Northern Blotting for Target Gene Expression: A Comprehensive Guide from Principles to Advanced Applications
This article provides a comprehensive guide to Northern blotting for monitoring target gene expression, tailored for researchers, scientists, and drug development professionals.
Northern Blotting for Target Gene Expression: A Comprehensive Guide from Principles to Advanced Applications
Abstract
This article provides a comprehensive guide to Northern blotting for monitoring target gene expression, tailored for researchers, scientists, and drug development professionals. It covers foundational principles, from RNA separation and probe hybridization to the technique's unique advantage of providing transcript size and integrity information. The content details optimized methodological protocols, including recent modifications for enhanced sensitivity and specific applications in biomedical research. It offers practical troubleshooting and optimization strategies for common challenges like RNA degradation and background noise. Finally, the article presents a critical comparative analysis, positioning Northern blotting against modern techniques like qPCR and RNA-Seq, and validating its enduring role as a gold standard in gene expression validation for pharmaceutical quality control and basic research.
The Foundational Principles of Northern Blotting: Understanding RNA Analysis
Northern blotting remains a cornerstone technique in molecular biology for the direct detection and quantification of specific RNA molecules within a complex mixture. First developed in 1977, this method allows researchers to study gene expression by providing information about both the abundance and size of RNA transcripts, enabling the identification of alternatively spliced variants and the monitoring of transcript turnover [1]. Despite the emergence of powerful techniques like RT-PCR and RNA sequencing, Northern analysis retains its relevance due to its direct visual confirmation of RNA integrity and specificity in detecting target sequences [2]. In target gene expression monitoring for drug development and basic research, Northern blotting serves as a robust validation tool that offers a direct relative comparison of message abundance between samples on a single membrane, making it indispensable for confirming transcript identity and integrity [2] [3].
The fundamental principle of Northern blotting involves the separation of RNA molecules by size through denaturing gel electrophoresis, followed by transfer to a solid membrane support and subsequent hybridization with labeled sequence-specific probes. This multi-step process preserves the spatial separation achieved during electrophoresis, allowing for accurate size determination of detected transcripts while providing semi-quantitative data on expression levels [4]. For researchers investigating gene expression patterns in different tissues, developmental stages, or experimental conditions, Northern blotting provides a reliable method to validate findings from high-throughput screenings and is particularly valuable for studying the biogenesis of different RNA forms, including primary transcripts, precursors, and mature RNAs [3].
Core Principles and Methodological Framework
Theoretical Foundation of Northern Analysis
The theoretical foundation of Northern blotting hinges on two key molecular principles: size-based separation of nucleic acids and specific hybridization between complementary sequences. The process begins with the denaturation of RNA secondary structures to ensure linear molecules that migrate through gels according to their molecular weight rather than structural conformation [4]. This is typically achieved using denaturing agents such as formaldehyde or glyoxal in the gel system, which prevent RNA folding by disrupting hydrogen bonds [2]. Following electrophoresis, the separated RNA fragments are transferred and immobilized onto a solid membrane, preserving the distribution pattern established during separation.
Hybridization, the core detection principle, relies on the precise base-pairing rules of nucleic acids. When a labeled probe with sequence complementarity to the target RNA is applied under appropriate conditions, it forms stable duplexes specifically with its target sequence. The stringency of hybridization and subsequent washing steps determines the specificity of detection, with higher stringency conditions permitting only perfectly matched or highly similar sequences to remain hybridized [1]. This specificity enables researchers to distinguish between closely related transcripts and detect specific splice variants with high confidence, a particular advantage over methods that may amplify non-specific products.
Electrophoretic Separation of RNA
The initial critical step in Northern blotting involves the separation of RNA samples according to size using denaturing gel electrophoresis. Denaturing conditions are essential to eliminate RNA secondary structures that would otherwise affect migration through the gel [4]. Two primary denaturing systems are commonly employed: formaldehyde-containing agarose gels and glyoxal/DMSO systems. Formaldehyde gels are widely used and provide reliable denaturation, though they require the use of a fume hood due to safety concerns. The NorthernMax-Gly system, utilizing glyoxal/DMSO denaturation, offers an alternative that eliminates formaldehyde handling while potentially providing sharper bands [2].
The electrophoresis process typically uses agarose gels for most mRNA analyses, while polyacrylamide gels are preferred for smaller RNA species (<200 nucleotides) such as microRNAs due to their superior resolution in lower size ranges [5] [3]. The gel concentration can be adjusted based on the expected size of the target RNA, with lower percentage gels (1-1.2%) providing better separation for larger transcripts and higher percentages offering improved resolution for smaller fragments. Before sample loading, RNA is mixed with denaturing loading buffer and typically heated to 65°C for 10-15 minutes to ensure complete denaturation [6] [4].
Table 1: Electrophoresis Conditions for Northern Blotting
| Parameter | Standard Conditions | Alternative Conditions | Small RNA Analysis |
|---|---|---|---|
| Gel Type | 1.2% Agarose-formaldehyde | Glyoxal/DMSO agarose gel | 8-15% Polyacrylamide-urea |
| Denaturant | 2.2 M Formaldehyde | 1% Glyoxal | 7 M Urea |
| Running Buffer | 1× MOPS | 1× MOPS or TBE | 0.5-1× TBE |
| Voltage/Time | 125V for 3 hours | 90V for 2-3 hours | 200-300V for 2-3 hours |
| RNA Load | 5-30 μg total RNA | 5-30 μg total RNA | 10-50 μg total RNA |
| Visualization | Ethidium bromide pre-stain | SYBR Green post-stain | Ethidium bromide post-stain |
Membrane Transfer and Immobilization
Following electrophoretic separation, the RNA must be transferred from the gel to a solid support membrane while maintaining the spatial distribution achieved during separation. The transfer process can be accomplished through several methods, each with distinct advantages. Capillary transfer represents the most traditional approach, where buffer moves upward through the gel by capillary action, carrying RNA to the membrane placed on top [4]. This passive method requires minimal equipment but typically takes several hours to overnight for completion.
More efficient transfer methods include vacuum blotting and electroblotting, which actively drive the RNA from the gel to the membrane. Vacuum blotting systems can complete transfers in 60-90 minutes with improved efficiency, especially for larger RNA fragments [6]. Electroblotting is particularly preferred for polyacrylamide gels used in small RNA analysis, where electrical current facilitates the transfer [3]. The choice of membrane is critical, with positively charged nylon membranes being preferred over nitrocellulose due to their superior nucleic acid binding capacity and mechanical robustness [2] [5].
After transfer, RNA must be immobilized on the membrane to prevent washing away during subsequent hybridization and washing steps. This is typically achieved through UV crosslinking (120-150 mJ/cm²) or baking at 80°C for 1-2 hours [7] [4]. These treatments create covalent linkages between the RNA and membrane matrix, ensuring permanent fixation of the nucleic acid pattern. For small RNAs, specialized crosslinking methods using EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide) have been developed to improve retention efficiency [3].
Probe Design and Hybridization
The detection of specific RNA species depends on hybridization with complementary labeled probes, with careful probe design being crucial for assay success. Several probe types can be employed, including DNA probes (random-primed or PCR-generated), RNA probes (prepared by in vitro transcription), and synthetic oligonucleotides [2]. RNA probes (riboprobes) generally offer highest sensitivity due to their higher thermal stability and the efficiency of in vitro transcription labeling systems [2].
Probes can be labeled using radioactive isotopes (³²P, ³³P) or non-radioactive systems (digoxigenin, biotin, fluorescein). While radioactive labeling provides high sensitivity and broad dynamic range, non-radioactive alternatives have improved significantly and offer greater safety and stability [8] [3]. The hybridization process involves incubating the membrane with the labeled probe in a specialized buffer containing components to minimize non-specific binding, typically for several hours to overnight at temperatures optimized for the specific probe-target combination.
Table 2: Probe Labeling and Hybridization Options
| Probe Type | Labeling Method | Sensitivity | Advantages | Limitations |
|---|---|---|---|---|
| Random-primed DNA | Random hexamer labeling with Klenow fragment | Moderate (3-5x less than RNA) | Easy protocol, stable probes | Lower sensitivity |
| In vitro transcribed RNA | RNA polymerase incorporation of labeled NTPs | High (gold standard) | Highest sensitivity, stringent washes | RNA probe stability |
| Oligonucleotide | End-labeling with T4 polynucleotide kinase | Variable | Target-specific, no cloning needed | Lower signal for small probes |
| Radioactive (³²P) | Isotope incorporation | Very high (detection of rare transcripts) | High sensitivity, quantitative | Safety concerns, short half-life |
| Non-radioactive (DIG/Biotin) | Antibody-based detection | High (approaching radioactivity) | Safe, stable, long shelf life | Potential for higher background |
Advanced Applications and Protocol Implementation
Specialized Applications in Gene Expression Research
Northern blotting has evolved to address specialized research needs, particularly in the study of non-coding RNAs and transcript processing. For microRNA (miRNA) analysis, Northern blotting remains particularly valuable as it can distinguish between the premature primary transcripts (pri-miRNAs), precursor forms (pre-miRNAs), and mature miRNAs, providing insights into miRNA biogenesis and processing [3]. This capability is crucial for investigating dysregulated miRNA expression in diseases such as cancer, where altered miRNA levels can serve as diagnostic markers or therapeutic targets.
In drug development research, Northern blotting provides critical validation of gene expression changes observed in response to compound treatment. The technique's ability to detect splice variants makes it invaluable for characterizing how pharmaceutical interventions might affect alternative splicing patterns, which could underlie therapeutic mechanisms or side effects [4] [1]. Furthermore, Northern analysis can monitor transcript stability through time-course experiments after transcriptional inhibition, offering insights into post-transcriptional regulatory mechanisms that might be targeted by drugs.
The development of liquid hybridization assays represents a significant advancement, where hybridization occurs in solution before electrophoresis, dramatically reducing processing time while increasing sensitivity, especially for small RNAs [3]. This approach combines the specificity of solution hybridization with the size resolution of gel electrophoresis, making it particularly suitable for high-sensitivity detection of low-abundance transcripts in precious samples.
Detailed Experimental Protocol
RNA Extraction and Quality Assessment
Begin with high-quality RNA extraction using TRIzol or column-based methods, maintaining strict RNase-free conditions throughout [7]. Treat samples with DNase I to eliminate genomic DNA contamination. Assess RNA quality by spectrophotometry (A260/A280 ratio ≥1.8) and by running a small aliquot on a denaturing gel to verify integrity through sharp ribosomal RNA bands. For mRNA analysis, poly(A)+ selection can be performed using oligo(dT) cellulose or magnetic beads to enrich for messenger RNA, increasing detection sensitivity for low-abundance transcripts [5].
Denaturing Gel Electrophoresis
Prepare a 1.2% agarose gel by dissolving agarose in 1× MOPS buffer, then cooling to 60°C before adding formaldehyde to a final concentration of 2.2 M (working in a fume hood) [6] [4]. Assemble the gel cast with comb and allow to solidify. For the RNA samples, mix 15-20 μg of total RNA (or 2-5 μg of poly(A)+ RNA) with 2× RNA loading buffer (containing formamide and ethidium bromide). Denature samples at 65°C for 10-15 minutes, then place on ice. Load samples alongside an appropriate RNA ladder for size determination. Run the gel in 1× MOPS running buffer at 100-125V for approximately 3 hours, monitoring dye front migration.
Capillary Transfer to Membrane
After electrophoresis, rinse the gel briefly in RNase-free water to remove excess formaldehyde. Set up a capillary transfer system using a glass dish filled with 20× SSC transfer buffer [4]. Place a platform on the dish and cover with a wick made of filter paper saturated with transfer buffer. Place the gel on the wick, removing all air bubbles. Pre-wet a nylon membrane in RNase-free water, then in 20× SSC, and place carefully on the gel. Complete the stack with pre-wetted filter papers, paper towels, and a weight, then allow transfer to proceed for 12-16 hours.
RNA Immobilization and Crosslinking
Following transfer, disassemble the stack and mark the membrane orientation. Rinse the membrane briefly in 2× SSC to remove residual gel particles, then air-dry. Immobilize the RNA using UV crosslinking at 120-150 mJ/cm² (optimal energy should be determined empirically for specific membranes) [7]. Alternatively, bake the membrane at 80°C for 1-2 hours between filter papers. Crosslinked membranes can be stored desiccated at room temperature for several months before hybridization.
Probe Preparation and Hybridization
For a 300-500 bp DNA probe, label using random primed labeling with [α-³²P]dCTP or with digoxigenin-11-dUTP following manufacturer protocols [7]. Purify labeled probes using spin columns to remove unincorporated nucleotides. Prehybridize the membrane for 1-2 hours at 42°C (for DNA probes) or 68°C (for RNA probes) in appropriate hybridization buffer (e.g., ULTRAhyb or DIG Easy Hyb) [2]. Denature double-stranded DNA probes by boiling for 10 minutes, then add to fresh hybridization buffer and incubate membrane with probe solution for 14-16 hours at the appropriate temperature with gentle agitation.
Post-Hybridization Washes and Detection
After hybridization, perform stringency washes to remove non-specifically bound probe [6]. For DNA probes, start with 2× SSC/0.1% SDS at room temperature for 5-10 minutes, followed by 0.1-1× SSC/0.1% SDS at 42-65°C (depending on desired stringency) for 15-30 minutes each. For radioactive probes, expose the washed membrane to a phosphorimager screen or X-ray film at -80°C for several hours to days. For non-radioactive detection, proceed with antibody conjugation and chemiluminescent substrate incubation according to manufacturer instructions, then expose to X-ray film or capture with a digital imaging system.
Research Reagent Solutions and Technical Considerations
Essential Research Reagents and Materials
Successful Northern blotting requires careful selection of reagents and materials optimized for RNA work. The following table details key solutions and their specific functions in the experimental workflow:
Table 3: Essential Research Reagents for Northern Blotting
| Reagent/Material | Function/Purpose | Examples/Alternatives |
|---|---|---|
| TRIzol/ Guanidinium Thiocyanate | RNA isolation by denaturing proteins and inhibiting RNases | TRIzol, Qiazol, RNA STAT-60 |
| DNase I (RNase-free) | Removal of genomic DNA contamination | Turbo DNase, RNase-Free DNase Set |
| Formaldehyde/Glyoxal | Denaturing agent preventing RNA secondary structure | Glyoxal (NorthernMax-Gly system) |
| MOPS Buffer | Electrophoresis buffer maintaining appropriate pH | SOPBS buffer as alternative |
| Positively Charged Nylon Membrane | Nucleic acid binding support for transfer | Hybond-N+, BrightStar-Plus, Zeta-Probe |
| Formamide | Hybridization buffer component lowering melting temperature | Deionized, molecular biology grade |
| Salmon Sperm DNA | Blocking agent reducing non-specific probe binding | Cot-1 DNA for repetitive sequences |
| SSC Buffer | Salt buffer for transfers and washes controlling stringency | SSPE as alternative buffer |
| Digoxigenin-11-dUTP | Non-radioactive label for probe detection | Biotin-16-dUTP, Fluorescein-12-dUTP |
| ULTRAhyb Buffer | Commercial hybridization solution enhancing sensitivity | DIG Easy Hyb, PerfectHyb Plus |
Troubleshooting and Optimization Strategies
Several technical challenges commonly arise in Northern blotting that require systematic troubleshooting. Poor signal intensity may result from inefficient transfer, probe degradation, or insufficient RNA loading. Verify transfer efficiency by staining the gel post-transfer with ethidium bromide; significant residual RNA indicates incomplete transfer. Check probe specific activity and ensure appropriate exposure times. High background often stems from inadequate blocking, insufficient washing stringency, or membrane contamination. Increase blocking agent concentration, perform more stringent washes at higher temperatures, and ensure proper membrane handling.
RNA degradation manifests as smearing on the gel and membrane, particularly in the lower molecular weight region. Maintain strict RNase-free conditions throughout the procedure, using dedicated RNase-free reagents and equipment. Uneven blotching on the membrane typically indicates incomplete transfer with air bubbles or uneven contact during capillary transfer. Ensure careful assembly of the transfer stack with particular attention to removing all air bubbles between gel and membrane.
For optimal results, perform pilot experiments to determine the ideal RNA load for your target abundance, probe concentration, and washing stringency. Keep detailed records of all parameters, including batch numbers of key reagents, as subtle lot-to-lot variations can affect results. When moving between different RNA targets or sample types, be prepared to re-optimize conditions, particularly hybridization and washing temperatures.
Northern blotting maintains its position as a fundamental technique in gene expression analysis, providing unambiguous data on RNA size and integrity that complements newer technologies. The core principles of electrophoresis, transfer, and hybridization form a robust framework that continues to evolve with improvements in sensitivity, safety, and specificity. For researchers monitoring target gene expression in basic research and drug development, Northern analysis offers a reliable, quantitative approach with well-established protocols and interpretable results. While requiring careful attention to technical details and RNA quality, its direct visualization of specific transcripts provides validation confidence that remains invaluable in molecular biology research.
Despite the development of newer molecular techniques, Northern blotting maintains a vital role in gene expression analysis for monitoring target genes. This method provides a unique combination of advantages, including direct information on transcript size and the ability to detect alternatively spliced isoforms and partially homologous sequences, making it an indispensable validation tool in modern research and drug development. This application note details the protocol, key advantages, and market context that secure Northern blotting's continued relevance in the molecular scientist's toolkit.
Northern blotting, developed in 1977, is a cornerstone molecular biology technique used for the detection and analysis of specific RNA sequences within a complex sample [9] [10]. Its fundamental principle involves the separation of RNA molecules by size using denaturing gel electrophoresis, followed by transfer to a solid membrane and subsequent hybridization with a labeled sequence-specific probe [9] [4]. While often considered a "classical" method, it remains a robust and highly informative procedure for monitoring the transcription and abundance of target genes, providing data that is both qualitative and quantitative [9] [2].
The global market for Northern blotting products, valued at an estimated USD 170.4 million in 2025 and projected to reach USD 252.3 million by 2035, is a testament to its sustained utility in life science research [11]. This growth, registering a compound annual growth rate (CAGR) of 4.0%, is driven by the technique's vital role in RNA profiling, understanding disease mechanisms, and drug response pathways [11]. Its application is widespread, with the pharmaceutical and biotechnology industries constituting the largest end-user segment (50.6% revenue share in 2025), underscoring its importance in therapeutic development [11].
Key Advantages in Modern Research
Northern blotting offers a set of unique benefits that make it particularly valuable for researchers and drug development professionals focused on gene expression.
Direct Sizing of Transcripts: Unlike RT-qPCR or RNA-Seq, Northern blotting directly reveals the molecular size of RNA transcripts [2]. This capability is crucial for identifying alternatively spliced variants and RNA processing intermediates, providing insights into the functional diversity of gene products that sequence abundance alone cannot offer [10] [2].
Detection of RNA Isoforms and Processing Intermediates: The technique can distinguish between different isoforms of an RNA molecule, such as pre-miRNA and mature miRNA, or various pre-rRNA intermediates [10] [12]. This is essential for studying RNA maturation and turnover, as well as for characterizing aberrant processing in disease states.
High Specificity and Validation Power: Northern hybridization is exceptionally versatile, allowing the use of DNA, RNA, or oligonucleotide probes, and can tolerate sequences with only partial homology (e.g., cDNA from a different species) [2]. Its high specificity makes it a gold-standard method for validating results obtained from high-throughput techniques like microarrays or RNA sequencing, ensuring that observed expression changes are genuine [13] [2].
Quantitative Capability: When combined with appropriate controls and detection methods (e.g., phosphor imaging), Northern blotting provides reliable quantitative data for comparing mRNA abundance between samples on a single membrane [13] [2]. This allows for direct relative comparison of message abundance across different treatments, tissues, or developmental stages.
Table 1: Key Advantages and Research Applications of Northern Blotting
| Advantage | Technical Basis | Research/Drug Development Application |
|---|---|---|
| Direct Transcript Sizing | Electrophoretic separation by molecular weight prior to detection. | Identification of alternative splicing patterns, validation of transcript integrity, and detection of abnormal RNA species [2] [4]. |
| Isoform Detection | Visualizes multiple hybridizing bands on a membrane. | Studying RNA processing (e.g., pre-rRNA maturation, pre-miRNA to miRNA processing) and characterizing isoform-specific expression in different tissues or disease states [10] [12]. |
| High Specificity | Stringent hybridization and washing conditions with sequence-specific probes. | Confirmation of gene expression data from RNA-Seq or microarrays; critical for validating targets in preclinical drug development [13] [2]. |
| Quantitative Analysis | Signal intensity is proportional to the abundance of the target RNA. | Monitoring changes in gene expression levels in response to drug treatments, pathogens, or during cellular differentiation [9] [10]. |
Northern Blotting Market Context
The steady growth of the Northern blotting market reflects its embedded role in the molecular biology workflow. The reagents segment alone is projected to hold a 28.9% market revenue share in 2025, indicating strong, recurring demand for consumables that support this technique [11]. Geographically, emerging research hubs are showing particularly strong growth, with China and India leading with CAGRs of 5.4% and 5.0%, respectively [11].
Table 2: Global Northern Blotting Market Outlook (2025-2035)
| Metric | Value |
|---|---|
| Market Value (2025) | USD 170.4 million [11] |
| Projected Value (2035) | USD 252.3 million [11] |
| Forecast CAGR (2025-2035) | 4.0% [11] |
| Leading Product Segment (2025) | Reagents (28.9% share) [11] |
| Leading Application Segment (2025) | Academic Research (41.2% share) [11] |
| Leading End-User (2025) | Pharmaceutical & Biotechnology Industries (50.6% share) [11] |
Detailed Protocol for Northern Analysis
The following protocol, adapted from standard procedures and commercial kits, provides a reliable workflow for Northern blot analysis of a target gene [9] [2] [4].
RNA Extraction and Integrity Assessment
The initial and most critical step is obtaining high-quality, intact RNA.
- Procedure: Extract total RNA from cells or tissues using a validated method, such as organic solvent (phenol-chloroform) extraction or silica-based column purification [9]. Treat the extracted RNA with DNase to eliminate genomic DNA contamination.
- Quality Control: Assess RNA concentration and purity using a spectrophotometer (A260/A280 ratio ~1.9-2.1). Verify integrity by running 1-2 µg of RNA on a denaturing agarose gel. Intact total RNA should display sharp 28S and 18S ribosomal RNA bands, with the 28S band approximately twice the intensity of the 18S band [10] [2].
- mRNA Enrichment (Optional): For low-abundance transcripts, isolate poly(A)+ mRNA from total RNA using an oligo(dT)-cellulose column or magnetic beads [9]. This enrichment increases the assay's sensitivity by removing most non-coding RNAs [9].
