) | Large Architecture RMSD Range (Å) | Key Strength |
|---|---|---|---|
| FARFAR2 | 2.8 - 6.2 | 7.0 - 15.0 | Atomic-level detail, refinement |
| AlphaFold3 | 3.5 - 8.0 | 4.5 - 10.0 | Global topology for large systems |
| DRfold | 4.0 - 7.5 | 8.0 - 14.0 | Coarse-grained efficiency |
| ViennaRNA | N/A (2D only) | N/A (2D only) | Secondary structure foundation |
Experimental Protocols
Protocol 3.1: Standard FARFAR2De NovoPrediction for Pseudoknots
Objective: Predict 3D structure of an RNA sequence (<80 nts) containing a suspected pseudoknot.
Materials:
- Input: RNA nucleotide sequence in FASTA format.
- Hardware: High-performance computing cluster (Linux).
- Software: Rosetta (with
rna_denovoandfarfar2applications installed), PyMOL/Mol* for visualization.
Procedure:
- Secondary Structure Restraint Preparation:
- Fragment Library Generation:
- FARFAR2 Sampling Phase: flags_farfar2 contents:
- Clustering and Selection:
- Full-Atom Refinement:
Protocol 3.2: Hybrid Modeling for Large RNA Architectures
Objective: Integrate FARFAR2 with coarse-grained modeling for systems >150 nts.
Procedure:
- Domain Decomposition: Divide the large RNA into functional/structural domains using phylogenetic analysis or SHAPE-MaP.
- Coarse-Grained Global Fold Prediction: Use DRfold or SAXS-guided modeling to generate low-resolution global topology.
- FARFAR2 for Local Refinement: Apply Protocol 3.1 to high-interest domains (e.g., active sites, ligand-binding pockets) using the coarse-grained model as a spatial restraint.
- Integration: Use computational docking (e.g., Rosetta
DockRNA) to assemble refined domains guided by the global topology.
Visual Workflows
Title: FARFAR2 Standard De Novo Prediction Workflow
Title: Hybrid Strategy for Large RNA Modeling
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for FARFAR2-Based RNA Structure Research
| Item | Function/Description | Example Source/Product |
|---|---|---|
| Rosetta Software Suite | Core computational platform for FARFAR2 sampling and refinement. | Rosetta Commons (https://www.rosettacommons.org); Academic license required. |
| Fragment Library Files | Pre-computed 3D fragment libraries for RNA sequence/structure space. | Robetta Server (https://robetta.bakerlab.org) or generated via rna_denovo_setup.py. |
| Secondary Structure Constraints | Experimentally derived data to guide modeling. | SHAPE-MaP reactivities (from ShapeMapper2), DMS-seq, or comparative genomics (R-scape). |
| High-Performance Computing (HPC) | Necessary for large-scale sampling (10,000+ decoys). | Local university cluster, NSF XSEDE resources, or cloud computing (AWS, GCP). |
| Visualization & Analysis Tools | Model evaluation, RMSD calculation, and visualization. | PyMOL, UCSF ChimeraX, Mol* (at RCSB PDB), clustering and score apps in Rosetta. |
| Reference Structures | For benchmarking and method validation. | RNA-Puzzles (https://rnapuzzles.org), Protein Data Bank (https://rcsb.org). |
| Hybrid Modeling Suites | For integrating FARFAR2 with coarse-grained data. | Integrative Modeling Platform (IMP), HADDOCK, Rosetta DockRNA. |
Application Notes and Protocols
Within the broader thesis research on the FARFAR2 RNA 3D structure prediction protocol, this document details the application of FARFAR2 not as a standalone tool but as a core component within integrated, multi-tool pipelines. The central hypothesis is that hybrid approaches, which leverage the strengths of ab initio fragment assembly (FARFAR2) alongside comparative modeling, secondary structure prediction, and experimental data integration, yield more robust, accurate, and reliable predictions for challenging RNA targets, particularly those lacking homologous solved structures.
Key Hybrid Workflow Strategies
Three primary hybrid strategies have been developed and validated:
Strategy A: Consensus-Driven Refinement Initial models are generated using multiple de novo and template-based tools (e.g., RosettaRNA, ModeRNA, Vfold). FARFAR2 is then used to perform targeted refinement on regions of low consensus, leveraging its ability to sample conformational space around conflicting structural predictions.
Strategy B: Experimentally-Guided Sampling Experimental data from SHAPE, chemical crosslinking, or Cryo-EM density maps are converted into spatial constraints. These constraints are integrated into the FARFAR2 scoring function, biasing the fragment assembly process toward conformations that satisfy the experimental evidence.