Denaturing Gel Electrophoresis
RNA samples are separated by size under denaturing conditions to prevent secondary structures from affecting migration.
- Gel Preparation: Prepare a 1.0-1.5% agarose gel containing 2.2 M formaldehyde as a denaturant [10] [4]. Formaldehyde masks the secondary structure of RNA, ensuring separation is based primarily on molecular weight.
- Sample Preparation: Mix 5-30 µg of total RNA (or a proportional amount of mRNA) with a denaturing loading buffer containing formaldehyde and formamide (or a commercial glyoxal/DMSO solution) [2]. Denature the samples at 65°C for 10-15 minutes before loading.
- Electrophoresis: Run the gel in a formaldehyde-containing running buffer at 3-5 V/cm. Include an RNA ladder in a separate lane for accurate size determination of the target transcript [2] [4].
Blotting and Immobilization
The separated RNA is transferred from the gel to a solid support for hybridization.
- Transfer: Soak the gel in transfer buffer (typically 20x SSC) to remove formaldehyde. Set up a capillary transfer system, using a high-salt buffer (e.g., 20x SSC) to passively elute the RNA from the gel and onto a positively charged nylon membrane over 6-18 hours [9] [4]. For a faster transfer (1-2 hours), active methods like vacuum or electroblotting can be used [9].
- Immobilization: After transfer, immobilize the RNA on the membrane by crosslinking using UV light (1200 J/m²) or by baking at 80°C under vacuum for 1-2 hours [9] [2]. This covalent attachment prevents the RNA from washing off during subsequent steps.
Probe Preparation and Hybridization
A labeled probe complementary to the target RNA is used for specific detection.
- Probe Labeling: Generate a high-specific-activity probe. Radiolabeled (³²P) probes, synthesized by random-priming of DNA or in vitro transcription of RNA, offer high sensitivity [2] [14]. For non-radioactive detection, label probes with digoxigenin (DIG) or biotin using enzymatic reactions [13] [2]. Locked Nucleic Acid (LNA)-modified oligonucleotide probes provide a significant increase in sensitivity and specificity, especially for short RNAs like miRNAs [14].
- Pre-hybridization and Hybridization: Incubate the membrane in a pre-hybridization buffer containing blocking agents (e.g., Denhardt's solution, salmon sperm DNA) to minimize non-specific probe binding [9] [4]. Replace the buffer with a fresh hybridization solution containing the denatured, labeled probe. Hybridize at a temperature optimized for the probe type and stringency (typically 42-65°C for several hours to overnight) [2] [4].
Washing and Detection
Remove non-specifically bound probe and detect the signal from the target RNA.
- Washing: Perform a series of washes with SSC/SDS buffers, gradually increasing stringency (e.g., from 2x SSC to 0.1x SSC) to reduce background while retaining specific signal [2] [4].
- Detection: For radiolabeled probes, expose the membrane to a phosphor storage screen and image using a phosphor imager (preferred for quantitation and dynamic range) or to X-ray film [13] [2]. For non-radioactive probes, use chemiluminescent or colorimetric substrates for detection. Fluorescently labeled probes can be detected directly on compatible imaging systems [13].
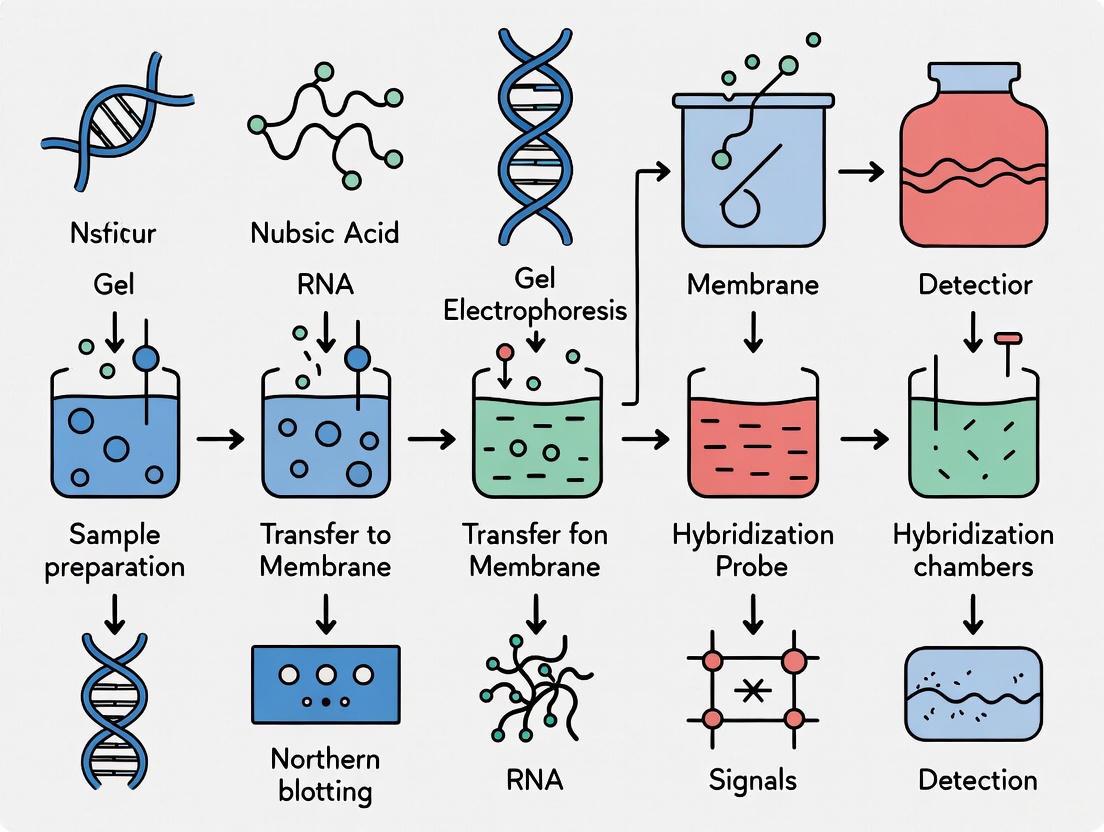
Diagram 1: Northern blotting workflow.
The Scientist's Toolkit: Essential Reagents and Equipment
Successful Northern blotting relies on a set of core reagents and instruments. Key players in the market providing these solutions include ThermoFisher Scientific, Sigma Aldrich Co, Qiagen Inc, Perkin Elmer Inc., and Pall Corporation [11].
Table 3: Key Research Reagent Solutions for Northern Blotting
| Item | Function/Description | Examples/Notes |
|---|---|---|
| RNA Isolation Kits | Obtain high-quality, DNase-free total or poly(A)+ RNA from various sample types. | Guanidium isothiocyanate-phenol based (TRIzol) or silica-membrane column methods [9] [2]. |
| Denaturing Gels | Separate RNA by size while inhibiting secondary structure formation. | Formaldehyde-agarose gels (for mRNAs) or Urea-polyacrylamide gels (for small RNAs like miRNAs) [10] [4] [12]. |
| Blotting Membranes | Solid support for immobilizing RNA after transfer. | Positively charged nylon membranes are preferred due to high nucleic acid binding affinity and robustness [9] [2]. |
| Labeled Probes | Sequence-specific detection of target RNA. | Radiolabeled (³²P), or non-radioactive (DIG, Biotin). LNA-modified probes offer enhanced sensitivity [13] [2] [14]. |
| Hybridization Buffers | Optimized solution for probe hybridization. | Commercial ultrasensitive hybridization buffers can significantly increase signal-to-noise ratio and reduce hybridization time [2]. |
| Detection System | Visualization and documentation of specific signals. | Phosphor imager (for radioactivity), CCD-based imager for chemiluminescence/fluorescence (e.g., Azure Sapphire FL) [13]. |
Northern blotting remains a profoundly relevant and powerful technique for monitoring target gene expression. Its unique capacity to provide direct information on transcript size and integrity, coupled with its high specificity and quantitative potential, makes it an essential component of the molecular biologist's arsenal. As the market data and continued technical refinements demonstrate, this robust methodology will continue to be a critical tool for validating high-throughput data and answering fundamental questions in gene regulation, particularly in pharmaceutical development and advanced research settings.
Northern blotting remains a foundational technique for the direct detection and analysis of specific RNA molecules, providing critical information about gene expression levels, transcript size, and RNA processing [2] [15]. Despite the emergence of newer technologies like RT-PCR and RNA-Seq, Northern blotting maintains its relevance as a gold standard for validation studies due to its ability to provide direct relative comparisons of message abundance between samples on a single membrane without amplification biases [16] [15]. The technique involves multiple sequential steps: RNA separation by denaturing gel electrophoresis, transfer to a solid support, immobilization, and hybridization with labeled probes [2]. The quality and appropriateness of the three essential components—gels, membranes, and probes—significantly influence the sensitivity, specificity, and overall success of any Northern analysis experiment. This application note provides detailed guidance on selecting and optimizing these critical components within the context of target gene expression monitoring for research and drug development applications.
Gel Systems for RNA Separation
Denaturing Agarose Gels
The primary function of the gel matrix in Northern blotting is to separate RNA molecules by size under conditions that minimize secondary structure. Denaturing agarose gels are the most common platform for separating RNA molecules in the range of hundreds to thousands of nucleotides [2] [15].
Table 1: Comparison of Denaturing Gel Systems for Northern Blotting
| Gel Parameter | Formaldehyde Gels | Glyoxal/DMSO Gels | Polyacrylamide/Urea Gels |
|---|---|---|---|
| Denaturant | 2.2 M Formaldehyde | 1% Glyoxal, 50% DMSO | 7-8 M Urea |
| Typical Use | General mRNA analysis (0.5-10 kb) | General mRNA analysis | Small RNAs, miRNAs (<100 nt) |
| Gel Concentration | 0.8%-1.5% agarose | 0.8%-1.2% agarose | 6%-15% polyacrylamide |
| Safety Considerations | Requires fume hood; toxic | Reduced toxicity; no fume hood required | Standard laboratory precautions |
| Advantages | Well-established protocol | Sharper bands; safer procedure | High resolution for small fragments |
| Disadvantages | Health hazards; longer destaining | Requires prepared reagents | More complex gel preparation |
Formaldehyde, typically at a concentration of 2.2 M in the gel, serves as the traditional denaturant by reacting with amino groups of nucleotide bases to prevent RNA secondary structure formation [17] [15]. However, the NorthernMax-Gly system utilizing glyoxal/DMSO offers a safer alternative that eliminates the need for fume hood use while potentially providing sharper bands [2]. For optimal results with agarose gels, practical considerations include pouring gels as thin as possible to facilitate efficient transfer while providing sufficient well depth to accommodate sample volume [18]. Agarose concentrations should generally not exceed 1.2% to prevent impeded transfer, though lower concentrations (0.8%-1.0%) are preferable for larger RNA molecules [18].
Specialized Gel Applications
For analysis of small RNA species such as microRNAs (20-25 nucleotides) or their precursors (60-120 nucleotides), denaturing polyacrylamide gels (6%-15%) with urea provide superior resolution compared to agarose systems [15] [3]. These gels require electrophoretic instead of capillary transfer but offer the sensitivity necessary to detect low-abundance small RNAs, which is particularly valuable in studies of regulatory RNAs in drug development research [3].
Monitoring Electrophoresis and Transfer
Incorporating ethidium bromide (EtBr) directly into RNA samples at 10 µg/mL enables real-time monitoring of RNA integrity during electrophoresis and assessment of transfer efficiency post-blotting [18]. While EtBr may cause slight alterations in RNA migration and a minor decrease in signal intensity, the benefits of procedural monitoring generally outweigh these disadvantages for most applications [18]. The presence of intact 28S and 18S ribosomal RNA bands, with the 28S band approximately twice the intensity of the 18S band, serves as a key quality indicator for total RNA samples [15].
Figure 1: Northern Blotting Workflow. The complete process from RNA sample preparation through detection of specific sequences.
Membrane Selection and Transfer Methodologies
Membrane Types and Properties
The selection of an appropriate membrane is critical for successful Northern blotting, as it must effectively bind nucleic acids while maintaining structural integrity throughout hybridization and washing procedures.
Table 2: Membrane Types for Northern Blotting
| Membrane Type | Binding Mechanism | Advantages | Disadvantages | Recommended Applications |
|---|---|---|---|---|
| Positively Charged Nylon | Electrostatic and hydrophobic interactions | High binding capacity; durable for multiple reprobing; compatible with various detection methods | Higher background potential; requires blocking | Standard mRNA detection; multiple reprobing |
| Neutral Nylon | Hydrophobic interactions | Reduced background compared to charged nylon | Lower binding capacity | High-abundance targets |
| Nitrocellulose | Hydrophobic interactions | Low background | Brittle; lower binding capacity; not reusable | Limited use with modern protocols |
Positively charged nylon membranes are strongly recommended for most Northern blot applications due to their high binding capacity (400-500 µg/cm²) and durability through multiple rounds of stripping and reprobing (up to 5-8 cycles) [2] [16]. The positive charge facilitates strong electrostatic interactions with the negatively charged phosphate backbone of nucleic acids, promoting efficient retention even through stringent washing conditions [2]. The BrightStar-Plus positively charged nylon membrane is specifically optimized for use with NorthernMax kits and provides exceptional sensitivity with minimal background [2].
RNA Transfer and Immobilization
Efficient transfer of RNA from gels to membranes represents a critical step that directly impacts sensitivity and quantitative accuracy. Both capillary and electroblotting methods are employed, each with distinct advantages.
Capillary transfer, utilizing a stack of dry paper towels to draw transfer solution through the gel and membrane, represents the most accessible method [18]. The setup can be upward or downward, with downward transfer potentially offering faster and more efficient transfer [18]. To optimize capillary transfer:
- Cut all materials (membrane, filter paper, paper towels) precisely to gel dimensions to prevent "short-circuiting" where transfer solution bypasses the gel and membrane [18]
- Avoid introducing bubbles between layers, particularly between gel and membrane, by rolling a pipette gently over each layer during assembly [18]
- Position the gel with the bottom of the wells facing the membrane to minimize migration distance for RNA molecules [18]
Electroblotting utilizes an electric field to drive RNA from the gel to the membrane and can be more rapid and efficient when manufacturer's instructions are followed precisely [18]. The NorthernMax protocol incorporates a rapid, alkaline transfer method that efficiently moves RNA, especially larger transcripts, onto the membrane in just 2 hours compared to overnight transfers required by some standard protocols [2].
Following transfer, RNA must be immobilized on the membrane to prevent elution during subsequent hybridization and washing steps. Ultraviolet (UV) crosslinking is the preferred method, creating covalent bonds between the membrane and nucleic acid bases [18] [2]. Alternative methods include baking at 80°C for 30 minutes to 2 hours, though this may be less efficient for some membrane types [18]. For small RNA detection, chemical crosslinking with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) has been reported to improve retention compared to UV crosslinking [3].
Probe Design and Labeling Strategies
Probe Types and Selection Criteria
The selection of an appropriate hybridization probe fundamentally determines the sensitivity and specificity of Northern detection. Three primary probe types offer distinct characteristics suitable for different applications.
Table 3: Comparison of Probe Types for Northern Blotting
| Probe Type | Synthesis Method | Sensitivity | Stability | Stringency | Key Applications |
|---|---|---|---|---|---|
| RNA Probes (Riboprobes) | In vitro transcription | Very High (10-100x DNA) | High | High (RNase washes possible) | Low-abundance messages; homologous probing |
| DNA Probes | Random priming, PCR, oligonucleotide synthesis | Moderate to High | Moderate | Moderate | General purpose; heterologous probing |
| Oligonucleotide Probes | Chemical synthesis | Lower (unless modified) | High | High | Known sequences; miRNA detection |
RNA probes (riboprobes), synthesized by in vitro transcription, generally provide the highest sensitivity—up to 10-fold greater than random-primed DNA probes under standard hybridization conditions [2]. This enhanced sensitivity, combined with the ability to perform stringent RNase washes to reduce background, makes riboprobes ideal for detecting low-abundance messages [2]. The Strip-EZ RNA System facilitates efficient probe removal for multiple reprobing without damaging the membrane-bound RNA [18].
DNA probes can be generated by random priming, PCR, or oligonucleotide synthesis and offer convenience and flexibility [15]. While traditionally less sensitive than riboprobes, the sensitivity gap narrows significantly when using optimized hybridization buffers like ULTRAhyb, which can increase sensitivity up to 100-fold compared to standard hybridization solutions [2]. DNA probes are particularly valuable for heterologous probing across species where sequence divergence requires adjustment of stringency conditions [16].
Labeling and Detection Methods
Both radioactive and nonradioactive detection methods provide viable options for Northern blotting, with selection dependent on safety requirements, sensitivity needs, and equipment availability.
Radioactive labeling with ³²P provides exceptional sensitivity, capable of detecting fewer than 100,000 target molecules on a blot [2]. The high specific activity and direct detection of radioactive emissions make this approach particularly valuable for low-abundance targets or when using heterologous probes with reduced hybridization efficiency [16]. However, safety concerns, regulatory requirements, and waste disposal issues have motivated development of nonradioactive alternatives.
Nonradioactive detection systems utilizing haptens such as digoxigenin (DIG) or biotin, coupled with enzyme-mediated colorimetric or chemiluminescent detection, offer safer alternatives with increasingly competitive sensitivity [3]. Chemiluminescent detection, using alkaline phosphatase or horseradish peroxidase-conjugated antibodies with appropriate substrates, can approach the sensitivity of radioactive methods while providing faster results and eliminating radiation hazards [15] [3]. Recent advances demonstrate that optimized nonradioactive protocols can detect mRNA and small RNA species using biotinylated probes with sensitivity comparable to radiolabeled approaches [3].
Hybridization and Washing Optimization
Post-hybridization washing conditions critically influence the balance between signal retention and background reduction. Traditional protocols employ sequential low and high stringency washes, but modified approaches using quantitatively controlled moderate-stringency washes until background radioactivity reaches 20-50 counts per second can maximize retention of specifically bound probes, significantly improving detection sensitivity for low-expression genes [16].
The composition of hybridization buffers substantially impacts sensitivity. Specialty formulations like ULTRAhyb Ultrasensitive Hybridization Buffer dramatically increase sensitivity compared to standard buffers, particularly for DNA probes where signal intensity can approach that achieved with RNA probes [2]. For many messages, hybridization can be completed in just 2 hours when using optimized buffers, though overnight hybridization may still be necessary for very low-abundance targets [2].
Research Reagent Solutions
Table 4: Essential Research Reagents for Northern Blotting
| Reagent Category | Specific Examples | Function | Technical Notes |
|---|---|---|---|
| RNA Isolation | TRIzol Reagent, RNA extraction kits | High-quality RNA extraction | Integrity critical; A260/A280 >1.8 |
| Denaturing Gels | NorthernMax-Gly Kit, formaldehyde, glyoxal | RNA separation by size | Prevent RNA secondary structure |
| Membranes | BrightStar-Plus, positively charged nylon | RNA immobilization | High binding capacity; multiple reprobing |
| Hybridization Buffers | ULTRAhyb Ultrasensitive Hybridization Buffer | Signal enhancement | Up to 100x sensitivity increase |
| Labeling Systems | MAXIscript Kit, DECAprime II Kit | Probe generation | Radioactive or nonradioactive options |
| Detection | Chemiluminescent substrates, X-ray film | Signal detection | Alternative to radioactive detection |
| Crosslinking | UV crosslinkers, baking ovens | RNA immobilization | UV preferred over baking |
| Size Markers | RNA Millennium Markers, ribosomal RNA | Size determination | 28S (5kb) and 18S (2kb) rRNA as internal markers |
The strategic selection and optimization of gels, membranes, and probes fundamentally determines the success of Northern blotting experiments for target gene expression monitoring. Denaturing agarose gels with formaldehyde or glyoxal provide effective RNA separation, while positively charged nylon membranes offer optimal binding capacity and durability for multiple reprobing. Probe selection represents a critical decision point, with RNA probes providing maximum sensitivity for low-abundance targets, while DNA probes offer practical advantages when used with optimized hybridization systems. By carefully considering these essential components within the context of their specific research objectives, scientists and drug development professionals can implement Northern blotting protocols that deliver reliable, sensitive, and quantitative gene expression data for both basic research and applied pharmaceutical applications.
Northern blotting remains a foundational technique in molecular biology for the specific detection and analysis of RNA molecules. This hybridization-based method provides critical information about RNA expression, size, and integrity, complementing modern high-throughput technologies in both basic research and drug development contexts [11]. Despite the emergence of newer transcriptomic platforms, Northern blotting maintains its relevance due to its ability to deliver highly specific and qualitative information on RNA size and abundance, making it invaluable for validating results from sequencing technologies [11]. For researchers monitoring target gene expression, particularly in studies of cancer, infectious diseases, and genetic disorders, this technique offers robust, reproducible data that continues to inform therapeutic development and molecular diagnostics [19] [11].
The Northern Blotting Workflow
The following diagram illustrates the complete Northern blotting procedure from sample preparation to detection:
Detailed Methodological Protocols
RNA Extraction and Denaturation
Principles and Significance: Isolating intact, high-quality RNA is the most critical step in Northern blotting, as RNA integrity directly impacts detection accuracy and experimental reproducibility. The extraction process must effectively separate RNA from DNA, proteins, and other cellular components while preserving full-length transcripts.
Detailed Protocol:
- Homogenization: Begin with immediate homogenization of tissue or culture samples in denaturing buffer containing guanidinium thiocyanate to inactivate RNases [20]. Maintain samples on ice throughout processing.
- RNA Isolation: Use acid-phenol:chloroform extraction at pH 4.7-4.8 to preferentially partition RNA into the aqueous phase while DNA and proteins remain in the organic phase or interface [20].
- Quantification and Purity Assessment: Precisely measure RNA concentration using spectrophotometry (A260/A280 ratio of 1.8-2.0 indicates pure RNA). Verify integrity by running a small aliquot on a denaturing agarose gel with sharp ribosomal RNA bands.
- Denaturation: Prepare samples using formaldehyde (or glyoxal/DMSO) as denaturing agents to maintain RNA in a linear state [20]. For microRNA or small RNA detection (<200 nucleotides), polyacrylamide gels with urea as a denaturing agent are preferred [20]. Heat samples to 65°C for 10-15 minutes, then immediately place on ice to prevent RNA secondary structure formation.
Technical Considerations: Always use RNase-free reagents, tubes, and equipment. For tissues rich in RNases (pancreas, spleen), increase the concentration of denaturing agents. Include RNA ladder and ribosomal RNA markers for size determination during electrophoresis [20].
Gel Electrophoresis and Membrane Transfer
Principles and Significance: Denaturing gel electrophoresis separates RNA molecules by size, while membrane transfer immobilizes the separated RNA for subsequent hybridization analysis. This combination enables accurate size determination of target transcripts.
Detailed Protocol:
- Gel Preparation: Prepare a 1.0-1.2% denaturing agarose gel containing 2.2M formaldehyde in MOPS or phosphate buffer [20]. For higher resolution of small RNAs (<200 nucleotides), use 8-12% polyacrylamide gels with 7M urea [20].
- Electrophoresis Conditions: Load 5-20μg of total RNA or 0.5-2μg of poly(A)+ RNA per lane. Run gel at 3-5V/cm in recirculating buffer to maintain constant pH. Include an RNA ladder and ethidium bromide staining for visualization [20].
- Capillary Transfer: Assemble transfer stack in this order (from bottom to top): sponge, filter paper, gel, nylon membrane, filter paper, paper towels, weight [20]. Use 20× SSC (3M NaCl, 0.3M sodium citrate) as transfer buffer. Allow transfer to proceed for 12-16 hours [20].
- Alternative Transfer Methods: For improved efficiency, use vacuum or electroblotting systems, which reduce transfer time to 30-90 minutes and increase yield for larger RNA fragments.
Technical Considerations: Ensure complete denaturation of RNA samples before loading. Handle formaldehyde-containing gels in a fume hood. For capillary transfer, prevent air bubbles between gel and membrane as they block transfer. Always mark the gel orientation on the membrane.
Probe Design, Hybridization, and Detection
Principles and Significance: Hybridization with specific probes enables selective detection of target RNA sequences. Proper probe design and hybridization conditions determine the sensitivity and specificity of Northern blot detection.
Detailed Protocol:
- Probe Selection: Choose from three probe types: (1) single-stranded DNA probes (25+ complementary basepairs), (2) RNA probes (in vitro transcribed), or (3) oligonucleotide probes (synthesized DNA or RNA) [20].
- Probe Labeling: Radiolabel with ³²P using nick translation, random priming, or in vitro transcription for maximum sensitivity [21]. Alternatively, use non-radioactive labels (alkaline phosphatase, horseradish peroxidase) with chemiluminescent detection [20].
- Membrane Fixation: After transfer, immobilize RNA on nylon membrane by UV crosslinking (120,000 μJ/cm²) or baking at 80°C for 1-2 hours [20].
- Prehybridization and Hybridization: Prehybridize membrane for 2-4 hours at 42-65°C in buffer containing formamide (reduces annealing temperature), Denhardt's solution, salmon sperm DNA, and SDS [20]. Add labeled probe and hybridize for 12-16 hours.
- Stringency Washes: Perform sequential washes starting with 2× SSC/0.1% SDS at room temperature, progressing to 0.1× SSC/0.1% SDS at 50-65°C [20]. Adjust wash stringency based on probe specificity requirements.
- Signal Detection: For radioactive probes, expose membrane to X-ray film or phosphorimager screen (1 hour to several days) [20]. For chemiluminescent detection, incubate with substrate and expose for seconds to minutes.
Technical Considerations: Optimize hybridization temperature based on probe length and GC content. Include positive and negative controls. Rehybridize membranes with housekeeping gene probes (e.g., GAPDH, β-actin) for normalization.
The Scientist's Toolkit: Essential Research Reagents
Table 1: Key reagents and materials for Northern blotting experiments
| Reagent/Material | Function/Purpose | Technical Notes |
|---|---|---|
| Formaldehyde | RNA denaturant for gel electrophoresis | Prevents RNA secondary structure formation; handle in fume hood [20] |
| Agarose/Polyacrylamide | Matrix for size-based RNA separation | Agarose for standard RNA; PAGE for small RNAs [20] |
| Nylon Membrane | Solid support for RNA immobilization | Positively charged membranes enhance RNA retention [20] |
| Formamide | Hybridization buffer component | Lowers probe annealing temperature; use high-grade deionized [20] |
| Radioactive Probes ([³²P]-labeled) | High-sensitivity RNA detection | ³²P provides superior sensitivity for low-abundance transcripts [21] [20] |
| Non-radioactive Probes | Alternative detection method | Alkaline phosphatase or HRP systems with chemiluminescence [20] |
| RNA Ladder | Molecular size standards | Essential for determining transcript size [20] |
| Transfer Buffer (20× SSC) | Medium for capillary RNA transfer | High salt concentration promotes RNA binding to membrane [20] |
Advanced Applications and Quantitative Analysis
Advanced Applications in Modern Research
Northern blotting continues to evolve with applications in cutting-edge research areas. The technique has been adapted for studying ribosome-associated noncoding RNAs, particularly tRNA-derived fragments (tDRs), which have emerged as key regulators of translation under stress conditions [21]. The recently developed tDR-quant method employs electroporation of radiolabeled tDRs into yeast spheroplasts, followed by polysome profiling and radioactivity detection to quantitatively assess tDR-ribosome interactions in vivo [21]. This approach has revealed that tDR interactions with ribosomes are stress- and dose-dependent, primarily associating with the 40S subunit but also with 60S, monosomes, and polysomes under specific conditions [21].
Market Outlook and Technical Evolution
The continued relevance of Northern blotting is reflected in market analyses projecting steady growth. The global Northern blotting market is expected to grow at a CAGR of 4.0-5.8%, reaching USD 252.3-862.3 million by 2035 [19] [11]. This growth is driven by the technique's vital role in gene expression studies and RNA analysis across molecular biology and biomedical research, particularly in drug discovery, clinical diagnostics, and academic research [11].
Table 2: Northern blotting market outlook and application segments
| Parameter | Value/Ranking | Context and Significance |
|---|---|---|
| Projected Market Value (2035) | USD 252.3-862.3 million | Steady growth reflects continued relevance in molecular biology [19] [11] |
| Leading Application Segment | Academic Research (41.2%) | Dominance in fundamental RNA expression studies and validation [11] |
| Dominant End User | Pharmaceutical & Biotechnology (50.6%) | Critical role in drug discovery and therapeutic development [11] |
| Key Product Segment | Reagents & Consumables (28.9-45.7%) | Recurring demand for high-quality, standardized reagents [19] [11] |
| Fastest-growing Region | Asia-Pacific (CAGR 6.9%) | Expanding biomedical research infrastructure and funding [19] |
Troubleshooting and Optimization Strategies
Common Challenges and Solutions:
- RNA Degradation: Always use RNase-free conditions. Include denaturing agents throughout initial steps. Check RNA integrity by electrophoresis before proceeding.
- High Background: Increase stringency of washes. Ensure adequate prehybridization with blocking agents. Optimize probe concentration to reduce non-specific binding.
- Weak or No Signal: Check probe labeling efficiency. Increase RNA loading amount. Extend exposure time. Verify probe specificity and integrity.
- Uneven Blotting: Eliminate air bubbles during transfer. Ensure even weight distribution in capillary transfer. Use fresh transfer buffer.
Quantitative Considerations: For expression analysis, include multiple housekeeping genes for normalization. Ensure signals are within the linear range of detection. Perform biological and technical replicates to assess variability.
Northern blotting remains an essential technique in the molecular biologist's toolkit, providing unambiguous data on RNA size, integrity, and expression levels that complements next-generation sequencing technologies. The detailed workflow presented here—from careful RNA isolation through specific detection—enables researchers to obtain reliable, reproducible results for target gene expression monitoring. As the field advances with improved sensitivity reagents, automated platforms, and novel applications like noncoding RNA analysis, Northern blotting continues to adapt and maintain its relevance in both basic research and drug development contexts. Its enduring value lies in its ability to provide direct, quantitative evidence of RNA expression that remains the gold standard for validation of transcriptional regulation.
Advanced Northern Blot Protocols and Applications in Research & Development
Optimized RNA Isolation and Quality Assessment
Within gene expression monitoring research, Northern blotting remains a definitive method for directly detecting and quantifying specific mRNA molecules, providing invaluable information on transcript size and abundance [2] [22]. The success of this technique is critically dependent on the initial quality and integrity of the isolated RNA [2]. Degraded RNA, a common challenge during isolation, severely compromises the ability to quantitate expression and can lead to complete experimental failure [2] [23]. This application note details optimized protocols for obtaining high-yield, high-quality RNA, ensuring reliable and reproducible results for Northern blotting and subsequent gene expression analysis.
Critical Principles for RNA Integrity
The inherent lability of RNA necessitates strict adherence to RNase-free techniques throughout the isolation process. RNases are ubiquitous enzymes that can rapidly degrade RNA; therefore, all equipment and work surfaces must be decontaminated using specific RNase deactivation reagents, and researchers must always wear gloves [2] [22]. The foundational principle is that even a single cleavage in a fraction of target mRNA molecules can significantly diminish the detected signal on a Northern blot, directly impacting quantitative accuracy [2].
Optimized RNA Isolation Protocols
The choice of isolation method depends on the starting biological material. Below are two optimized protocols for challenging sample types: plant tissues rich in secondary metabolites and frozen whole blood.
Modified SDS-Based Protocol for Challenging Plant Tissues (e.g.,Musa spp.)
Plant tissues often contain high levels of polysaccharides, polyphenols, and other secondary metabolites that co-precipitate with and degrade RNA [24]. This modified SDS-based method effectively combats these challenges.
Experimental Workflow:
The following diagram illustrates the key stages of the optimized SDS-based RNA isolation protocol.
Detailed Methodology:
- Sample Preparation: Collect fresh or frozen tissue (e.g., leaf, root) and immediately grind 100 mg to a fine powder in liquid nitrogen using a pre-cooled mortar and pestle [24].
- Homogenization: Transfer the powdered tissue to a microcentrifuge tube containing 1 mL of pre-warmed (65°C) SDS extraction buffer. The optimized buffer composition is critical and should contain: 2% SDS (w/v), 100 mM Tris-HCl (pH 8.0), 25 mM EDTA, and 2% polyvinylpyrrolidone (PVP). Mix by vigorous vortexing until the solution is homogeneous [24].
- Incubation: Incubate the homogenate at 65°C for 10 minutes with occasional mixing [24].
- Organic Extraction: Add an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1). Mix thoroughly by inversion and centrifuge at 12,000 × g for 15 minutes at 4°C. Carefully transfer the upper aqueous phase to a new tube. Repeat the extraction with an equal volume of chloroform to remove residual phenol [24].
- RNA Precipitation: To the clear aqueous phase, add 1/4 volume of 10 M LiCl solution to achieve a final concentration of 2 M. Mix well and incubate overnight at 4°C. Precipitating with LiCl, instead of isopropanol, selectively precipitates RNA and reduces polysaccharide contamination [24].
- RNA Pellet Washing: Centrifuge at 12,000 × g for 30 minutes at 4°C to pellet the RNA. Carefully decant the supernatant and wash the pellet with 1 mL of 70% ethanol (prepared with DEPC-treated water). Centrifuge again for 10 minutes, discard the ethanol, and air-dry the pellet for 5-10 minutes.
- Resuspension: Dissolve the dried RNA pellet in 30-50 µL of RNase-free/DEPC-treated water [24].
High-Quality RNA from Frozen EDTA Blood
Frozen blood collected in conventional EDTA tubes is notoriously difficult for RNA extraction due to thaw-induced hemolysis and RNase release [23]. The following novel protocol reverses this degradation.
Detailed Methodology: EDTA-mixed thawing-Nucleospin (EmN) Protocol
- Thawing with Lysis Buffer: Remove the frozen EDTA blood sample from -80°C. Crucially, immediately add 1.3 mL of Nucleospin (Macherey-Nagel) lysis buffer directly to the frozen blood pellet. Vortex briefly to mix. The thawing process must occur in the presence of this RNA-stabilizing buffer [23].
- Complete Homogenization: Continue vortexing until the sample is completely thawed and homogenous. This ensures immediate cell lysis and RNase inhibition upon thawing.
- RNA Purification: Follow the manufacturer's instructions for the Nucleospin Blood RNA kit for the subsequent purification steps, including possible DNase digestion [23].
- Elution: Elute the purified RNA in a suitable volume of RNase-free water.
Performance Data: This EmN protocol yields RNA with an average RNA Integrity Number (RIN) of 7.3, comparable to the gold-standard PAXgene method, but with a 5-fold higher average yield (4.7 µg/mL blood vs. 0.9 µg/mL) [23].
Comprehensive RNA Quality Assessment
Rigorous assessment is mandatory before proceeding to sensitive techniques like Northern blotting. The following table summarizes the key metrics and their ideal values.
Table 1: Comprehensive Assessment of RNA Quality and Integrity
| Assessment Method | Parameter Measured | Optimal Value/Range | Interpretation and Significance |
|---|---|---|---|
| Spectrophotometry (NanoDrop) | A260/A280 Ratio | 1.8 - 2.1 [24] | Indicates purity; values outside this range suggest protein or phenol contamination. |
| A260/A230 Ratio | 2.0 - 2.3 [24] | Indicates purity from salts, carbohydrates, or organic solvents; a low ratio signals contamination. | |
| Agarose Gel Electrophoresis | Ribosomal RNA Bands | Sharp, distinct 28S and 18S bands [22] | Visual assessment of integrity. The 28S band should be approximately twice as intense as the 18S band. |
| Bioanalyzer (Agilent 2100) | RNA Integrity Number (RIN) | > 8.0 for sequencing [25]; > 7.0 is acceptable for Northern blot [23] | A quantitative score (1-10) of RNA integrity; higher values indicate less degradation. |
| Qubit RNA IQ Assay | RNA IQ Value | 7.8 - 9.9 [24] | A fluorometric-based metric for RNA integrity and quality, complementary to RIN. |
The Scientist's Toolkit: Essential Reagents for RNA Isolation
Table 2: Key Research Reagent Solutions for RNA Isolation and Quality Control
| Reagent / Kit | Function and Application |
|---|---|
| TRIzol Reagent | Monophasic solution of phenol and guanidine isothiocyanate for effective cell lysis and simultaneous dissolution of biological material and RNase inhibition [24] [25]. |
| Nucleospin Blood RNA Kit | Silica-membrane based kit designed for RNA purification from difficult blood samples; central to the EmN protocol [23]. |
| Modified SDS Buffer | Extraction buffer optimized for challenging plant tissues; SDS disrupts membranes, while EDTA chelates Mg2+ to inhibit RNases, and PVP binds polyphenols [24]. |
| PAXgene Blood RNA System | Integrated system of blood collection tubes and purification kits for prospective RNA stabilization at the point of collection [23]. |
| LiCl (Lithium Chloride) | Preferentially precipitates RNA over polysaccharides, making it ideal for plants and other polysaccharide-rich samples [24]. |
| DNase I (RNase-free) | Enzyme used to digest and remove contaminating genomic DNA from the RNA preparation, essential for accurate gene expression analysis [22]. |
| Agilent 2100 Bioanalyzer | Microfluidics-based system providing electrophoretic traces and RIN for objective, high-throughput RNA quality assessment [23] [25]. |
High-quality, intact RNA is the non-negotiable foundation for successful Northern blotting. The optimized protocols detailed here, complemented by rigorous quality control, provide a reliable pathway to robust gene expression data. By selecting the appropriate isolation method for their sample type and adhering to these stringent quality assessment metrics, researchers can ensure their Northern blotting experiments for target gene monitoring yield clear, quantifiable, and publication-ready results.
Within the framework of target gene expression monitoring research, Northern blotting remains a foundational technique for the specific detection and analysis of RNA molecules. This application note provides a detailed protocol for two critical stages of the Northern blot: denaturing agarose gel electrophoresis, which separates RNA molecules by size while preventing secondary structure formation, and the subsequent efficient blotting of the separated RNA onto a solid membrane for detection [26] [20]. The integrity of RNA throughout this process is paramount for obtaining accurate and reproducible data on transcript abundance and size [27].
Materials and Reagents
Reagents for Denaturing Gel Electrophoresis
The following reagents are essential for preparing and running a denaturing formaldehyde agarose gel [27].
- Agarose: High-quality agarose for gel formation.
- Formaldehyde (37%): Used as a denaturing agent to prevent RNA secondary structure. Warning: Formaldehyde is toxic and should be handled in a chemical fume hood [27].
- 10X MOPS Running Buffer: Composed of 0.4 M MOPS (pH 7.0), 0.1 M sodium acetate, and 0.01 M EDTA [27].
- RNA Sample Loading Buffer: A denaturing buffer, such as Formaldehyde Load Dye, typically containing formamide, EDTA, and tracking dyes (e.g., bromophenol blue and xylene cyanol) [27].
- Ethidium Bromide or Alternative Stain: For post-electrophoresis visualization of RNA. Ethidium bromide can be added directly to the loading buffer at a final concentration of 10 µg/ml or the gel can be stained afterward. More sensitive alternatives like SYBR Gold or SYBR Green II are available for low-abundance samples [27].
Reagents for Efficient Blotting
The following materials are required for the capillary transfer of RNA from the gel to a membrane.
- Solid Support Membrane: Positively charged nylon membranes are generally preferred due to their high binding affinity and robustness for nucleic acids [26].
- Blotting Paper: Whatman filter paper or equivalent for creating the transfer stack.
- Transfer Buffer: Standard saline citrate (SSC) buffer is commonly used (e.g., 20x SSC: 3 M NaCl, 0.3 M sodium citrate, pH 7.0).
- UV Crosslinker: For immobilizing RNA onto the membrane after transfer by creating covalent linkages [26].
Methodology
Protocol: Denaturing Agarose Gel Electrophoresis of RNA
This protocol is modified from established molecular biology methods and is designed to assess RNA integrity and yield [27].
Step 1: Prepare the Denaturing Gel
- Combine 1 g of agarose with 72 mL of deionized water. Heat until the agarose is completely dissolved.
- Cool the agarose solution to approximately 60°C.
- In a fume hood, add 10 mL of 10X MOPS running buffer and 18 mL of 37% formaldehyde to the cooled agarose. Mix thoroughly.
- Pour the gel into a casting tray with an appropriate comb and allow it to solidify.
Step 2: Prepare the RNA Sample
- For a standard analysis, combine 1-3 µg of total RNA with 0.5 to 3 volumes of Formaldehyde Load Dye. Use a higher ratio (e.g., 3 volumes) for complete denaturation required in Northern blots [27].
- If not pre-mixed, ethidium bromide can be added to the sample to a final concentration of 10 µg/ml.
- Heat denature the samples at 65–70°C for 5–15 minutes. Use 5 minutes for simple quality checks and 15 minutes for Northern blot analysis [27]. Immediately place on ice after heating.
Step 3: Electrophoresis
- Place the solidified gel in the electrophoresis tank and submerge it in 1X MOPS running buffer.
- Load the denatured RNA samples and an appropriate RNA molecular weight marker (e.g., RNA Millennium Markers) into the wells [27].
- Run the gel at 5–6 V/cm (measured between the electrodes) until the bromophenol blue dye has migrated 2–3 cm into the gel or up to two-thirds of the gel length [27].
Step 4: Visualization and Quality Assessment
- Visualize the gel on a UV transilluminator.
- Intact total RNA from eukaryotic samples will display sharp 28S and 18S ribosomal RNA bands. A 2:1 intensity ratio (28S:18S) indicates high-quality, non-degraded RNA [27]. Partially or completely degraded RNA will appear as a smear or will lack these distinct bands (see Table 1 for troubleshooting).
Protocol: Efficient Capillary Blotting for Northern Analysis
This section describes the traditional capillary transfer method, which uses a passive flow of buffer to move RNA from the gel to a membrane [26] [20].
Step 1: Set Up the Capillary Transfer System
- Place a platform in a large dish filled with transfer buffer (e.g., 20x SSC).
- Lay a wick (a long piece of blotting paper) over the platform, ensuring its ends are submerged in the buffer.
- Carefully place the gel on top of the wick, avoiding air bubbles.
- Surround the gel with Parafilm to prevent short-circuiting the flow of buffer.
- Place the nylon membrane, cut to the exact size of the gel, on top of the gel.
- Place several sheets of dry blotting paper on the membrane, followed by a stack of paper towels.
- Place a glass plate and a moderate weight (e.g., a 500 g weight) on top to ensure good contact.
Step 2: Transfer and Immobilize RNA
- Allow the capillary transfer to proceed for several hours or overnight.
- After transfer, disassemble the stack. RNA can be visualized on the membrane by briefly exposing it to UV light if the gel contained ethidium bromide.
- Immobilize the RNA onto the membrane by UV cross-linking (typically at 254 nm) or by baking at ~80°C [26] [20]. The membrane can now be used for hybridization or stored dry.
The following workflow diagram illustrates the complete Northern blotting procedure from RNA separation to detection.
The Scientist's Toolkit: Key Research Reagent Solutions
The following table details essential reagents and their functions for a successful Northern blotting experiment, compiled from the referenced protocols [27] [26] [20].
Table 1: Essential Reagents for Northern Blotting
| Reagent | Function/Description | Key Considerations |
|---|---|---|
| Denaturing Agent (Formaldehyde) | Prevents formation of RNA secondary structures during electrophoresis, ensuring migration by molecular weight [27]. | Toxic; requires use in a fume hood. Glyoxal is an alternative denaturant [27]. |
| RNA Loading Dye | Provides density for gel loading; contains dyes (bromophenol blue) to track migration; often includes formamide for denaturation [27]. | Commercial formulations (e.g., NorthernMax) are optimized for safety and performance [27]. |
| Nylon Membrane | Solid support for immobilizing RNA after blotting; positive charge enhances nucleic acid binding [26] [20]. | Preferred over nitrocellulose for its robustness and higher binding affinity for RNA [26]. |
| Blocking Agent | Used in pre-hybridization to block non-specific binding sites on the membrane, reducing background noise [26]. | Examples include denatured salmon sperm DNA or other proprietary blocking solutions [26]. |
| Hybridization Probe | Labeled DNA or RNA sequence complementary to the target RNA; allows specific detection via autoradiography or chemiluminescence [26] [20]. | Can be radioactive or non-radioactive (e.g., chemiluminescent); must be designed for the specific target [20]. |
Troubleshooting and Quantitative Data Interpretation
Accurate interpretation of results and troubleshooting common issues are critical skills. The table below summarizes key quantitative and qualitative data points.