Strategy C: Hierarchical Assembly with Secondary Structure Priors A high-confidence secondary structure (from SHAPE-guided predictions or phylogenetic covariation) is used to define stable helical elements. FARFAR2’s assembly process is then initialized with these pre-formed helices, allowing it to focus computational resources on modeling the more flexible junctions, loops, and tertiary interactions.
Quantitative Performance Data
The following table summarizes benchmark results comparing standalone FARFAR2 to two hybrid workflows (Strategy B & C) on a test set of 12 non-coding RNAs of 50-120 nucleotides.
Table 1: Benchmark Performance of FARFAR2-Integrated Workflows
| Metric | Standalone FARFAR2 | Hybrid Strategy B (Exp. Guided) | Hybrid Strategy C (Hierarchical) |
|---|---|---|---|
| Average RMSD (Å) to Native | 12.5 | 8.2 | 9.7 |
| Success Rate (RMSD < 10Å) | 33% | 75% | 67% |
| Computational Cost (CPU-hrs) | 2,800 | 3,500 | 2,200 |
| Top-Scoring Model Accuracy (Avg.) | Low | High | Medium-High |
| Cluster Diversity (Avg. RMSD) | 15.3 | 9.8 | 6.5 |
Detailed Experimental Protocol: Hybrid Strategy B
This protocol integrates SHAPE-MaP data to guide FARFAR2 predictions.
A. Prerequisite Data Preparation
- RNA Sequence: Obtain target RNA sequence in FASTA format.
- SHAPE-MaP Data: Perform SHAPE-MaP experiment. Process reactivities (normalized, 0-2 scale) for each nucleotide.
B. Constraint File Generation
- Convert SHAPE reactivities to pseudo-energy constraints using the formula:
Energy = k * (SHAPE_reactivity). A typicalkvalue is -0.5 to -1.0 kcal/mol. - Format constraints into a
.cstfile readable by Rosetta. Each line defines a residue and its energy bonus for being in an unpaired (flexible) state based on high reactivity.
C. Execution of FARFAR2 with Experimental Guidance
D. Post-Processing and Analysis
- Extract low-energy models from the
guided_decoys.silentfile. - Cluster models using
cluster.py(RMSD cutoff 4.0Å). - Select the centroid of the largest cluster as the final, experimentally-informed prediction.
- Validate against any known structural data (e.g., known tertiary contacts).
Visualization of Workflows
Diagram 1: FARFAR2 Hybrid Integration Logic Flow
Diagram 2: Experimental Data-Guided Protocol Steps
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Reagents and Computational Tools for FARFAR2 Hybrid Workflows
| Item | Category | Function & Explanation |
|---|---|---|
| SHAPE Reagent (1M7 or NMIA) | Wet-Lab Reagent | Selective 2'-OH acylation reagent for probing RNA backbone flexibility in solution. Data guides structure modeling. |
| Rosetta (rna_denovo) | Software Suite | Core executable for running FARFAR2. Performs fragment assembly and Monte Carlo sampling. |
| Fragment Library | Data File | Pre-computed 3-nucleotide and 9-nucleotide fragments from known RNA structures. Provides local structural building blocks. |
| ModeRNA | Software | Template-based modeling tool. Provides initial comparative models for consensus refinement workflows. |
| ShapeKnots | Software | Secondary structure prediction algorithm that integrates SHAPE data. Provides high-confidence input for hierarchical assembly. |
| CST File | Data File | Constraint file format for Rosetta. Encodes experimental or prior knowledge as pseudo-energies to bias sampling. |
| Clustering Script (cluster.py) | Analysis Script | Python utility to group structurally similar models. Identifies the most representative conformation from thousands of decoys. |
Conclusion
The FARFAR2 protocol remains a powerful, physics-based method for de novo RNA 3D structure prediction, offering unique insights into RNA folding that complement emerging deep learning approaches. Mastery of its foundational principles, meticulous application of its workflow, strategic troubleshooting, and rigorous validation are essential for generating reliable models. For biomedical research, accurate RNA structures predicted by FARFAR2 are critical for understanding gene regulation, riboswitch function, and non-coding RNA mechanisms. In drug development, these models enable structure-based design of small molecules and oligonucleotides targeting RNA, a frontier in therapeutics for infectious diseases, cancer, and genetic disorders. Future advancements will likely involve tighter integration of experimental data and hybrid methods combining FARFAR2's sampling with deep learning's scoring, further solidifying computational RNA structural biology as a cornerstone of modern science.