Table 2: Troubleshooting Guide and Data Interpretation
| Observation | Interpretation | Potential Solution |
|---|---|---|
| Faint or No Bands | Low RNA abundance or transfer inefficiency. | Ensure at least 200 ng of RNA is loaded for ethidium bromide staining; use more sensitive stains (e.g., SYBR Gold) for low-yield samples [27]. |
| Smeared RNA on Gel | RNA degradation. | Check RNA integrity prior to loading; ensure all reagents and equipment are RNase-free [27] [26]. |
| High Background on Membrane | Non-specific binding of the probe. | Increase the stringency of washes; ensure adequate blocking during pre-hybridization [26]. |
| Intact Total RNA (Eukaryotic) | Sharp 28S and 18S rRNA bands with a 2:1 intensity ratio [27]. | This is the ideal result, indicating high-quality RNA. |
| Poor Transfer Efficiency | RNA remains in the gel after blotting. | Check gel thickness; ensure proper setup of the capillary stack without air bubbles; consider vacuum blotting for faster, more reproducible transfers [26]. |
Probe Design, Labeling (Radioactive and Non-Radioactive), and Hybridization
Within the framework of target gene expression monitoring, the Northern blot remains a cornerstone technique for the specific detection and quantification of RNA transcripts. Its utility extends beyond mere confirmation of gene expression to providing critical data on transcript size and the identification of alternatively spliced variants, which are often essential in drug development research [2]. The core of this method's specificity and sensitivity lies in the effective execution of three interdependent processes: the design of specific probes, their subsequent labeling, and the final hybridization with the target RNA. This protocol details comprehensive methodologies for both radioactive and non-radioactive approaches, enabling researchers to select the optimal balance of sensitivity, safety, and convenience for their experimental needs in gene expression analysis.
Probe Design and Synthesis
The foundation of a successful Northern blot experiment is a well-designed probe. The choice of probe type—whether DNA, RNA, or oligonucleotide—directly impacts the assay's sensitivity, specificity, and signal-to-noise ratio.
- DNA Probes: These can be double-stranded (e.g., cDNA fragments) or single-stranded. Double-stranded DNA probes are often generated via random-primed labeling or asymmetric PCR [2]. While DNA probes are generally less sensitive than RNA probes, their performance is significantly enhanced when used with optimized hybridization buffers like ULTRAhyb, making them a viable option [2].
- RNA Probes (Riboprobes): Synthesized via in vitro transcription from a linearized plasmid template, riboprobes offer superior sensitivity [28] [2]. This is due to their higher thermodynamic stability when forming RNA:RNA hybrids with the target mRNA, which also permits the use of more stringent washing conditions to minimize background [28]. Riboprobes are therefore ideal for detecting low-abundance transcripts.
- Oligonucleotide Probes: These short, single-stranded DNA sequences (typically 20-50 nucleotides) are designed to be complementary to a specific region of the target RNA [29]. They are particularly useful for detecting small RNA species and for distinguishing between closely related sequences, such as members of a miRNA family [30].
Table 1: Comparison of Northern Blot Probe Types
| Probe Type | Typical Length | Sensitivity | Key Advantages | Ideal For |
|---|---|---|---|---|
| DNA (random-primed) | >100 bp | Moderate [2] | Easy to produce, stable | Detecting abundant transcripts |
| RNA (Riboprobe) | >100 bp | High [28] [2] | Very high sensitivity and specificity, allows stringent washes | Detecting low-abundance mRNAs |
| Oligonucleotide | 20-50 nt | Varies with label | High specificity for small RNAs, can distinguish single-base changes | microRNAs, siRNAs, and other small non-coding RNAs [30] |
The following workflow outlines the critical decision points and subsequent steps in the probe preparation process:
Probe Labeling Technologies
After synthesis, probes must be labeled to enable detection. The choice between radioactive and non-radioactive labels is a critical one, impacting sensitivity, safety, cost, and probe stability.
Radioactive Labeling
Radioactive labeling, particularly with Phosphorus-32 (³²P), remains the gold standard for maximum sensitivity, and is often essential for detecting low-abundance transcripts or small RNAs [30].
- Labeling Methods:
- Oligonucleotide Probes: Typically end-labeled using T4 Polynucleotide Kinase (T4 PNK) and [γ-³²P]ATP. This reaction transfers the radioactive phosphate group to the 5' end of the DNA oligonucleotide [30].
- DNA and RNA Probes: These are usually uniformly labeled during synthesis by incorporating nucleotides labeled with α-³²P (e.g., [α-³²P]dCTP or [α-³²P]UTP) [2].
- Protocol: End-Labeling of an Oligonucleotide Probe with T4 PNK
- Reaction Setup: In a nuclease-free microcentrifuge tube, combine the following on ice:
- 100 ng of your oligonucleotide probe
- 5 μL of [γ-³²P]ATP (e.g., 3000 Ci/mmol)
- 2 μL of 10X T4 PNK Reaction Buffer
- 1 μL (10 units) of T4 PNK Enzyme
- Nuclease-free water to a final volume of 20 μL.
- Incubation: Incubate the reaction at 37°C for 30-60 minutes.
- Termination: Heat-inactivate the enzyme at 65°C for 10 minutes.
- Purification: Purify the labeled probe from unincorporated [γ-³²P]ATP using a size-exclusion chromatography column (e.g., P-30 column), following the manufacturer's instructions [30]. This step is crucial to reduce background signal.
- Reaction Setup: In a nuclease-free microcentrifuge tube, combine the following on ice:
Non-Radioactive Labeling
Non-radioactive methods have advanced significantly, offering excellent sensitivity with improved safety and probe stability. Common labels include digoxigenin (DIG) and biotin [8] [28].
- Labeling Methods:
- PCR-based Labeling: DIG- or biotin-labeled nucleotides can be directly incorporated into DNA probes during a PCR amplification reaction [7].
- In Vitro Transcription Labeling: Riboprobes can be synthesized in the presence of DIG- or biotin-labeled UTP to generate a labeled RNA probe [28] [2].
- Oligonucleotide Probes: Commercially synthesized oligonucleotides can be purchased with a hapten (e.g., DIG, biotin) already conjugated to the 5' or 3' end.
- Protocol: Labeling a DNA Probe by PCR with DIG
- Reaction Setup: On ice, prepare a 50 μL PCR reaction containing:
- 1X PCR Buffer
- Your template DNA (e.g., 2 μL of cDNA)
- Forward and Reverse primers (e.g., 5 μL of each at 10 μM)
- PCR DIG Labeling Mix (from Roche, contains DIG-dUTP) [7]
- DNA Polymerase (e.g., 0.75 μL of enzyme mix)
- Nuclease-free water to 50 μL.
- Amplification: Run PCR using standard cycling conditions for your template and primers.
- Verification & Purification: Analyze a small aliquot of the PCR product on an agarose gel to confirm successful labeling and amplification. Purify the remaining product using a standard PCR cleanup kit.
- Reaction Setup: On ice, prepare a 50 μL PCR reaction containing:
Table 2: Comparison of Radioactive and Non-Radioactive Labeling Methods
| Parameter | Radioactive (e.g., ³²P) | Non-Radioactive (e.g., DIG, Biotin) |
|---|---|---|
| Sensitivity | Very High (gold standard) [30] | High, sufficient for many applications [28] |
| Resolution | Excellent | Good |
| Probe Stability | Short (days, due to decay) | Long (months to years) [31] |
| Safety & Regulation | Requires special handling, training, and waste disposal [29] | Much safer, minimal regulatory concerns [31] |
| Detection Time | Hours to days (film exposure) | Minutes to hours (chemiluminescence) |
| Cost | Low (per experiment), high (waste disposal) | Higher (reagents), low (waste disposal) |
Hybridization and Detection
Hybridization is the process where the labeled probe binds to its complementary RNA target sequence immobilized on the membrane. The conditions of this step are critical for achieving high specificity.
Pre-hybridization and Hybridization
- Pre-hybridization (Blocking): Before adding the probe, the membrane must be incubated in a pre-hybridization buffer containing blocking agents (e.g., Denhardt's solution, salmon sperm DNA, or proprietary mixtures). This step coats the membrane to prevent the probe from binding non-specifically, thereby reducing background noise [2] [32].
- Hybridization: Replace the pre-hybridization buffer with fresh hybridization buffer containing the heat-denatured (to make it single-stranded) labeled probe. Incubate the membrane in a sealed bag or hybridization tube in a rotating hybridization oven. The use of specialized buffers like ULTRAhyb can increase sensitivity up to 100-fold and allow for shorter hybridization times (e.g., 2 hours for abundant messages) [2].
- Washing: After hybridization, a series of washes are performed to remove unbound and weakly bound probe. Washes typically start with low stringency (e.g., 2X SSC, 0.1% SDS) at room temperature and progress to higher stringency (e.g., 0.1X SSC, 0.1% SDS) at higher temperatures (e.g., 42-68°C) [7] [2]. The optimal wash stringency must be determined empirically for each probe.
Detection
The detection method depends on the label used.
- Radioactive Probes: The washed membrane is exposed to an X-ray film or, more commonly, a phosphorimager screen. The screen is then scanned, and the data is quantified using densitometry software [30].
- Non-Radioactive Probes:
- For DIG-labeled probes, the membrane is incubated with an anti-DIG antibody conjugated to Alkaline Phosphatase (AP). After washing, the membrane is incubated with a chemiluminescent AP substrate (e.g., CDP-Star). The light emitted is captured on X-ray film or a digital imaging system [7].
- For biotin-labeled probes, detection involves streptavidin conjugated to Horseradish Peroxidase (HRP), followed by a chemiluminescent substrate [12] [30].
The complete workflow from hybridization to detection is summarized below:
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Probe Labeling and Hybridization
| Item | Function/Description | Example Products / Notes |
|---|---|---|
| DIG Labeling Kit | A complete set of reagents for incorporating digoxigenin into DNA or RNA probes via PCR or in vitro transcription. | Roche DIG Labeling Kit [7] |
| T4 Polynucleotide Kinase (T4 PNK) | Enzyme used for end-labeling oligonucleotides with radioactive ³²P. | Common component of molecular biology reagent suites [30] |
| ULTRAhyb Hybridization Buffer | An optimized, proprietary buffer used during pre-hybridization and hybridization to significantly enhance sensitivity and reduce hybridization time. | Ambion ULTRAhyb Buffer [2] |
| Anti-DIG-AP Fab Fragments | Antibody conjugate that binds specifically to digoxigenin. The Alkaline Phosphatase (AP) enzyme component produces a light signal upon reaction with a substrate. | Roche Anti-digoxigenin-AP fab fragments [7] |
| CDP-Star / CSPD | A chemiluminescent substrate for Alkaline Phosphatase. Upon dephosphorylation, it emits light that can be captured on film. | Roche CDP-Star [7] |
| Positively Charged Nylon Membrane | The solid support to which RNA is transferred and immobilized. Positively charged membranes bind nucleic acids more efficiently, increasing sensitivity. | Hybond-N+; BrightStar-Plus [2] [30] |
| Size-Exclusion Purification Columns | Used to purify labeled probes from unincorporated labeled nucleotides, which is critical for achieving low background. | P-30 Columns for oligonucleotides [30] |
Within the framework of target gene expression monitoring research, Northern blotting remains a foundational technique, valued for its ability to provide direct, qualitative information on RNA size, integrity, and abundance. [33] [11] This application note details the core protocol of Northern blotting and contrasts it with emerging, ultra-sensitive RNA detection technologies that are expanding the frontiers of molecular diagnostics. While traditional Northern blotting continues to be indispensable for validating transcriptomic data, especially in academic and pharmaceutical research, recent advancements are pushing detection limits to the single-molecule level, thereby opening new avenues for clinical diagnostics and therapeutic development. [34] [35] [36]
Experimental Protocols
Detailed Northern Blotting Protocol for Gene Expression Analysis
The following protocol, adapted from a peer-reviewed bio-protocol, enables the detection of specific mRNA transcripts (e.g., ATF3, ATF4, GADD153) in cell culture models, such as during viral infection. [7]
A. RNA Extraction
- Cell Lysis and Homogenization: Culture and treat cells in 100-mm dishes. At desired time points, rinse cells with 10 mL of PBS and lyse directly in the dish using 1 mL of TRIzol reagent for 5 minutes at room temperature. [7]
- Phase Separation: Transfer the lysate to a microcentrifuge tube. Add 0.2 mL of chloroform per 1 mL of TRIzol, shake vigorously for 15 seconds, and incubate for 3 minutes at room temperature. Centrifuge at 12,000 × g for 15 minutes at 4°C. [7]
- RNA Precipitation and Wash: Transfer the upper aqueous phase to a new tube. Mix with 0.5 mL of 100% isopropanol per 1 mL of TRIzol used initially. Incubate at room temperature for 10 minutes and precipitate the RNA by centrifuging at 12,000 × g for 10 minutes at 4°C. Wash the resulting RNA pellet with 1 mL of 70% ethanol and centrifuge at 7,500 × g for 5 minutes. [7]
- RNA Dissolution and Storage: Air-dry the pellet and dissolve it in 100 µL of RNase-free water by incubating at 65°C for 15 minutes. Determine the concentration and purity using a spectrophotometer (e.g., NanoDrop) and store the RNA at -80°C. [33] [7]
B. Probe Preparation by DIG Labeling
- cDNA Synthesis: Use 2 µg of total RNA, denatured with an oligo(dT) primer at 65°C for 10 minutes. Synthesize first-strand cDNA in a 20 µL reaction containing 10 mM dNTPs, 20 units of Rnasin, reverse transcriptase buffer, and 50 units of reverse transcriptase at 43°C for 1 hour. [7]
- PCR Labeling: Amplify the target cDNA using sequence-specific primers (e.g., for ATF4: forward 5’-CCGTCCCAAACCTTACGATC-3’, reverse 5’-ACTATCCTCAACTAGGGGAC-3’). Perform the PCR in a 50 µL reaction containing PCR buffer, DIG labeling mix, primers, enzyme mix, and the cDNA template. Use the following thermocycling conditions: initial denaturation at 95°C for 2 min; followed by cycles of 95°C for 10 sec, 60°C for 30 sec, and 68°C for 30 sec. [7]
C. Gel Electrophoresis and Blotting
- Gel Preparation: Prepare a 1.3% agarose gel containing formaldehyde (e.g., using MOPS buffer) to denature the RNA and prevent secondary structure formation. [33] [7]
- RNA Separation and Staining: Load the RNA samples (typically 5-20 µg of total RNA) mixed with RNA loading buffer. Perform electrophoresis until adequate separation is achieved. Visualize the RNA bands by staining with ethidium bromide or a safer alternative under UV light to assess RNA integrity and equal loading. [33]
- Capillary Transfer: Set up a capillary transfer system using a glass plate, tissue paper stack, and a Hybond-N+ nylon membrane. Transfer the RNA from the gel to the membrane overnight via upward capillary action using 20x SSC buffer. [7] Immobilize the RNA on the membrane by crosslinking with UV light (~254 nm) or by baking at ~80°C. [33]
D. Hybridization and Detection
- Pre-hybridization: Incubate the membrane in DIG Easy Hyb buffer at the hybridization temperature for 30 minutes to 1 hour to block non-specific binding sites. [7]
- Hybridization: Add the denatured DIG-labeled DNA probe directly to the pre-hybridization buffer and incubate the membrane with the probe overnight under continuous agitation in a hybridization oven. [7]
- Stringency Washes and Signal Detection: Wash the membrane twice with 2x SSC containing 0.1% SDS at room temperature, followed by two washes with 0.1x SSC containing 0.1% SDS at 68°C. Detect the hybridized probe using an anti-digoxigenin antibody conjugated to alkaline phosphatase and a chemiluminescent substrate (e.g., CDP-Star). Expose the membrane to X-ray film or a digital imager to visualize the signal. [7]
Protocol for Ultrasensitive miRNA Detection Using Metasurface Biosensors
This protocol leverages a novel CRISPR/Cas13a-based system and metasurface technology for single-miRNA detection, representing a significant advancement beyond traditional blotting. [34] [35]
A. Sample Preparation and RT-PCR
- miRNA Reverse Transcription: Use synthesized single-strand miRNAs (e.g., hsa-miR-15a-5p) as targets. Perform reverse transcription (RT) using specific primers to generate cDNA. [35]
- PCR Amplification: Amplify the cDNA using a two-primer system and up to 45 PCR cycles to achieve the necessary amplification for single-molecule detection. Suppressing false reactions during PCR is critical for accuracy. [35]
B. Metasurface Fluorescence Detection
- Biosensor Functionalization: Immobilize cysteine-streptavidin (Cys-SA) binding molecules on the all-dielectric metasurface biosensor chip. [35]
- Amplicon Capture and Imaging: Incubate the biosensor with the PCR amplicons, which are biotinylated. The biotin-streptavidin interaction captures the amplicons on the metasurface. Employ a microfluidic system for automated liquid handling. Use LED-light excitation to induce fluorescence signals from the captured amplicons, which are significantly enhanced by the metasurface. Capture the fluorescence images with a CCD camera. [35]
The following workflow diagram illustrates the key steps and logical relationship of this advanced detection method.
The Scientist's Toolkit: Key Research Reagent Solutions
The following table catalogs essential materials and reagents used in the Northern blotting protocol, along with their critical functions in the experimental workflow. [7]
Table 1: Key Research Reagent Solutions for Northern Blotting
| Item | Function/Application | Example |
|---|---|---|
| TRIzol Reagent | A monophasic solution of phenol and guanidine isothiocyanate for the effective disruption of cells and simultaneous inactivation of RNases during total RNA isolation. [7] | Life Technologies, Cat# 15596-018 |
| DNase I (RNase-free) | Enzyme used to digest and remove contaminating genomic DNA from RNA preparations, preventing false positives in subsequent hybridization steps. [33] | Not specified in results |
| Oligo(dT) Primers | Sequences used to isolate mRNA via its poly-A tail or to prime reverse transcription for cDNA synthesis. [33] [7] | 1st Base Biochemicals |
| DIG Labeling Kit | A non-radioactive system for generating high-sensitivity DNA probes labeled with digoxigenin for hybridization and detection. [7] | Roche, Cat# 11175025910 |
| Nylon Membrane | A positively charged membrane with high nucleic acid binding affinity and robustness, used to immobilize RNA after transfer. [33] | Hybond-N+ (Amersham) |
| Anti-Digoxigenin-AP | Antibody fragment conjugated to alkaline phosphatase, which binds to the DIG-labeled probe for chemiluminescent detection. [7] | Roche, Cat# 11093274910 |
| CDP-Star | A highly sensitive chemiluminescent substrate for alkaline phosphatase. Upon dephosphorylation, it emits light, which is captured on X-ray film. [7] | Roche, Cat# 12041677001 |
Data Presentation: Market Outlook and Performance Metrics
The continued relevance of Northern blotting is reflected in its steady market growth, while the performance of emerging RNA detection technologies demonstrates a clear trajectory towards unprecedented sensitivity.
Table 2: Northern Blotting Market Overview and Forecast [11]
| Metric | Value |
|---|---|
| Market Value (2025) | USD 170.4 Million |
| Market Forecast (2035) | USD 252.3 Million |
| Forecast CAGR (2025-2035) | 4.0% |
| Leading Product Segment (2025) | Reagents (28.9% revenue share) |
| Leading Application Segment (2025) | Academic Research (41.2% revenue share) |
| Leading End-User Segment (2025) | Pharmaceutical & Biotechnology Industries (50.6% revenue share) |
Table 3: Comparative Performance of RNA Detection Techniques
| Technique | Key Feature | Reported Limit of Detection (LOD) | Key Advantage |
|---|---|---|---|
| Northern Blotting | Detects RNA size and abundance [33] | Not quantitatively specified | Qualitatively distinguishes between RNA isoforms [33] [11] |
| Standard CRISPR/Cas13a | RNA-targeting CRISPR system [34] | Baseline sensitivity | Foundation for further development [34] |
| CARRD (Novel CRISPR) | Room-temperature, anti-tag mediated [34] | 10 attomolar (10,000x more sensitive than standard Cas13a) [34] | Simplicity and sensitivity for resource-limited settings [34] |
| Metasurface Biosensor | All-dielectric nanostructured sensor [35] | Sub-attomolar (Single-molecule detection) [35] | Unprecedented sensitivity for miRNA biomarkers [35] |
| RT-qPCR | Fluorescence-based quantitative PCR [35] | ~2 femtomolar (for miRNAs) [35] | Gold standard for quantification |
| Droplet Digital PCR | Fractionation and statistical analysis [35] | 5 copies/test [35] | Absolute quantification without standards |
The landscape of RNA detection spans from the robust, time-tested Northern blot to futuristic platforms capable of single-RNA detection. Northern blotting maintains its vital role in gene expression validation, supported by a growing market and continuous reagent innovation. [11] Concurrently, emerging technologies like the CARRD test and metasurface biosensors are revolutionizing clinical diagnostics by achieving unparalleled sensitivity and operational simplicity. [34] [35] Together, these techniques provide a powerful, multi-tiered toolkit for researchers and clinicians, driving advances from basic molecular biology to personalized medicine and next-generation diagnostics. [36]
Recent Modifications for Enhanced Sensitivity and Reproducibility
Within the framework of target gene expression monitoring research, Northern blotting remains a cornerstone technique for the direct detection and analysis of specific RNA molecules. Despite the advent of high-throughput methodologies, its capacity to provide qualitative and semi-quantitative data on transcript size, integrity, and abundance ensures its continued relevance in molecular biology [31]. The technique has undergone significant refinements to address its inherent limitations, particularly concerning sensitivity and reproducibility. These modifications have transformed Northern blotting from a traditionally labor-intensive protocol to a more robust, reliable, and sensitive application essential for rigorous gene expression analysis, validation of sequencing data, and biomarker discovery in drug development [11] [31]. This article outlines the latest technical advancements, providing detailed protocols and resources to empower researchers in leveraging Northern blotting for critical research and diagnostic applications.
Market Outlook and Technical Evolution
The Northern blotting market reflects the technique's enduring value in life sciences research and diagnostics. The market is projected to grow from an estimated value of USD 170.4 million in 2025 to USD 252.3 million by 2035, registering a compound annual growth rate (CAGR) of 4.0% [11]. This growth is underpinned by the technique's vital role in gene expression studies, RNA analysis, and the expanding field of transcriptomic research, particularly in oncology, virology, and genetic disorders [11].
Table 1: Global Northern Blotting Market Forecast and Key Segments (2025-2035)
| Metric | Value / Projection |
|---|---|
| Market Value (2025) | USD 170.4 million [11] |
| Market Value (2035) | USD 252.3 million [11] |
| Forecast CAGR (2025-2035) | 4.0% [11] |
| Leading Product Segment (2025) | Reagents (28.9% revenue share) [11] |
| Dominant Application Segment (2025) | Academic Research (41.2% revenue share) [11] |
| Dominant End User (2025) | Pharmaceutical & Biotechnology Industries (50.6% revenue share) [11] |
| High-Growth Country | China (CAGR 5.4%) [11] |
The market is characterized by a rising demand for standardized, reproducible, and sensitive blotting systems. The reagents segment continues to dominate, driven by the recurring need for high-quality components in the blotting workflow [11]. Concurrently, the technical evolution of Northern blotting has been marked by a shift from radioactive detection to safer, non-radioactive alternatives like digoxigenin (DIG) and biotin-labeled probes, enhancing both safety and probe stability [31]. Furthermore, the development of optimized hybridization buffers and high-affinity membranes has significantly improved signal-to-noise ratios, pushing the boundaries of detection sensitivity [2] [31].
Key Modifications for Enhanced Performance
Advancements in Probe Technology and Hybridization
The sensitivity and specificity of Northern blotting are fundamentally dictated by the choice of probe and the efficiency of hybridization.
Locked Nucleic Acid (LNA) Probes: The incorporation of LNA nucleotides into oligonucleotide probes represents a major breakthrough, particularly for the detection of small RNAs such as microRNAs (miRNAs). LNAs possess a bridged sugar backbone that locks the structure, conferring unprecedented thermal stability upon hybridization with complementary RNA [14]. This results in a significant increase in melting temperature (Tm) by +1 to +10°C per LNA monomer compared to DNA probes [14]. This enhanced affinity translates directly into a documented 10-fold increase in sensitivity for detecting mature miRNAs, enabling the use of lower RNA quantities and the study of low-abundance transcripts [14]. LNA probes also offer superior specificity, allowing for the discrimination of single-nucleotide mismatches [14].
High-Efficiency RNA Probes: While DNA probes are common, research demonstrates that antisense RNA probes, synthesized via in vitro transcription, can provide up to a 10-fold increase in sensitivity over random-primed DNA probes [2]. RNA probes can be hybridized under more stringent conditions, reducing background noise and cross-hybridization issues [2].
Optimized Hybridization Buffers: The development of specialized, commercial hybridization buffers has dramatically improved assay performance. For instance, ULTRAhyb Ultrasensitive Hybridization Buffer can increase sensitivity up to 100-fold compared to standard formamide-based buffers [2]. These optimized solutions facilitate complete hybridization with minimal background, enabling the detection of as few as 10,000 to 100,000 target molecules on a blot and reducing required hybridization times to just 2 hours for many messages [2].
Refinements in Gel Electrophoresis and Transfer
The initial steps of Northern blotting are critical for preserving RNA integrity and ensuring efficient transfer, both of which directly impact reproducibility.
Alternative Denaturing Agents: While formaldehyde remains a common denaturant, the glyoxal/DMSO system offers several advantages. It eliminates the safety concerns associated with formaldehyde fumes, removes the need to run gels in a fume hood, and can produce sharper RNA bands, improving resolution [2]. Streamlined commercial kits based on this system have reduced sample denaturation and processing times [2].
Advanced Transfer Methodologies: The traditional passive capillary transfer, which can take overnight, is being supplanted by more efficient active transfer methods.
- Vacuum Blotting: This method uses a vacuum pump to rapidly draw transfer buffer through the gel, completing the transfer in approximately 1-2 hours [37]. This not only saves time but also produces tighter bands and enhances reproducibility by preventing diffusion of RNA fragments [2] [37].
- Rapid Alkaline Transfer: Some commercial systems incorporate a rapid, slightly alkaline downward transfer that can complete in about 2 hours, significantly shortening the protocol without compromising RNA immobilization on positively charged nylon membranes [2].
Improved Detection and Immobilization
Non-Radioactive Detection Systems: The shift from radioactive isotopes to non-radioactive labels like digoxigenin (DIG) and biotin has been a pivotal advancement [31]. These systems are safer, offer longer probe stability, and are compatible with highly sensitive chemiluminescent and fluorescent detection methods. When coupled with anti-DIG antibodies conjugated to horseradish peroxidase (HRP) and enhanced chemiluminescence (ECL) substrates, sensitivity comparable to radioactive methods can be achieved [38].
Membrane and Crosslinking Innovations: Positively charged nylon membranes are now preferred due to their high nucleic acid binding capacity and robustness through multiple washing and stripping cycles [2] [37]. UV crosslinking is the preferred method for immobilizing RNA onto these membranes, as it creates stable covalent bonds and is more reliable and consistent than baking [2] [39].
| Modification | Traditional Approach | Recent Enhancement | Impact on Performance |
|---|---|---|---|
| Probe Technology | DNA oligonucleotides, random-primed DNA | LNA-modified oligonucleotides, in vitro transcribed RNA probes | ↑↑ Sensitivity (up to 10-fold), ↑ Specificity, higher Tm [2] [14] |
| Hybridization Buffer | Standard formamide buffer | Commercial optimized buffers (e.g., ULTRAhyb) | ↑↑ Sensitivity (up to 100-fold), ↓ Background, ↓ Hybridization time [2] |
| Gel Denaturation | Formaldehyde | Glyoxal/DMSO | ↑ Safety, ↑ Resolution (sharper bands), no fume hood required [2] |
| Blotting Method | Capillary Transfer (Overnight) | Vacuum Blotting, Rapid Alkaline Transfer (1-2 hours) | ↑ Throughput, ↑ Reproducibility, ↓ Protocol time [2] [37] |
| Detection Method | Radioactive (^32P) | Non-radioactive (DIG, Biotin) with Chemiluminescence | ↑ Safety, ↑ Probe stability, comparable sensitivity [31] [38] |
| Membrane Type | Nitrocellulose | Positively Charged Nylon | ↑ RNA binding capacity, ↑ Durability for reprobing [2] [37] |
Detailed Experimental Protocol for High-Sensitivity Northern Blotting
This protocol integrates the modern modifications detailed above to achieve high sensitivity and reproducibility, suitable for detecting low-abundance mRNAs and small RNAs.
The following diagram illustrates the integrated high-sensitivity Northern blotting workflow:
Protocol Steps
Step 1: RNA Isolation and Integrity Assessment
- Procedure: Extract total RNA using a guanidium thiocyanate-phenol-based method (e.g., TRI Reagent) or a silica-membrane column kit. For mRNA studies, perform poly-A+ selection using oligo(dT) columns or beads [37]. Treat samples with DNase I to remove genomic DNA contamination [37].
- Quality Control: Quantify RNA using a spectrophotometer (NanoDrop) and assess integrity by running 100-500 ng on a denaturing agarose gel. Sharp, intact 18S and 28S ribosomal RNA bands indicate high-quality RNA [39] [37]. CRITICAL: Maintain RNase-free conditions throughout by using RNase-free reagents, gloves, and dedicated equipment [2].
Step 2: Denaturing Gel Electrophoresis
- Procedure:
- Prepare a 1-1.2% denaturing agarose gel. For the Glyoxal system, use a NorthernMax-Gly kit or prepare a gel with 1x MOPS buffer and add 1.48% formaldehyde as the denaturant [39].
- Denature 5-30 μg of total RNA (or 1-5 μg of mRNA) in glyoxal/DMSO loading dye or formaldehyde loading dye at 75°C for 10 minutes [2] [39]. Include an RNA ladder (e.g., Millennium Markers) for size determination [2].
- Perform electrophoresis at 125 V for approximately 3 hours in the appropriate running buffer (1x MOPS for formaldehyde gels; no recirculation needed for glyoxal) [2] [6].
- Visualization: Stain the gel with Ethidium Bromide or GelRed and visualize under UV light to confirm equal loading and RNA integrity before transfer [39].
Step 3 Efficient Transfer and Immobilization
- Procedure:
- Rapid Transfer: Set up a vacuum blotter or a passive, alkaline downward capillary transfer system [2]. For vacuum blotting, transfer in 10x SSC buffer at 5 inches of Hg for 90 minutes [6]. For capillary transfer, use the same buffer for a 2-hour rapid transfer or overnight standard transfer [2] [4].
- Immobilization: After transfer, disassemble the setup and UV crosslink the damp nylon membrane at 254 nm with 120-200 J/cm² to covalently attach the RNA to the membrane [39] [6]. The membrane can be used immediately or stored dry at 4°C.
Step 4: High-Sensitivity Hybridization and Detection
- Procedure:
- Pre-hybridization: Wet the membrane in SSC and place it in a hybridization bottle. Add sufficient ULTRAhyb buffer (10-15 ml for a standard membrane) to cover it. Pre-hybridize in a hybridization oven for 45 minutes at 68°C for DNA/RNA probes or 42°C for oligonucleotide probes [2] [39].
- Probe Preparation and Hybridization:
- LNA Oligonucleotide Probe: Label 10 pmol of an LNA-modified oligonucleotide with [γ-³²P]ATP using T4 Polynucleotide Kinase or use a non-radioactive 5'-end label (e.g., Biotin or DIG) [14]. Denature the radioactive probe at 95°C for 1 minute before adding.
- RNA Probe: Generate a high-specific-activity antisense RNA probe by in vitro transcription using the MAXIscript kit with [α-³²P]UTP or a non-radioactive labeled NTP [2] [39].
- Add the denatured probe directly to the prehybridization buffer and hybridize for 2 hours to overnight at the appropriate temperature (e.g., 34-45°C for LNA probes, 68°C for RNA probes) [2] [14].
- Stringency Washes: Pour off the hybridization buffer and perform washes to remove non-specifically bound probe.
- Detection:
- For radioactive probes, expose the washed and sealed membrane to a phosphor storage screen for several hours to days and visualize using a phosphorimager (e.g., Typhoon FLA) [39].
- For DIG-labeled probes, block the membrane, incubate with an HRP-conjugated anti-DIG antibody, and develop with ECL detection reagent. Capture the chemiluminescent signal using a digital imager (e.g., Image Quant LAS-4000) [38].
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of a high-sensitivity Northern blot depends on a suite of specialized reagents and kits. The following table details essential solutions for the modernized protocol.
Table 3: Key Research Reagent Solutions for Enhanced Northern Blotting
| Item | Function and Rationale | Example Products / Components |
|---|---|---|
| High-Sensitivity Hybridization Buffer | Prehybridization and hybridization solution formulated to maximize signal-to-noise ratio, enabling faster hybridization and detection of low-abundance targets. | ULTRAhyb Ultrasensitive Hybridization Buffer [2] [39] [38] |
| LNA-Modified Oligonucleotide Probes | Synthetic DNA probes incorporating Locked Nucleic Acid monomers to dramatically increase thermal stability and specificity of hybridization, crucial for miRNA and short RNA detection. | Custom LNA probes (e.g., from Exiqon) [14] |
| In Vitro Transcription Kit | For synthesizing high-specific-activity RNA probes, which offer superior sensitivity over traditional DNA probes. | MAXIscript T7 Kit [2] [39] |
| Optimized Denaturing Gel Systems | Complete kits providing RNase-free reagents for streamlined gel-based separation of RNA under denaturing conditions, with improved safety profiles. | NorthernMax-Gly Kit (Glyoxal system) [2] |
| Positively Charged Nylon Membrane | Membrane with high binding capacity for nucleic acids, essential for robust retention of RNA during hybridization and stringent washing. | BrightStar-Plus, Immobilon-Ny+, Hybond-N [2] [39] [38] |
| Non-Radioactive Labeling & Detection System | A complete system for safe and sensitive probe labeling and detection, often offering stability comparable to radioactive methods. | DIG Oligonucleotide Labeling Kit, HRP-conjugated anti-DIG antibody, ECL Detection Reagent [38] |
The collective advancements in probe chemistry, buffer systems, and transfer methodologies have profoundly enhanced the sensitivity and reproducibility of Northern blotting. The integration of LNA technology, optimized hybridization buffers, and efficient vacuum transfer moves this classical technique firmly into the modern research landscape. By adopting these detailed, application-focused protocols, researchers in drug development and academic research can confidently employ Northern blotting as a robust, reliable, and highly sensitive method for target gene expression monitoring, ensuring the acquisition of high-quality, reproducible data.
Troubleshooting Northern Blotting: Expert Strategies for Optimization
Preventing RNA Degradation and Ensuring Sample Integrity
Within gene expression studies, particularly those utilizing Northern blotting, the integrity of RNA is the foundational pillar upon which reliable data is built. RNA molecules are inherently labile, and their degradation poses a significant threat to experimental accuracy, potentially leading to erroneous quantification and misinterpretation of gene expression levels [40]. Effective prevention of RNA degradation is therefore not merely a preliminary step but a critical, continuous process that ensures sample quality from cell lysis to final detection. This document outlines standardized protocols and key considerations for maintaining RNA integrity, framed within the context of Northern blotting for target gene expression monitoring, to support researchers in obtaining robust and reproducible results.
The RNA Degradation Challenge
Ribonucleic acids (RNA) are highly susceptible to degradation by ubiquitous ribonucleases (RNases), enzymes that are remarkably stable and require no cofactors to function [29]. The challenges are particularly acute in Northern blot analysis, where even slight degradation can drastically affect data quality and quantitation, as the technique directly measures the size and abundance of intact RNA sequences [40].
The table below summarizes the major risk factors and their impact on RNA samples:
Table 1: Major Risks to RNA Integrity and Their Consequences
| Risk Factor | Impact on RNA Sample | Downstream Effect on Northern Blot |
|---|---|---|
| Ubiquitous RNases(from skin, dust, surfaces) | Strand cleavage and fragmentation. | Smearing on the gel, multiple bands, reduced signal intensity, and inaccurate size estimation. |
| Temperature Fluctuations | Accelerated chemical degradation and enzymatic activity. | Loss of full-length target mRNA, leading to poor hybridization and unreliable quantification. |
| Repeated Freeze-Thaw Cycles | Physical shearing and breakdown of RNA strands. | Degraded RNA appearing as a low molecular weight smear instead of discrete ribosomal RNA bands. |
| Metal Ions & Contaminants | Can catalyze non-enzymatic RNA hydrolysis. | General degradation, high background noise, and inconsistent blotting results. |
| Extended Processing Time | Increased exposure to potential RNases and oxidative damage. | Reduced sensitivity and failure to detect low-abundance transcripts. |
Core Principles for RNA Integrity
Adherence to the following core principles is essential for preventing RNA degradation across all stages of an experiment, from sample collection to analysis.
Rigorous RNase-Free Technique
The first line of defense is establishing an RNase-free workspace. This involves decontaminating surfaces, pipettes, and equipment with dedicated RNase decontamination solutions. Researchers must always wear gloves and use nuclease-free, disposable plasticware [41] [29]. All solutions should be prepared using RNase-free water and reagents.
Temperature Control
Maintaining low temperatures is critical for preserving RNA integrity. Cell and tissue samples should be processed on ice whenever possible, and RNA extracts should be stored at -80°C for long-term preservation [41]. Work with thawed RNA samples should be performed on ice, and the number of freeze-thaw cycles must be minimized, ideally by aliquoting RNA stocks.
Use of RNase Inhibitors
For samples inherently rich in RNases (e.g., from pancreas, lung, or spleen) or during prolonged protocols, the inclusion of RNase inhibitors in wash and resuspension buffers is strongly recommended [41]. This is also critical for preparations of single nuclei suspensions [41]. Commercial RNase inhibitors are highly effective at neutralizing a broad spectrum of RNases.
Protocols for Sample Handling and Integrity Assessment
Protocol: Preparation of a High-Quality Single Cell Suspension for RNA Analysis
This protocol is adapted for preparing cells prior to RNA extraction, ensuring a high-quality starting material [41].
- Harvesting and Washing: Collect cells and pellet them by gentle centrifugation. Discard the supernatant and wash the cell pellet with a cold, RNase-free buffer such as phosphate-buffered saline (PBS) containing 0.04% BSA.
- Resuspension: Gently resuspend the final cell pellet in an appropriate volume of cold buffer. When working with mostly dissociated cells, any pipet mixing should be performed gently with factory-made wide-bore pipet tips to minimize mechanical shearing. Avoid cutting tips to create wide-bore versions at the bench.
- RNase Inhibition: If working with RNase-rich cell types (e.g., granulocytes, pancreatic cells) or preparing nuclei, add an RNase inhibitor to the resuspension buffer.
- Debris Removal: If the suspension contains large aggregates or abundant debris, filter the sample using 40 µm Flowmi tip strainers to prevent clogging and ensure sample homogeneity.
- Viability Assessment: Assess cell viability using a dye exclusion method like Trypan Blue. Low viability can lead to high background RNA from dead cells, complicating data interpretation. If viability is low, consider a magnetic bead-based cleanup (e.g., Miltenyi’s Dead Cell Removal Kit) or flow sorting.
- Storage or Immediate Use: Process the cell suspension for RNA extraction immediately. If immediate processing is not possible, cryopreservation is an option, though significant cell death is expected upon thawing.
Protocol: RNA Integrity Check via Agarose Gel Electrophoresis
Assessing RNA integrity is a critical quality control step before proceeding to Northern blotting or other sensitive downstream applications [40].
- Gel Preparation: Prepare a denaturing agarose gel, typically containing formaldehyde, to prevent RNA secondary structure formation and ensure separation by molecular weight.
- Sample Loading: Mix a small aliquot of the isolated RNA (e.g., 100-500 ng) with an RNA loading dye. Include an RNA ladder/marker in one well to determine fragment sizes.
- Electrophoresis: Run the gel at a constant voltage in an appropriate buffer (e.g., MOPS) until the dye front has migrated sufficiently.
- Visualization: Stain the gel with a nucleic acid stain such as ethidium bromide or SYBR Safe and visualize under UV light.
- Interpretation: Intact, high-quality total RNA will display two sharp, clear bands corresponding to the 18S and 28S ribosomal RNA subunits. The ratio of intensity (28S:18S) should be approximately 2:1. A smear of RNA below these bands or a change in the ratio indicates significant degradation.
Northern Blotting: A Detailed Methodology
The Northern blot remains a foundational technique for detecting specific RNA sequences, providing direct information about transcript size and abundance [40] [29]. The protocol below details the key steps, with an emphasis on preventing degradation throughout the process.
- RNA Separation: The RNA sample is first separated by size using denaturing agarose gel electrophoresis. This step is crucial for resolving intact RNA fragments. RNA molecules can form streaks rather than distinct bands if degraded [40].
- Capillary Transfer: The separated RNA fragments are transferred from the gel onto a solid support membrane, typically made of nylon or nitrocellulose, via capillary action. The separation pattern is preserved during this transfer.
- Immobilization: The RNA is permanently fixed to the membrane to prevent washing away in subsequent steps. This is achieved by crosslinking under UV light or, for nitrocellulose membranes, by baking at 80°C for 30 minutes to 2 hours [29].
- Staining (Optional but Recommended): After crosslinking, the membrane can be stained with methylene blue to visualize the position of ribosomal RNA (rRNA). This provides a benchmark for estimating the location of the target mRNA and assessing transfer efficiency [29].
- Hybridization: The blot is incubated with a labeled, single-stranded DNA or RNA probe that is complementary to the target RNA sequence. Hybridization is typically performed in a specialized buffer (e.g., Church buffer or SSC-based buffer) at a controlled temperature [29].
- Washing and Detection: The membrane is washed with buffer (e.g., SSC) several times to remove any unbound probe. The bound, labeled probe is then detected. If a radioactive label is used, it can be visualized directly on X-ray film or with a digital imaging system. For non-radioactive probes, an enzyme-mediated colorimetric or chemiluminescent reaction is used [40] [29].
Table 2: Essential Research Reagent Solutions for Northern Blotting
| Reagent / Material | Function / Purpose | Key Considerations |
|---|---|---|
| RNase Inhibitor | Neutralizes contaminating RNases during sample prep and reactions. | Essential for RNase-rich tissues and nuclei preparations [41]. |
| Denaturing Gel(e.g., Formaldehyde-/Urea-gel) | Separates RNA by size while preventing secondary structure formation. | Critical for accurate size determination; requires a fume hood for formaldehyde [29]. |
| Nylon/Nitrocellulose Membrane | Solid support for immobilizing separated RNA for probe hybridization. | Nylon membranes are more durable for re-probing. |
| Labeled DNA/RNA Probe | Hybridizes to complementary target RNA sequence for detection. | Can be radioactive (high sensitivity) or fluorescent; requires hybridization buffer (e.g., Church buffer) [29]. |
| Hybridization Buffer(e.g., Church Buffer) | Provides optimal ionic and chemical conditions for probe-target binding. | Pre-made solutions are available to ensure consistency and save time [29]. |
| Wash Buffer(e.g., SSC Buffer) | Removes non-specifically bound probe after hybridization to reduce background. | Stringency (salt concentration and temperature) is adjusted to control specificity. |
| Methylene Blue Stain | Visualizes ribosomal RNA bands on the membrane post-transfer. | Provides a quick assessment of RNA integrity and transfer efficiency [29]. |
Optimizing Hybridization and Wash Conditions for Specificity
Northern blotting remains a cornerstone technique in molecular biology for the detection and quantification of specific RNA sequences, providing critical validation for data obtained from high-throughput transcriptomic analyses [2] [16]. Despite the emergence of more sensitive PCR-based methods, Northern blotting maintains unique advantages for target gene expression monitoring, including the ability to determine transcript size, detect alternative splicing variants, and provide direct relative quantification of message abundance across samples on a single membrane [2] [15]. The technique's reliability stems from its direct detection approach without amplification steps, thereby avoiding artifacts that can complicate data interpretation in methods like RT-PCR [16].
The specificity and sensitivity of Northern blotting are predominantly determined during the hybridization and post-hybridization wash steps, where precise conditions dictate the balance between signal intensity and background noise [2] [42]. This application note provides detailed protocols and optimization strategies to maximize detection specificity while maintaining sensitivity, with particular emphasis on stringency control for research and drug development applications.
Principles of Hybridization Specificity
The fundamental principle of Northern blotting involves the sequence-specific hybridization of labeled nucleic acid probes to target RNA molecules immobilized on a membrane [2] [15]. Successful hybridization depends on the formation of stable hydrogen bonds between complementary base pairs, a process influenced by multiple factors including temperature, ionic strength, probe composition, and base pair mismatch tolerance [42].
Stringency refers to the conditions that affect the stability of nucleic acid hybrids, with high stringency promoting only perfectly matched sequences to remain hybridized while destabilizing partial matches [42]. The key relationship between temperature, salt concentration, and stringency follows this principle: increasing temperature and decreasing salt concentration increases stringency, thereby enhancing detection specificity [42]. This occurs because higher thermal energy disrupts hydrogen bonds in mismatched hybrids, while lower salt concentrations reduce electrostatic shielding between phosphate backbones, increasing repulsion and further destabilizing imperfect matches [42].
Optimization Strategies
Hybridization Buffer Selection
The composition of hybridization buffer significantly impacts signal sensitivity and background levels. Optimal buffers contain denaturing agents (e.g., formamide), ionic strength regulators, blocking agents, and detergents [2].
Table 1: Hybridization Buffer Components and Their Functions
| Component | Function | Optimal Concentration |
|---|---|---|
| Formamide | Lowers probe-RNA annealing temperature, preventing RNA degradation | 50% in standard buffers [15] |
| SSC or SSPE | Provides ionic strength to neutralize nucleic acid repulsion | 5-6X SSPE or equivalent [14] |
| Blocking Agents | Prevent nonspecific probe binding to membrane | Denhardt's solution, salmon sperm DNA [14] |
| Detergents | Reduce background staining | SDS (0.5-1%) [14] |
Specialized commercial hybridization buffers like ULTRAhyb Ultrasensitive Hybridization Buffer can increase sensitivity up to 100-fold compared to standard formamide buffers, enabling detection of as few as 10,000 target molecules [2]. These optimized formulations push hybridization to completion without increasing background, potentially reducing required hybridization times to just 2 hours for abundant messages [2].
Probe Design and Selection
Probe characteristics profoundly influence hybridization efficiency and specificity. Northern blots can be probed with radioactively or nonisotopically labeled RNA, DNA, or oligodeoxynucleotide probes [2], each with distinct advantages:
RNA probes (riboprobes) typically offer 10-fold greater sensitivity than random-primed DNA probes and can be hybridized and washed under more stringent conditions due to their higher thermal stability, resulting in lower background and reduced cross-hybridization [2]. RNA probes are synthesized by in vitro transcription using kits such as the MAXIscript Kit [2].
DNA probes generated by asymmetric PCR demonstrate 3-5 fold greater sensitivity than random-primed probes [2]. The quickest method for obtaining high specific activity DNA probes is a 10-minute random-priming reaction using commercial labeling kits [2].
LNA-modified oligonucleotides represent a significant advancement for detecting small RNAs such as microRNAs. These bicyclic high-affinity RNA analogues exhibit unprecedented thermal stability when hybridized with their RNA targets, increasing sensitivity by at least 10-fold compared to DNA probes while maintaining high specificity [14]. The increased melting temperature (approximately +2 to +10°C per incorporated LNA monomer) allows for more stringent washing conditions, effectively discriminating single nucleotide mismatches [14].
Table 2: Probe Selection Guide for Specific Applications
| Probe Type | Best Applications | Sensitivity | Specificity |
|---|---|---|---|
| Random-primed DNA | General purpose, abundant transcripts | Moderate | Good with optimization |
| Asymmetric PCR DNA | Low to moderate abundance transcripts | High (3-5X random-primed) | Good with optimization |
| RNA probes | Low abundance transcripts, high background | Very high (10X random-primed) | Excellent |
| LNA oligonucleotides | microRNAs, small RNAs | Extreme for small targets | Exceptional (discriminates mismatches) |
Wash Stringency Optimization
Post-hybridization washes are critical for removing unbound and nonspecifically bound probes while retaining specific hybrids. Traditional protocols employ sequential low and high stringency washes for scheduled times, but modified approaches using quantitatively controlled moderate-stringency washes have demonstrated improved sensitivity for low-expression genes [16].
Table 3: Standard Wash Conditions for Different Probe Types
| Probe Type | Low Stringency Wash | High Stringency Wash | Modified Approach |
|---|---|---|---|
| Standard DNA | 2X SSC, 0.1% SDS, 34-45°C [14] | 0.1X SSC, 0.1% SDS, 65°C [14] | - |
| RNA probes | 2X SSC, 0.1% SDS, room temperature | 0.1X SSC, 0.1% SDS, 65°C or higher | - |
| LNA probes | 2X SSC, 0.1% SDS, 34-45°C [14] | 0.1X SSC, 0.1% SDS, 65°C [14] | - |
| Modified Protocol | - | - | Monitor radioactivity to 20-50 cps with moderate-stringency [16] |
The quantitatively controlled wash approach abandons the traditional sequential low and high stringency regimen in favor of moderate-stringency washes continued until radioactivity levels decrease to 20-50 counts per second, maximizing retention of specifically bound radiolabeled probes [16]. This method has proven particularly valuable when using heterologous probes with partial target sequence homology [16].
Detailed Experimental Protocols
Protocol 1: Standard Northern Hybridization with High Stringency Washes
This protocol provides a foundation for detecting moderate to high abundance transcripts with DNA or RNA probes [2] [15].
Materials and Reagents:
- ULTRAhyb Ultrasensitive Hybridization Buffer or equivalent [2]
- Nylon positively charged membrane [2]
- SSC buffer (20X stock: 3M NaCl, 0.3M sodium citrate) [7]
- SDS solution (10%)
- Labeled probe (DNA, RNA, or oligonucleotide)
Procedure:
- Prehybridization: Immerse membrane in hybridization buffer (0.1 mL/cm²) and prehybridize for 15 minutes to 2 hours at 42°C [2].
- Hybridization: Replace buffer with fresh hybridization buffer containing denatured probe. Hybridize for 2 hours to overnight at 42°C [2].
- Low Stringency Wash: Wash membrane twice for 5 minutes each with 2X SSC, 0.1% SDS at room temperature [14].
- High Stringency Wash: Wash membrane twice for 15 minutes each with 0.1X SSC, 0.1% SDS at 65°C [14].
- Detection: Proceed with appropriate detection method based on probe label (autoradiography, chemiluminescence, or fluorescence) [2] [13].
Protocol 2: Modified Northern Blot with Quantitatively Controlled Washes
This modified protocol enhances sensitivity for low-expression genes and is particularly useful with heterologous probes [16].
Materials and Reagents:
- Standard hybridization reagents as in Protocol 1
- Geiger counter or radiation monitor for radioactive probes
Procedure:
- Follow prehybridization and hybridization steps as described in Protocol 1.
- Moderate-Stringency Washes: Wash membrane with 0.5X SSC, 0.1% SDS at 55-60°C, monitoring radioactivity retained on the filter after each wash cycle [16].
- Quantitative Control: Continue washing until radioactivity levels decrease to 20-50 counts per second, indicating removal of non-specifically bound probe while retaining specific hybrids [16].
- Detection: Proceed with standard detection methods.
Protocol 3: LNA-Modified Oligonucleotide Probes for Small RNA Detection
This specialized protocol optimizes Northern blotting for microRNA and other small RNA detection using high-affinity LNA probes [14].
Materials and Reagents:
- LNA-modified oligonucleotide probes (every third nucleotide position substituted by LNA) [14]
- 12% polyacrylamide gel containing 8M urea [14]
- Nytran N membrane or equivalent
- Hybridization buffer: 50% formamide, 0.5% SDS, 5X SSPE, 5X Denhardt's solution, 20 μg/mL sheared salmon sperm DNA [14]
Procedure:
- Electrophoresis: Separate total RNA on denaturing 12% polyacrylamide gel containing 8M urea [14].
- Transfer: Transfer RNA to membrane by capillary method and fix by UV crosslinking [14].
- Prehybridization: Prehybridize filters for at least 30 minutes at hybridization temperature [14].
- Hybridization: Hybridize with 32P-end-labeled LNA probes at 34-45°C for 12-16 hours [14].
- Stringent Washes: Wash membranes at high stringency in 0.1X SSC, 0.1% SDS at 65°C twice for 5 minutes each [14].
- Detection: Detect using phosphor imaging or X-ray film [14].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for Northern Blot Optimization
| Reagent/Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Hybridization Buffers | ULTRAhyb Ultrasensitive Hybridization Buffer [2] | Increases sensitivity up to 100-fold; enables 2-hour hybridizations for many messages |
| Membranes | BrightStar-Plus positively charged nylon membranes [2] | Minimizes background and maximizes signal; compatible with alkaline transfer methods |
| Probe Labeling Systems | MAXIscript Kit (RNA probes) [2], DECAprime II Kit (DNA probes) [2] | RNA probes offer highest sensitivity; DNA probes faster to generate |
| High-Affinity Probes | LNA-modified oligonucleotides [14] | 10-fold increased sensitivity for miRNAs; exceptional mismatch discrimination |
| Detection Systems | Chemiluminescence (CDP-Star) [7], Phosphor imaging [13] | Non-radioactive alternatives with good sensitivity and linear range |
| Stringency Control | SSC buffer (20X stock) [7], SDS solutions | Precise salt concentration control for wash optimization |
Troubleshooting and Technical Notes
Assessing Hybridization Specificity
When optimizing hybridization conditions, several indicators can help assess specificity:
- Clean background with sharp bands: Indicates appropriate stringency conditions
- Multiple bands: May suggest cross-hybridization with related sequences or detection of alternative splice variants
- Smearing: Often indicates probe degradation or insufficient stringency
- Unexpected band sizes: Could result from RNA degradation or non-specific binding
Addressing Common Problems
High Background: Increase stringency by raising wash temperature or decreasing salt concentration; ensure adequate blocking during prehybridization; check probe specificity and concentration [2] [42].
Weak Signal: Reduce stringency by lowering wash temperature or increasing salt concentration; verify probe quality and labeling efficiency; extend hybridization time or increase probe concentration; consider switching to more sensitive probe type (e.g., RNA or LNA) [2] [14].
Inconsistent Results: Standardize RNA quality and quantity; ensure consistent membrane handling; use fresh hybridization and wash buffers; control temperature accurately during hybridization and washes.
Optimizing hybridization and wash conditions is paramount for achieving specific and reproducible results in Northern blot analysis. The strategic manipulation of stringency through temperature and salt concentration control enables researchers to tailor detection conditions to their specific experimental needs, from identifying closely related transcripts to detecting low-abundance messages. The implementation of modified protocols, such as quantitatively controlled washing and specialized high-affinity probes, extends the technique's utility in modern molecular research and drug development. When properly optimized, Northern blotting remains an invaluable tool for target gene expression monitoring, providing robust validation of data obtained through high-throughput methodologies.
Northern blotting remains a cornerstone technique for the detection and quantification of specific RNA molecules, providing invaluable data for target gene expression monitoring in research and drug development. Despite the emergence of high-throughput technologies, Northern blotting maintains its relevance as a gold standard for validation due to its unique ability to provide information about transcript size, integrity, and alternative splicing events [1] [16]. The technique involves separating RNA samples via denaturing gel electrophoresis, transferring them to a solid membrane support, and detecting specific sequences through hybridization with labeled probes [15]. However, researchers frequently encounter three persistent challenges that can compromise data quality: background noise that obscures specific signals, faint hybridization signals that hinder quantification, and uneven transfer that creates inconsistent results. This application note addresses these critical problems with targeted protocols and solutions to ensure the reliability of gene expression data in pharmaceutical and basic research applications.
Troubleshooting Background Noise
Background noise manifests as nonspecific signal across the membrane, reducing the signal-to-noise ratio and making specific bands difficult to identify and quantify. This problem typically originates from improper hybridization conditions, probe quality issues, or membrane handling problems [43].
Common causes and solutions include:
- Blotchy signal across the membrane: This pattern often results from poor quality membranes, dried-out membranes, or mishandling (e.g., oil from skin, powder from gloves) [43]. Always use forceps to handle membranes from the edges and ensure membranes remain properly hydrated throughout the procedure.
- Speckling across the membrane: This problem frequently stems from probe preparations with poor incorporation or particulate matter in hybridization buffers [43]. Remove unincorporated nucleotides from labeled probes through purification, and filter probe solutions through a 0.22-micron filter to remove particulates.
- Smear through the lane: This specific pattern indicates suboptimal hybridization conditions or excessive probe concentration [43]. For nonisotopic probes, use approximately 10 pM DNA probes or 0.1 nM RNA probes, and optimize hybridization temperature by starting high and gradually decreasing until specific signal is obtained.
Table 1: Troubleshooting Guide for Background Noise in Northern Blotting
| Problem Pattern | Primary Causes | Recommended Solutions |
|---|---|---|
| Blotchy background | Membrane drying out, improper handling | Use high-quality nylon membrane, handle with forceps, ensure proper hydration |
| Speckled background | Particulates in probe or buffer, poor incorporation | Filter probes and buffers (0.22 µm), purify probes to remove unincorporated nucleotides |
| Lane-specific smearing | Low hybridization stringency, high probe concentration | Optimize hybridization temperature, use recommended probe concentrations (10 pM DNA, 0.1 nM RNA) |
| General high background | Inadequate blocking, contaminated reagents | Use fresh hybridization buffers (e.g., ULTRAhyb), ensure proper blocking, use RNase-free reagents |
Protocol: Optimized Hybridization and Washing to Minimize Background
The following modified protocol implements controlled posthybridization washes to maximize signal-to-noise ratio while retaining specific signals [16]:
Prehybridization: Incubate membrane in ULTRAhyb Ultrasensitive Hybridization Buffer for 15-30 minutes at 68°C [2]. This blocking step is critical for preventing nonspecific probe binding.
Hybridization: Add labeled probe directly to fresh hybridization buffer (do not pipet directly onto membrane). For DNA probes, use approximately 10 pM concentration; for RNA probes, use 0.1 nM concentration [43]. Hybridize for 2 hours to overnight at 68°C with gentle agitation.
Modified Posthybridization Washes [16]:
- Perform initial rinse at room temperature with 2× SSC/0.1% SDS to remove excess probe.
- Wash with moderate-stringency buffer (e.g., 0.2× SSC/0.1% SDS) at 42°C.
- Monitor radioactivity on the membrane during washing until levels decrease to 20-50 counts per second.
- Avoid prolonged washing under high-stringency conditions that can strip specific signals.
Detection: Proceed with appropriate detection method based on probe label (X-ray film, phosphorimager, or fluorescent imaging).
Enhancing Faint Signals
Strategies for Improving Detection Sensitivity
Faint signals present a significant challenge for detecting low-abundance transcripts, particularly in drug development research where quantitative accuracy is paramount. Sensitivity issues typically stem from suboptimal probe design, inefficient hybridization, or inadequate detection systems.
Key considerations for signal enhancement:
Probe Selection and Labeling: RNA probes (riboprobes) generally provide 10-fold greater sensitivity than DNA probes due to stronger hybridization and ability to withstand more rigorous washing [2]. For maximum sensitivity, use asymmetric PCR-generated DNA probes or in vitro transcribed RNA probes with high specific activity (>10⁹ cpm/µg for radioactive probes) [2]. Near-infrared fluorescent probes (irNorthern) offer sensitivity of approximately 0.05 fmol with additional benefits of probe stability and multiplexing capability [44].
Hybridization Buffers: Specialized buffers such as ULTRAhyb Ultrasensitive Hybridization Buffer can enhance hybridization signals 10-100 fold compared to standard formamide buffers by accelerating hybridization kinetics and reducing background [2].
Sample Enrichment: For low-abundance transcripts, use poly(A) selected RNA instead of total RNA to enrich mRNA species 30-100 fold, resulting in a similar increase in signal [43]. Alternatively, increase the amount of total RNA loaded per lane (up to 30 µg for standard NorthernMax protocols) [2].
Table 2: Comparison of Detection Methods for Northern Blotting
| Detection Method | Sensitivity | Advantages | Limitations |
|---|---|---|---|
| ³²P Radioactive | 0.005-0.01 fmol [44] | Highest sensitivity, well-established protocols | Safety concerns, short probe half-life (~14 days), regulatory requirements |
| Chemiluminescent | Varies by substrate | Improved safety, good sensitivity | Signal dependent on enzyme activity, less quantitative |
| Near-IR Fluorescent (irNorthern) | ~0.05 fmol [44] | Probe stability, multiplexing capability, quantitative | Slightly less sensitive than radioactive methods |
| Colorimetric | Lowest | Simple, no special equipment required | Least sensitive, not quantitative |
Protocol: High-Sensitivity Northern Blotting with irNorthern Probes
The near-infrared fluorescent Northern blotting (irNorthern) method provides a sensitive, stable alternative to radioactive detection with additional multiplexing capabilities [44]:
Probe Labeling:
- Synthesize ssDNA probe with azide-modified thymidine complementary to target sequence.
- Label using copper-free click chemistry to attach IRDye 800CW DBCO to the probe.
- Purify labeled probe and dilute to 1 nM in hybridization solution.
Electrophoresis and Transfer:
Hybridization and Detection:
- Prehybridize membrane in appropriate buffer for 15-30 minutes.
- Hybridize with IR dye-labeled probe (1 nM) for 2 hours at 68°C.
- Wash under moderate-stringency conditions.
- Detect using near-infrared imaging system with appropriate excitation/emission filters.
Resolving Uneven Transfer
Addressing Transfer Inconsistencies
Uneven transfer results in variable signal intensity across the membrane, compromising quantitative accuracy and making lane-to-lane comparisons unreliable. This problem typically stems from improper transfer setup, gel irregularities, or inefficient RNA migration from gel to membrane.
Common transfer problems and solutions:
- Poorly fitting transfer sandwich: Ensure the transfer stack is tight by using extra sponges if needed and appropriately thick filter paper [45]. All sandwich components must be clean, properly hydrated, and free from bubbles.
- Inefficient transfer of large RNAs: Large RNA species (>4 kb) may not transfer well due to their size [43]. Using a basic transfer buffer (e.g., NorthernMax One-Hour Transfer Buffer) will partially shear the RNA, facilitating more efficient transfer of larger transcripts.
- Incomplete transfer verification: Check transfer efficiency by including ethidium bromide in RNA samples or staining the gel after transfer [43]. RNA markers (e.g., Millennium Markers) are invaluable for demonstrating whether large RNAs have fully transferred.
Protocol: Optimized Capillary Transfer for Uniform Results
This protocol incorporates modifications to ensure consistent, uniform transfer of RNA species of all sizes:
Gel Preparation:
Transfer Setup:
- Create a capillary transfer system with strips of Parafilm around the outside edges of the gel to prevent short-circuiting [43].
- Use a slightly alkaline, downward elution method for faster transfer and tighter bands [2].
- Ensure all components (sponges, filter papers, membrane) are properly saturated with transfer buffer and free of bubbles.
Transfer Monitoring:
- Transfer for 1-2 hours using rapid transfer systems or overnight for traditional capillary transfer.
- After transfer, stain the gel with ethidium bromide to verify complete RNA transfer, particularly for larger RNA species.
- Crosslink RNA to membrane immediately after transfer using UV light or baking.
Integrated Workflow and Visualization
Comprehensive Northern Blotting Workflow
The following diagram illustrates the complete Northern blotting procedure with key decision points for troubleshooting common problems:
Troubleshooting Decision Pathway
When problems occur in Northern blotting, follow this systematic troubleshooting approach to identify and resolve issues:
Research Reagent Solutions
Table 3: Essential Reagents for Optimized Northern Blotting
| Reagent Category | Specific Products/Components | Function and Application Notes |
|---|---|---|
| Membranes | Positively charged nylon membrane | Optimal for RNA retention; higher binding capacity than nitrocellulose [2] |
| Hybridization Buffers | ULTRAhyb Ultrasensitive Hybridization Buffer | Enhances sensitivity 10-100×; reduces background; compatible with DNA/RNA probes [2] |
| Probe Labeling Systems | ³²P-dCTP (radioactive), IRDye 800CW DBCO (near-IR), Digoxigenin (DIG) | Choice depends on sensitivity requirements and safety considerations [44] [15] |
| Transfer Buffers | NorthernMax One-Hour Transfer Buffer | Alkaline buffer for rapid, efficient transfer; particularly beneficial for large RNAs [2] |
| RNA Size Markers | Millennium Markers (0.5-9 kb transcripts) | Accurate RNA sizing; detectable by ethidium bromide, radioactive, or nonradioactive probing [2] |
| Detection Substrates | Chemiluminescent substrates (HRP-based), Near-infrared fluorescent dyes | Signal generation for visualization and quantification [44] [46] |
Effective troubleshooting of background noise, faint signals, and uneven transfer in Northern blotting requires a systematic approach that addresses both technical execution and reagent selection. The protocols and solutions presented here provide researchers with targeted strategies to overcome these common challenges, thereby enhancing the reliability of gene expression data in target validation and drug development research. By implementing optimized hybridization conditions, selecting appropriate detection methods, and ensuring consistent transfer efficiency, scientists can maintain Northern blotting as a robust, quantitative tool for transcriptional analysis. The continued refinement of these methodologies ensures that Northern blotting remains relevant alongside newer genomic technologies, providing critical validation through its unique capacity to directly visualize RNA species without amplification biases.
Northern blotting remains a cornerstone technique for the specific detection and analysis of RNA within molecular biology research, particularly for studying gene expression patterns in various biological contexts. Despite the emergence of newer technologies like RT-PCR and microarrays, Northern blotting maintains critical advantages for quantitative analysis, including the ability to determine transcript size, detect alternative splicing variants, and provide direct visual confirmation of RNA integrity [47] [15]. For researchers and drug development professionals, these attributes are indispensable when validating gene expression data obtained from high-throughput screening methods or when characterizing novel RNA species.
The foundation of Northern blotting involves separating RNA samples via denaturing gel electrophoresis, transferring them to a solid membrane support, and hybridizing with labeled sequence-specific probes for detection [6] [13]. While traditionally considered a semi-quantitative method, advancements in probe technology, detection sensitivity, and normalization strategies have firmly established Northern blotting as a robust quantitative technique. Furthermore, the inherent capability of membranes to be stripped and re-probed for multiple targets makes Northern blotting exceptionally efficient for comprehensive gene expression studies, especially when working with a limited number of target genes or when confirming specific genetic pathways [47] [15]. This application note details advanced protocols and best practices to maximize the quantitative reliability and multiplexing potential of Northern blotting within a rigorous research framework.
Quantitative Data Analysis in Northern Blotting
Methodological Comparison for Quantitative Expression Analysis
The quantitative capability of Northern blotting is often validated through comparisons with other established methodologies. When performed with careful normalization and appropriate controls, Northern blotting demonstrates strong correlation with other quantitative techniques, though some variance is expected due to fundamental methodological differences.
Table 1: Comparison of Methodological Outcomes for Differential Gene Expression Analysis
| Gene Name | Northern Blot Ratio (PC3/PC3-M) | Microarray Ratio (PC3/PC3-M) | qRT-PCR Ratio (PC3/PC3-M) |
|---|---|---|---|
| Vimentin | 2.10 | 2.25 | 2.05 |
| Laminin Receptor 1 | 0.45 | 0.52 | 0.41 |
| Macrophage Migration Inhibitory Factor | 3.80 | 3.20 | 4.50 |
| Eukaryotic Translation Initiation Factor | 0.32 | 0.28 | 0.25 |
Data adapted from a study comparing differential gene expression between PC3 and PC3-M prostate cancer cell lines [48]. The expression ratios demonstrate general concordance across methods, though with notable outliers for certain genes.
Statistical analysis of such comparisons reveals important insights. One study found that correlation coefficients between Northern blotting and microarray data reached 0.72, while correlation between Northern blotting and qRT-PCR was 0.39. After excluding outlier genes, these correlations improved to 0.79 and 0.72, respectively [48]. This underscores that while Northern blotting generally provides reliable quantitative data, gene-specific effects can influence agreement between methods, potentially due to sequence-dependent variations in enzymatic steps required for microarray and qRT-PCR.
Key Considerations for Robust Quantitation
To ensure the quantitative accuracy of Northern blot data, researchers must address several critical experimental factors:
RNA Integrity and Quantification: The quality of input RNA is paramount. Even slight degradation can severely compromise quantitative accuracy, as a single cleavage in a fraction of target molecules will proportionally reduce the detected signal [47]. RNA integrity must be verified prior to blotting, typically by visual assessment of ribosomal RNA bands on an ethidium bromide-stained gel [15].
Probe Selection and Labeling: Probe design significantly impacts sensitivity and specificity. Radiolabeled probes (e.g., with ³²P) offer high sensitivity and are ideal for low-abundance transcripts [13]. Non-radioactive alternatives, such as those labeled with digoxigenin (DIG) or biotin, provide excellent safety and stability benefits while achieving sensitivity comparable to radioactive methods in optimized protocols [3] [13]. RNA probes (riboprobes) often provide greater sensitivity than DNA probes due to the formation of more stable RNA-RNA hybrids [47].
Signal Detection and Densitometry: Modern imaging systems, including phosphor imagers for radioactive detection and chemiluminescence or fluorescence imagers for non-radioactive detection, enable precise quantitation of band intensity [13]. The linear range of the detection system must be established to ensure that signals are accurately proportional to RNA abundance.
Normalization Strategies
Accurate normalization is the cornerstone of reliable quantitative Northern blot analysis. Common approaches include:
Housekeeping Genes: Traditional normalization relies on constitutively expressed "housekeeping" genes (e.g., GAPDH, actin). However, their expression can vary under experimental conditions, leading to misinterpretation [49]. It is crucial to validate the stability of reference genes for each specific experimental system.
Total RNA Staining: Staining the membrane with methylene blue or using ethidium bromide-stained ribosomal RNA bands from the gel can serve as a loading control. This method assumes equal loading and transfer efficiency but does not account for variations in RNA quality or integrity.
Multiprobe Normalization: For the highest quantitative accuracy, using a combination of multiple stable reference genes is recommended. Advanced algorithms, such as GrayNorm, can determine the optimal set of reference genes for a specific experimental context, thereby maximizing data accuracy [49]. This approach minimizes the risk of error introduced by fluctuating expression of any single reference gene.
Membrane Re-probing Protocols and Best Practices
The ability to sequentially probe a single Northern blot membrane for multiple targets dramatically enhances experimental efficiency, conserves precious RNA samples, and allows for direct internal comparison of different transcript levels. Success hinges on effective stripping protocols that remove bound probe without damaging the immobilized RNA.
Standard Re-probing Workflow
A generalized workflow for membrane re-probing involves several critical stages, from initial hybridization through to subsequent detections.
Detailed Stripping and Re-probing Protocol
Materials:
- Stripping buffer: 0.1% SDS, 5 mM Tris-HCl (pH 7.5), 0.1× SSC [47]
- Alternatively: 50% Formamide, 50 mM Tris-HCl (pH 8.0), 1% SDS [3]
- Hybridization oven or shaking water bath
- Standard hybridization buffers and detection reagents
Procedure:
Initial Probe Removal: After detection and documentation, pour off the detection reagents and wash the membrane briefly in distilled water. Incubate the membrane in a large volume of prewarmed stripping buffer (0.1% SDS, 5 mM Tris-HCl, 0.1× SSC) at 75-80°C for 30-60 minutes with constant agitation [47]. The use of formamide in stripping buffer can enhance probe removal efficiency, especially for strongly hybridized probes [3].
Efficiency Verification: Thoroughly rinse the membrane in wash buffer (e.g., 2× SSC) at room temperature. Re-expose the membrane to X-ray film (for radioactive probes) or subject it to the detection protocol (for chemiluminescence) to verify complete removal of the previous signal. This critical step ensures that subsequent signals originate exclusively from the new probe. If signal persists, repeat the stripping process.
Membrane Storage: Once stripped successfully, the damp membrane can be sealed in plastic wrap and stored at -20°C for future use, or it can proceed directly to the next hybridization.
Re-hybridization: Begin the standard hybridization protocol anew with the next labeled probe. Prehybridization is recommended before adding the new probe to block non-specific binding sites. With high-quality nylon membranes, this process can typically be repeated 3-5 times without significant loss of the target RNA [15].
Technical Considerations for Successful Re-probing
Membrane Choice: Positively charged nylon membranes are essential for re-probing experiments due to their superior nucleic acid binding capacity and mechanical durability through multiple processing cycles [47] [13]. Nitrocellulose membranes are less suitable as they become brittle and retain probes less efficiently.
Probe Design and Labeling: For experiments planned with multiple re-probing cycles, consider using probes with varying detection modalities (e.g., chemiluminescence for the first probe, fluorescence for the second) to eliminate cross-reactivity and simplify the stripping process [13].
Signal Fidelity: Always verify that the signal intensity for stable reference genes remains consistent across re-probing cycles. A significant drop may indicate RNA degradation or loss from the membrane, compromising quantitative comparisons.
Troubleshooting Common Challenges
Even with optimized protocols, researchers may encounter specific challenges during quantitative analysis and re-probing.
Table 2: Troubleshooting Guide for Quantitative Analysis and Re-probing
| Problem | Potential Cause | Solution |
|---|---|---|
| High background after stripping | Incomplete removal of previous probe | Increase stripping temperature or duration; use formamide-containing stripping buffer [3]. |
| Weak signal upon re-probing | RNA degradation or loss from membrane | Avoid excessive heating or overly harsh stripping conditions; ensure membrane does not dry out between probings. |
| Inconsistent reference gene signals | Reference gene instability or uneven loading | Validate reference genes under specific experimental conditions; use multiple reference genes or total RNA staining for normalization [49]. |
| Poor correlation with qRT-PCR data | Method-specific biases or differences in dynamic range | Verify RNA quality; ensure probes are specific and detection is within the linear range; consider inherent methodological differences [48]. |
| Fading signal over multiple re-probes | Cumulative RNA loss from membrane | Limit the number of re-probing cycles (optimally 3-5); detect low-abundance targets first [15]. |
The Scientist's Toolkit: Essential Reagents and Equipment
Successful implementation of advanced Northern blotting requires specific high-quality reagents and instruments.
Table 3: Research Reagent Solutions for Advanced Northern Blotting
| Item | Function/Purpose | Examples/Notes |
|---|---|---|
| Positively Charged Nylon Membrane | Solid support for RNA immobilization after transfer. Essential for multiple re-probing. | BrightStar-Plus membranes; compatible with alkaline transfer [47]. |
| Non-Radioactive Labeling Systems | Probe labeling for detection; safer alternative to radioactivity. | Biotin- or DIG-labeled nucleotides; detected via chemiluminescence [3] [13]. |
| High-Sensitivity Hybridization Buffers | Increases signal-to-noise ratio during hybridization, enhancing detection sensitivity. | ULTRAhyb Ultrasensitive Hybridization Buffer can increase sensitivity up to 100-fold [47]. |
| Stripping Buffer | Removes hybridized probes from the membrane between hybridizations. | Typically contains SDS and a denaturant like formamide; used at high temperature [3] [47]. |
| Versatile Imaging Systems | Detection and quantitation of signals from various labels (radioactive, chemiluminescent, fluorescent). | Azure Sapphire FL Imager; capable of phosphor, chemiluminescence, and fluorescence imaging [3] [13]. |
| RNA Markers | Accurate size determination of target transcripts on the blot. | Millennium RNA Markers; evenly spaced, single-stranded RNA transcripts [47]. |
Northern blotting continues to be an invaluable technique for precise gene expression analysis, particularly when quantitative accuracy and validation of transcript identity are required. By implementing the advanced protocols outlined for quantitative normalization and membrane re-probing, researchers can significantly enhance the reliability, efficiency, and informational yield of their experiments. The integration of modern probe technologies, sensitive detection platforms, and robust normalization strategies ensures that Northern blotting remains a powerful tool in the molecular biologist's arsenal, capable of providing critical insights into gene regulation mechanisms and supporting drug development pipelines.
Critical Controls for Reliable and Interpretable Results
Northern blotting remains a foundational technique in molecular biology for the detection and analysis of specific RNA molecules within complex samples. Despite the emergence of newer technologies like RT-PCR and RNA-Seq, Northern analysis provides unique advantages for gene expression studies, including the ability to determine transcript size, detect alternatively spliced variants, and validate RNA integrity [2] [50]. The technique involves separating RNA samples by size using denaturing gel electrophoresis, transferring them to a solid membrane support, and detecting specific sequences through hybridization with complementary labeled probes [15]. Within drug development and basic research, Northern blotting serves as a critical tool for validating gene expression patterns in response to therapeutic compounds, pathological conditions, or developmental cues [15] [51]. However, the reliability and interpretability of Northern blot results depend heavily on implementing appropriate controls throughout the experimental process. This application note details the essential controls and optimized protocols necessary for generating robust, reproducible Northern blot data in target gene expression monitoring research.
The Critical Role of Controls in Northern Analysis
Effective experimental controls are fundamental to distinguishing specific biological signals from technical artifacts in Northern blotting. The technique's versatility in assessing RNA abundance, size, and integrity also introduces multiple potential sources of variability that must be controlled [50]. Without proper controls, results can be compromised by RNA degradation, inefficient transfer, non-specific hybridization, or unequal loading. These issues are particularly problematic in quantitative comparisons between samples, such as assessing differential gene expression in diseased versus normal tissues or evaluating drug responses over time [15] [3]. Furthermore, the emergence of non-radioactive detection methods and applications for small RNA analysis has introduced additional considerations for control implementation [3] [52]. This section outlines the core control strategies that safeguard experimental integrity at each stage of the Northern blotting process, ensuring that observed signal variations genuinely reflect biological phenomena rather than technical inconsistencies.
Essential Control Strategies and Their Implementation
RNA Quality and Integrity Assessment
The foundation of any successful Northern blot experiment is high-quality, intact RNA. Even minimal degradation can severely compromise data quality and quantitation, as a single cleavage in 20% of a 4 kb target molecule will decrease the detected signal by 20% [2]. Ribosomal RNA bands serve as intrinsic quality markers and should appear as sharp bands on an ethidium bromide-stained gel, with the 28S rRNA band approximately twice the intensity of the 18S rRNA band in total RNA samples [15]. This assessment should be performed prior to blotting to avoid wasting resources on compromised samples. Additionally, measuring RNA concentration and purity via spectrophotometry (e.g., NanoDrop) provides quantitative data and ensures optimal loading amounts [7]. Working with RNA requires rigorous RNase-free techniques, using dedicated reagents and inhibitors such as DEPC to prevent sample degradation throughout the isolation and analysis process [15].
Loading and Transfer Controls
Accurate interpretation of gene expression changes requires normalization for variations in sample loading and transfer efficiency. Housekeeping genes with consistent expression across experimental conditions, such as GAPDH, β-actin, or 18S rRNA, are routinely used as loading controls [50]. Their constant expression level confirms that equivalent amounts of RNA were loaded in each lane and that observed differences in target gene signal reflect genuine expression changes rather than loading artifacts. To verify complete transfer of RNA from the gel to the membrane, the gel can be stained with ethidium bromide after transfer to confirm the absence of residual RNA [15]. Alternatively, visualizing ribosomal RNA bands on the membrane through methylene blue or SYBR Green staining provides direct evidence of successful transfer and equal loading across all lanes [2]. For precise quantitation, specially designed RNA ladders (e.g., Millennium Markers) should be run alongside experimental samples to enable accurate size determination of detected transcripts [2].
Hybridization Specificity and Signal Detection Controls
Specificity controls are essential for verifying that hybridization signals derive from the target RNA rather than non-specific probe binding. Including a negative control sample known to lack the target RNA (e.g., vehicle-treated cells or non-expressing tissues) identifies non-specific hybridization or background signals [15]. For probe validation, both sense RNA probes and no-probe controls can distinguish specific from non-specific hybridization [2]. When reprobing membranes for multiple targets, complete stripping of previous probes must be verified by exposing the membrane to film prior to adding the next probe [2]. For quantitative comparisons, dilution series of in vitro transcribed target RNA or synthetic oligonucleotides can generate standard curves for signal quantitation [3]. The sensitivity of detection systems must also be calibrated using known amounts of target RNA, particularly when employing non-radioactive detection methods such as chemiluminescence or digoxigenin labeling [3] [52].
Table 1: Essential Controls for Northern Blot Experiments
| Control Category | Specific Control | Purpose | Interpretation Criteria |
|---|---|---|---|
| RNA Quality | rRNA integrity on denaturing gel | Assess RNA degradation | Sharp 28S and 18S bands with 2:1 intensity ratio |
| Spectrophotometric analysis (A260/A280) | Determine RNA purity and concentration | A260/A280 ratio of ~2.0 indicates pure RNA | |
| Loading & Transfer | Housekeeping genes (GAPDH, β-actin, 18S rRNA) | Normalize for loading variations | Consistent signal across samples |
| Post-transfer gel staining | Confirm complete RNA transfer | Absence of residual RNA in gel | |
| RNA size markers | Determine transcript size | Accurate sizing of unknown transcripts | |
| Hybridization & Detection | Negative control sample (target absent) | Detect non-specific hybridization | No signal in negative control lane |
| No-probe control | Assess background from detection system | No signal in absence of probe | |
| Standard curve with known RNA amounts | Enable signal quantitation | Linear range of detection established | |
| Stripping verification when reprobing | Confirm probe removal before reprobing | No residual signal before adding new probe |
Specialized Controls for Small RNA Detection
The analysis of small RNAs such as microRNAs presents unique challenges that require specialized control strategies. Their small size (20-25 nt for mature miRNAs) and low abundance demand enhanced sensitivity, often achieved through optimized crosslinking methods [3]. Chemical crosslinking with 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide (EDC) has been shown to improve small RNA retention on membranes compared to conventional UV crosslinking [3] [52]. Specificity concerns are amplified with small RNAs, making appropriate probe design critical. Locked nucleic acid (LNA)-modified oligonucleotide probes significantly improve thermal stability and hybridization specificity for miRNA detection [52]. Given the multiple processing stages of small RNAs (primary, precursor, and mature forms), it is essential to include controls that distinguish between these different species, typically achieved through careful probe design and size determination using appropriate markers [3].
Comprehensive Northern Blot Protocol with Integrated Controls
RNA Isolation and Quality Control
Begin with extraction of total RNA using TRIzol reagent or specialized kits, maintaining RNase-free conditions throughout [7] [3]. Quantify RNA concentration and purity using a spectrophotometer, expecting A260/A280 ratios of approximately 2.0 for pure RNA [7]. Assess RNA integrity by running 1-2 µg of total RNA on a denaturing formaldehyde-agarose gel containing ethidium bromide. Under UV illumination, intact RNA should display sharp ribosomal bands with the 28S band approximately twice as intense as the 18S band [15]. This critical quality control step prevents proceeding with degraded samples that would yield unreliable results.
Gel Electrophoresis and RNA Transfer
Prepare a 1.3% formaldehyde-agarose denaturing gel using 1× MOPS buffer [7]. Denature RNA samples (typically 5-30 µg total RNA) in formamide-based loading buffer at 65°C for 10-15 minutes, then chill on ice. Include an RNA ladder in at least one lane for size determination. Run the gel at 5-6 V/cm until the dye front has migrated sufficiently (typically 2-3 hours) [2]. Following electrophoresis, transfer RNA to a positively charged nylon membrane using capillary, vacuum, or electrophoretic transfer methods. For optimal results with the capillary method, use a downward alkaline transfer system for 2 hours [2]. After transfer, immobilize RNA on the membrane using UV crosslinking (120 mJ/cm²) or baking at 80°C for 30-60 minutes [2]. To verify complete transfer and equal loading, stain the membrane with methylene blue or SYBR Green to visualize ribosomal RNA bands before proceeding with hybridization.
Probe Preparation and Hybridization
Generate specific probes for both target and housekeeping genes using RT-PCR with digoxigenin (DIG) labeling or alternative labeling methods [7]. For a 50 µL DIG-labeling reaction, combine: 32.25 µL ddH₂O, 5 µL PCR buffer, 5 µL PCR DIG labeling mix, 5 µL forward primer (10 pmol/µL), 5 µL reverse primer (10 pmol/µL), 0.75 µL enzyme mix, and 2 µL template cDNA [7]. Amplify using appropriate cycling conditions. Denature double-stranded DNA probes before hybridization. Prehybridize the membrane with DIG Easy Hyb buffer at the appropriate hybridization temperature (typically 42-68°C) for 30-60 minutes [7]. Replace with fresh hybridization buffer containing the denatured DIG-labeled probe (20-25 ng/mL) and incubate overnight at the appropriate temperature [7].
Washing, Detection, and Analysis
After hybridization, perform stringency washes to remove non-specifically bound probe: twice with 2× SSC, 0.1% SDS at room temperature for 5 minutes, followed by two washes with 0.1× SSC, 0.1% SDS at 68°C for 15 minutes each [7]. Detect hybridized probes using anti-digoxigenin-AP Fab fragments and CDP-Star chemiluminescent substrate according to manufacturer instructions [7]. Expose the membrane to X-ray film or capture images using a digital imaging system. For quantitative analysis, measure band intensities using densitometry software and normalize target gene signals to housekeeping gene controls. Include positive and negative control samples in each experiment to verify probe specificity and monitor background signals.
Table 2: Troubleshooting Common Northern Blot Issues
| Problem | Potential Causes | Solutions | Preventive Controls |
|---|---|---|---|
| High Background | Incomplete washing, insufficient blocking, probe concentration too high | Increase stringency of washes, optimize blocking conditions, dilute probe | Include no-probe control, test wash stringency |
| Faint or No Signal | RNA degradation, inefficient transfer, low probe sensitivity, insufficient exposure | Verify RNA quality, check transfer efficiency, use high-sensitivity probes (e.g., RNA probes), increase exposure time | Assess RNA integrity pre-blot, verify transfer with membrane staining, include positive control |
| Uneven Signal | Uneven transfer, air bubbles during transfer or hybridization, uneven membrane | Ensure even weight distribution in capillary transfer, remove air bubbles carefully, use fresh hybridization buffer | Visualize membrane after transfer, ensure proper setup |
| Multiple Bands | Alternative splicing, cross-hybridization with related sequences, RNA degradation | Increase hybridization stringency, design gene-specific probes, verify RNA quality | Include size markers, test probe specificity |
| Inconsistent Housekeeping Signal | True biological variation, inappropriate control gene, uneven loading | Validate housekeeping gene stability in your system, verify equal loading with additional method | Use multiple housekeeping genes, visualize rRNA |
Advanced Applications and Methodological Variations
Reverse Northern Blotting
The reverse Northern blot technique represents a significant methodological variation where the substrate nucleic acid affixed to the membrane is a collection of isolated DNA fragments, and the probe is RNA extracted from tissue and radioactively labeled [15]. This approach enables parallel screening of multiple genes and has evolved into gene expression profiling using DNA microarrays, where thousands of isolated DNA fragments are affixed to a substrate and hybridized with probes made from cellular RNA [15]. While this high-throughput method sacrifices size information for scale, it maintains the hybridization principles of traditional Northern analysis while dramatically increasing the number of genes that can be analyzed simultaneously.
Liquid Hybridization Assay
Recent advancements have introduced liquid hybridization (LH) as a faster, more sensitive alternative to conventional Northern blotting, particularly for detecting small RNAs and miRNAs [3]. In this protocol, RNA is hybridized with non-radioactive biotinylated probes in solution before electrophoresis, followed by visualization using chemiluminescent detection [3]. This method demonstrates sensitivity comparable to radiolabeled probes while using less than 10-100 times the total amount of RNA required for standard Northern blots [3]. The liquid hybridization approach maintains the specificity of Northern analysis while offering advantages in speed, cost-effectiveness, and safety through elimination of radioactive materials.
Small RNA Northern Blotting
The detection of small RNAs such as microRNAs requires specific modifications to standard Northern protocols. These include fractionation of RNA on denaturing polyacrylamide gels (rather than agarose), electrophoretic transfer to membranes, and specialized crosslinking methods using EDC to improve retention of small RNA molecules [3]. Locked nucleic acid (LNA)-modified oligonucleotide probes significantly enhance sensitivity and specificity for miRNA detection, sometimes improving sensitivity by 10-fold compared to traditional DNA probes [52]. These specialized protocols enable researchers to distinguish between primary, precursor, and mature forms of small RNAs, providing crucial insights into RNA processing and maturation [3].
Experimental Workflow and Visualization
The Northern blotting procedure involves multiple coordinated steps where controls ensure interpretable results. The following diagram summarizes the complete workflow with critical control points:
Diagram Title: Northern Blot Workflow with Control Points
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for Northern Blot Analysis
| Reagent/Category | Specific Examples | Function | Technical Notes |
|---|---|---|---|
| RNA Isolation | TRIzol reagent, guanidinium isothiocyanate | Cellular lysis, RNase inhibition, RNA recovery | Maintain RNase-free conditions; use DEPC-treated water [7] [3] |
| Gel Electrophoresis | Formaldehyde, agarose, MOPS buffer, glyoxal/DMSO | RNA denaturation and size separation | Formaldehyde requires fume hood; glyoxal system is safer alternative [2] |
| Membrane Transfer | Positively charged nylon membrane, transfer buffer | RNA immobilization for hybridization | Nitrocellulose incompatible with alkaline transfer buffers [2] |
| Probe Labeling | DIG labeling kit, radioactive 32P, biotin | Generate detection probes | DIG and biotin eliminate radioactivity hazards [7] [3] |
| Hybridization | ULTRAhyb buffer, DIG Easy Hyb, formamide | Enable specific probe-target binding | ULTRAhyb increases sensitivity up to 100-fold [2] |
| Detection | CDP-Star, anti-digoxigenin-AP, X-ray film | Visualize hybridized probes | Chemiluminescent methods faster than radioactive detection [7] |
| Size Standards | RNA ladders, Millennium Markers | Determine transcript size | Ribosomal RNA subunits can serve as endogenous size markers [15] [2] |
Implementing comprehensive control strategies throughout the Northern blotting process is essential for generating reliable and interpretable gene expression data. From initial RNA quality assessment to final signal detection, each step presents opportunities for technical variability that can compromise experimental conclusions. The controls detailed in this application note—including RNA integrity verification, appropriate normalization with housekeeping genes, hybridization specificity checks, and detection system validation—provide a framework for robust Northern analysis. As methodological variations continue to emerge, including liquid hybridization and small RNA detection protocols, the fundamental principle remains unchanged: rigorous controls transform Northern blotting from a simple descriptive technique into a powerful quantitative tool for gene expression analysis. By adhering to these standardized protocols and control measures, researchers can confidently apply Northern blotting to investigate gene regulation in development, disease, and therapeutic contexts.
Northern Blotting vs. Modern Techniques: Validation and Niche Applications
Northern blot analysis remains a cornerstone technique for gene expression validation, offering unparalleled reliability in the verification of data obtained from high-throughput methodologies. Despite the emergence of advanced transcriptional profiling technologies, northern blotting provides a robust, direct method for RNA detection and quantification, allowing researchers to discern between different RNA isoforms based on size and confirm gene expression patterns with high confidence [53] [44]. This application note details protocols and quantitative assessments that establish northern blotting as an essential validation tool in gene expression research and drug development.
In the post-genomic era, researchers increasingly rely on high-throughput techniques like microarrays and RNA sequencing for gene expression analysis. While these platforms generate vast datasets, they present significant challenges including inconsistent sequence fidelity, platform-specific variability, low probe specificity, and an inability to distinguish between different isoforms of a gene [53]. Northern blotting addresses these limitations by providing highly valid expression data that confirms transcript size, abundance, and integrity through direct membrane hybridization [53]. The technique's robustness stems from its direct visualization of target RNAs without amplification steps, making it particularly valuable for confirming drug-induced expression changes, validating biomarker candidates, and characterizing RNA processing intermediates in disease models.
Quantitative Performance Assessment
The validation capacity of northern blotting is evidenced through direct sensitivity comparisons with alternative detection methodologies. The following table summarizes key performance metrics for various northern blot approaches:
Table 1: Sensitivity Comparison of Northern Blot Methodologies
| Methodology | Detection Limit | Probe Stability | Multiplexing Capability | Safety Considerations |
|---|---|---|---|---|
| 32P Radioactive | 0.005-0.01 fmol [44] | ~14 days (half-life) [44] | Limited | Requires radioactive handling protocols |
| irNorthern (IR dye-labeled) | 0.02-0.05 fmol [44] | >8 months (stable at -80°C) [44] | Yes (multiple IR dyes) [44] | Non-radioactive |
| Biotin-Streptavidin IR | ~0.2 fmol (4x less sensitive than direct irNorthern) [44] | Dependent on streptavidin-IR conjugate | Limited | Non-radioactive |
| DIG-Chemiluminescent | ~0.05 fmol (similar to irNorthern) [44] | Several months with proper storage | Moderate | Non-radioactive |
Beyond sensitivity, northern blotting provides exceptional resolution for analyzing RNA length heterogeneity. High-resolution northern blotting can distinguish between miRNA isoforms differing by a single nucleotide, enabling precise characterization of RNA processing intermediates [54]. This resolution capability is crucial for validating siRNA reagents and monitoring processing fidelity in RNAi-based therapeutic development.
Experimental Protocols
RNA Isolation and Integrity Verification
Principle: High-quality, intact RNA is fundamental for reliable northern blot analysis. The protocol below details RNA extraction and quality assessment steps.
Materials and Reagents:
Procedure:
- Cell Lysis: Rinse cells with PBS and lyse in 1 ml TRIzol per 100-mm culture dish. Incubate for 5 minutes at room temperature [7].
- Phase Separation: Add 0.2 volumes of chloroform, shake vigorously for 15 seconds, incubate for 3 minutes at room temperature, and centrifuge at 12,000 × g for 15 minutes at 4°C [7].
- RNA Precipitation: Transfer the upper aqueous phase to a new tube, mix with an equal volume of isopropanol, and incubate for 10 minutes at room temperature [7].
- RNA Wash: Centrifuge at 12,000 × g for 10 minutes at 4°C to pellet RNA. Wash the pellet with 1 ml of 70% RNase-free ethanol and centrifuge at 7,500 × g for 5 minutes [7].
- RNA Resuspension: Air-dry the pellet and dissolve in 100 µl RNase-free H₂O by incubating at 65°C for 15 minutes [7].
- Quality Assessment: Determine RNA concentration and purity using spectrophotometry (NanoDrop). Store RNA at -80°C [7].
Validation: RNA integrity should be verified by denaturing agarose gel electrophoresis before proceeding to blotting. Sharp ribosomal RNA bands indicate intact RNA, while smearing suggests degradation.
High-Resolution Northern Blotting
Principle: This protocol enables high-resolution detection of RNAs ranging from 20-70 nucleotides, ideal for miRNA, siRNA, and precursor analysis [54].
Materials and Reagents:
- Formaldehyde [7]
- Agarose [7]
- 10× MOPS buffer [7]
- Hybond-N+ nylon membrane [7]
- DIG Easy Hyb solution [7]
- DIG Wash and Block Buffer Set [7]
- Anti-digoxigenin-AP Fab fragments [7]
- CDP-Star chemiluminescent substrate [7]
Procedure:
- Gel Electrophoresis:
- Prepare a 1.3% formaldehyde-agarose gel using 1× MOPS buffer [7].
- Denature 2-5 µg of total RNA in RNA loading buffer at 65°C for 15 minutes.
- Separate RNA by electrophoresis at 5 V/cm until adequate separation is achieved.
Membrane Transfer:
Probe Hybridization:
Signal Detection:
Troubleshooting: Optimize probe concentration and hybridization temperature to minimize background. Include positive and negative controls on each blot. For quantitative comparisons, ensure signals fall within the linear detection range.
Near-Infrared Fluorescent Northern Blot (irNorthern)
Principle: irNorthern utilizes near-infrared dye-labeled probes for sensitive, non-radioactive detection with capability for multiplexing [44].
Materials and Reagents:
Procedure:
- Probe Labeling:
- Hybridization and Detection:
Advantages: irNorthern probes maintain sensitivity after multiple uses and long-term storage (≥8 months at -80°C) [44]. The method enables multiplex detection of different RNA species using IR dyes with different emission wavelengths [44].
Workflow Visualization
The following diagram illustrates the complete northern blotting workflow, highlighting key validation checkpoints:
Northern Blotting Workflow for Data Validation
Research Reagent Solutions
The following essential materials are critical for successful northern blot experiments:
Table 2: Essential Research Reagents for Northern Blotting
| Reagent/Category | Specific Examples | Function and Application Notes |
|---|---|---|
| RNA Isolation Reagents | TRIzol reagent [7] | Maintains RNA integrity during extraction from cells and tissues |
| Membrane Systems | Hybond-N+ nylon membrane [7] | Optimal for RNA retention with minimal background during hybridization |
| Non-radioactive Labeling | DIG labeling kit [7] | Generates stable, sensitive probes for chemiluminescent detection |
| Advanced Probe Systems | IRDye 800CW DBCO with azide-modified probes [44] | Enables fluorescent detection with multiplexing capability and long probe stability |
| Hybridization Buffers | DIG Easy Hyb solution [7] | Provides optimized stringency conditions for specific probe binding |
| Detection Substrates | CDP-Star chemiluminescent substrate [7] | High-sensitivity substrate for alkaline phosphatase-based detection |
Northern blotting maintains its position as the gold standard for gene expression data validation through its unique combination of direct RNA visualization, size discrimination capability, and quantitative reliability. While newer high-throughput methods offer greater speed and parallel processing, northern blotting provides the essential validation framework that ensures transcriptional profiling data accurately reflects biological reality. The continued technical evolution of northern blotting, including non-radioactive detection methods and multiplexing capabilities, ensures its ongoing relevance in biomedical research and therapeutic development. For researchers requiring unequivocal confirmation of gene expression patterns, northern blotting remains an indispensable component of the molecular biology toolkit.
In the evolving landscape of molecular biology, techniques for analyzing gene expression have diversified significantly. Among these, Northern blotting stands as a historically pivotal method, first described in 1977 by Alwine, Kemp, and Stark [1] [15]. Despite the emergence of high-throughput technologies like quantitative polymerase chain reaction (qPCR) and DNA microarrays, Northern blotting retains a fundamental role in many laboratories, particularly for the validation of data generated by newer platforms [16] [55]. This analysis is often considered a gold standard for RNA detection due to its directness and reliability [16] [56]. The technique involves the separation of RNA samples via denaturing gel electrophoresis, transfer to a solid membrane, and subsequent detection of specific RNA molecules using labeled complementary DNA or RNA probes [1] [15].
The purpose of this application note is to provide a detailed comparative analysis of Northern blotting against qPCR and DNA microarrays. Framed within the broader context of target gene expression monitoring, we will dissect the technical specificities, optimal applications, and inherent limitations of each method. By integrating quantitative data, detailed protocols, and decision-making workflows, this document aims to serve as a comprehensive guide for researchers and drug development professionals in selecting the most appropriate gene expression analysis technique for their specific experimental needs.
Technical Comparison of Core Methodologies
The following table summarizes the core characteristics of Northern blotting, qPCR, and DNA microarrays, providing a clear, at-a-glance comparison of their key parameters.
Table 1: Core Methodological Characteristics of Northern Blotting, qPCR, and Microarrays
| Feature | Northern Blotting | qPCR | DNA Microarrays |
|---|---|---|---|
| Fundamental Principle | Size separation by gel electrophoresis, membrane transfer, and hybridization with a labeled probe [1] [15] | Reverse transcription of RNA to cDNA, followed by fluorescent-based quantitative PCR amplification [55] | Hybridization of labeled cDNA from a sample to thousands of immobilized DNA probes on a solid surface [57] |
| Key Output | Transcript size, abundance, and integrity; detects splicing variants [1] [15] [55] | Absolute or relative quantitation of specific transcript levels [55] | Relative expression levels of thousands of genes simultaneously (expression profiling) [58] [57] |
| Sample Throughput | Low to medium; suitable for a limited number of samples and genes [1] | High for a few genes across many samples [55] | High for thousands of genes across a moderate number of samples [58] |
| Information on Transcript Size | Yes, a key advantage [1] [55] | No | No |
| System Architecture | Targeted (closed system for specific genes) | Targeted (closed system for specific genes) | Genome-wide or pathway-focused (open system for discovery) [58] |
To complement this overview, a quantitative performance comparison is critical for experimental planning. The data below highlights the practical sensitivities and resource requirements of each technique.
Table 2: Quantitative Performance and Resource Comparison
| Performance Metric | Northern Blotting | qPCR | DNA Microarrays |
|---|---|---|---|
| Sensitivity | Low to moderate; can detect low-expression genes with protocol optimization [16] [55] | Extremely high; theoretically capable of detecting a single mRNA molecule [55] | Moderate; generally less sensitive than qPCR for low-abundance transcripts [59] [55] |
| Sample RNA Integrity | High-quality, intact RNA required [55] | Tolerant of partially degraded RNA [55] | Tolerant of partially degraded RNA [55] |
| Multiplexing Capability | Low; typically one probe per blot, though membranes can be stripped and re-probed [55] | Low per reaction; typically one or a few targets per tube | Very high; thousands of genes simultaneously [58] |
| Hands-on Time & Cost | Labor-intensive; lower equipment cost but higher hands-on time [49] | Faster; lower cost per sample for a few targets [49] | High initial setup cost; less hands-on time per data point after establishment [58] |
Detailed Experimental Protocols
A Modified Northern Blot Protocol for Enhanced Sensitivity
The traditional Northern blot protocol has been successfully modified to improve its sensitivity and reliability, making it more applicable for detecting low-expression genes [16].
Key Materials & Reagents:
- Total RNA: Requires high-quality, intact RNA. As little as 20 µg can be sufficient for low-expression genes with a modified protocol [16].
- Formaldehyde-Agarose Gel: A moderate concentration (e.g., 12%) provides adequate denaturation while facilitating subsequent blotting [16].
- Ethidium Bromide (EtBr): For pre-staining RNA to directly visualize RNA integrity and transfer efficiency during electrophoresis [16].
- Nylon Membrane: Positively charged nylon membranes are most effective for nucleic acid binding [15].
- Labeled Probe: Can be DNA, RNA, or oligonucleotides, labeled with radioisotopes (e.g., ³²P) or non-radioactive tags (e.g., biotin, digoxigenin) [15] [56].
- Hybridization Buffer: Contains formamide to lower the annealing temperature and prevent RNA degradation [15].
Step-by-Step Workflow:
- RNA Separation: Denature total RNA using a formaldehyde-agarose gel. The EtBr pre-staining allows for real-time monitoring of RNA quality and separation [16].
- Capillary Transfer: Transfer the size-separated RNA from the gel to a nylon membrane using a capillary blotting system with high-salt buffer [16] [15].
- Immobilization: Cross-link the RNA to the membrane using UV light or heat to create a permanent record of the gel separation [1] [15].
- Hybridization: Incubate the membrane with a labeled, sequence-specific probe. A staining jar placed in an enamel tray can be used for safe and efficient hybridization [16].
- Post-Hybridization Washes (Critical Modification): Instead of sequential low and high-stringency washes, perform washes under only moderate-stringency conditions. Monitor the membrane with a Geiger counter until radioactivity reaches 20-50 counts per second. This quantitatively controlled wash maximizes the retention of specifically bound probes, significantly enhancing detection sensitivity for low-abundance mRNAs [16].
- Detection: Detect the bound probe using autoradiography (for radioactive probes) or chemiluminescence (for non-radioactive probes). The membrane can be stripped and re-probed multiple times (up to 8 rounds reported) [16] [15].
Reverse Transcription-Quantitative PCR (RT-qPCR)
As a highly sensitive alternative, RT-qPCR is a widely used method for mRNA quantitation.
Key Materials & Reagents:
- RNA Sample: Can be used with partially degraded RNA, as it typically amplifies a short region (amplicon) [55].
- Reverse Transcriptase: Enzyme to synthesize cDNA from the RNA template.
- Sequence-Specific Primers: Designed for the gene of interest.
- Fluorescent Detection Chemistry: This includes DNA-binding dyes (e.g., SYBR Green) or sequence-specific fluorescent probes (e.g., TaqMan probes).
Step-by-Step Workflow:
- Reverse Transcription: Convert purified RNA into complementary DNA (cDNA) using a reverse transcriptase enzyme [55].
- Real-time PCR Amplification: Mix the cDNA with primers, nucleotides, and a fluorescent reporter in a thermocycler. The PCR reaction is run in real-time, with the fluorescence measured at the end of each cycle [55].
- Data Analysis: The cycle threshold (Ct), the point at which fluorescence crosses a background threshold, is determined. The Ct value is inversely proportional to the starting quantity of the target transcript. Absolute or relative quantitation is achieved by comparing Ct values to a standard curve or to internal control genes, respectively [55].
DNA Microarray Analysis
Microarrays are powerful tools for unbiased, genome-wide expression profiling.
Key Materials & Reagents:
- Microarray Chip: A solid support (usually a glass slide) containing thousands of immobilized DNA probes [57].
- Fluorophore-labeled cDNA: Sample RNA is reverse-transcribed into cDNA and labeled with fluorescent dyes (e.g., Cy3 and Cy5 for two-color arrays) [57].
- Hybridization Station: Equipment that provides controlled conditions for the hybridization reaction.
- Laser Scanner: A high-resolution scanner to detect the fluorescence signal from each probe spot on the array.
Step-by-Step Workflow:
- Target Preparation: Isolate RNA from test and control samples. Convert the RNA to cDNA and label each population with different fluorescent dyes [57].
- Hybridization: Mix the labeled cDNA targets and hybridize them to the microarray under stringent conditions [57].
- Washing and Scanning: Wash the array to remove non-specifically bound cDNA and then scan it with a laser to excite the fluorophores. The emitted fluorescence is measured for each spot [57].
- Data Analysis: Use specialized software to normalize the fluorescence intensities and calculate the relative abundance of each transcript between the two samples. Sophisticated statistical analyses are required to identify significantly differentially expressed genes from the vast dataset [58] [57].
Research Reagent Solutions
The following table catalogs essential reagents and their critical functions for successfully conducting a Northern blot analysis, serving as a quick-reference guide for laboratory preparation.
Table 3: Key Research Reagents for Northern Blotting
| Reagent / Material | Function / Explanation |
|---|---|
| Denaturing Agarose Gel | Typically contains formaldehyde or glyoxal/DMSO to denature RNA secondary structures, ensuring separation strictly by molecular weight [15]. |
| Positive-Charged Nylon Membrane | The solid support for transferred RNA; positive charge enhances binding of negatively charged nucleic acids, increasing sensitivity and retention [15]. |
| Hybridization Probes | Can be cDNA, RNA (riboprobes), or oligonucleotides (≥25 bases). RNA probes often offer higher specificity and sensitivity due to stronger hybridization [15]. |
| Labeling Molecules | Radioisotopes (³²P, high sensitivity) or non-radioactive tags (Biotin, Digoxigenin; safer and more stable). Chemiluminescent detection is common for non-radioactive probes [16] [15] [56]. |
| Crosslinking Agent | UV light or heat is used to covalently attach RNA to the membrane after transfer, preventing loss during hybridization and washes [1]. |
| Stringency Wash Buffers | Contain salts and detergents (e.g., SSC and SDS) to remove non-specifically and weakly bound probes from the membrane, reducing background noise [16] [15]. |
Analysis Workflow and Decision Logic
Selecting the optimal gene expression analysis method depends heavily on the research question's specific goals and constraints. The following decision diagram outlines a logical workflow to guide researchers to the most appropriate technique.
Diagram 1: Gene Expression Technique Selection Guide
Discussion and Concluding Remarks
The comparative analysis presented here underscores a central theme in modern molecular biology: there is no single "best" technique for gene expression analysis. Instead, Northern blotting, qPCR, and microarrays each occupy a distinct and valuable niche within the researcher's toolkit. Northern blotting provides a unique combination of quantitative data and structural information, confirming transcript size and identity, which is why it remains a gold standard for validating results from high-throughput screens [16] [49]. However, its lower sensitivity and throughput are clear limitations. qPCR excels in sensitivity, speed, and quantitative precision for focused studies on a limited number of genes, but it lacks the ability to provide information on transcript isoforms and is highly susceptible to amplification biases [55]. Microarrays offer an unparalleled breadth of analysis, allowing for the discovery of novel gene expression patterns across the entire genome, but they can be less sensitive than qPCR and require sophisticated data normalization and analysis to avoid false positives [58] [57].
The choice between these methods is therefore dictated by the experimental objective. For discovery-driven, hypothesis-generating research, microarrays are a powerful starting point. For the sensitive and absolute quantitation of a known, limited set of genes, qPCR is unmatched. When the research question requires confirmation of transcript integrity, size, or the presence of alternative splice variants—particularly for validating results from microarrays or RNA-Seq—Northern blotting remains an indispensable and reliable method. Furthermore, recent innovations, such as optimized non-radioactive detection and modified hybridization protocols, continue to enhance the sensitivity and safety of Northern blotting, ensuring its continued relevance in the molecular biology laboratory [16] [56]. Ultimately, these techniques are often most powerful when used in concert, leveraging the strengths of each to build a robust and validated understanding of gene expression.
Sensitivity and Specificity in the Era of RNA-Seq
The accurate measurement of gene expression is fundamental to advancing research in molecular biology and drug development. For decades, Northern blotting served as the foundational method for target gene expression monitoring, providing a benchmark for specificity with its ability to confirm transcript size and identity. However, the advent of RNA Sequencing (RNA-Seq) has revolutionized the field, offering unprecedented sensitivity and the ability to profile the entire transcriptome in a single assay. This application note examines the critical performance parameters of sensitivity and specificity for both Northern blotting and RNA-Seq, providing researchers with a structured comparison and detailed protocols to guide their experimental strategies within modern research contexts. The transition to RNA-Seq does not render Northern blotting obsolete but redefines its role as a confirmatory tool with high specificity, used to validate findings from high-throughput, discovery-focused RNA-Seq experiments [48] [15] [60].
Comparative Analysis of Method Performance
The choice between Northern blotting and RNA-Seq is guided by the experimental goals, with a fundamental trade-off between throughput and specificity. The quantitative performance of these methods is summarized in the table below.
Table 1: Quantitative Comparison of Northern Blotting and RNA-Seq
| Performance Metric | Northern Blotting | RNA-Seq |
|---|---|---|
| Throughput | Low (1 to a few genes per blot) [15] | High (entire transcriptome) [61] |
| Sensitivity | Low to Moderate [15] | Very High (broad dynamic range) [61] |
| Specificity | High (confirms transcript size) [15] [60] | High (but requires bioinformatic filtering) [62] |
| Quantitative Accuracy | Semi-quantitative [15] | Highly Quantitative [61] |
| Key Advantage | Detects transcript size and isoforms without prior knowledge [15] | Discovers novel transcripts, fusions, and SNPs [61] |
| Major Limitation | Requires large amounts of non-degraded RNA [15] | Computational complexity and cost [63] |
A study directly comparing differential gene expression measurements across methodologies found that while correlations are generally good, they are not perfect. The correlation coefficient (r) between Northern blot and microarray results was reported as 0.72, and between Northern blot and qRT-PCR as 0.39. After removing outlier genes, these correlations improved to 0.79 and 0.72, respectively, confirming that the assessment of differential gene expression is indeed dependent on the methodology used [48].
For variant detection from RNA-Seq data, tools like VarRNA demonstrate the method's power. This machine learning-based approach can identify about 50% of the variants detected by exome sequencing while also uncovering unique RNA variants absent in DNA data, highlighting its unique sensitivity to expressed alterations [62]. Another study noted that targeted RNA-Seq can uniquely identify variants with significant pathological relevance missed by DNA-seq, though it requires careful control of the false positive rate to ensure high accuracy [64].
Experimental Protocols
Detailed Protocol: Northern Blotting for Target Gene Validation
Northern blotting remains a valuable technique for validating the expression and size of specific transcripts. The following is a detailed protocol.
- RNA Extraction: Extract total RNA from cells or tissue using a phenol-chloroform method (e.g., TRIzol) or a silica-based column. Critical: Use RNase-free reagents and equipment to prevent degradation. Treat the extracted RNA with DNase to remove genomic DNA contamination. Assess RNA quality and quantity using a spectrophotometer and confirm integrity by electrophoresis on a denaturing agarose gel [60].
- mRNA Isolation (Optional): To increase sensitivity for low-abundance transcripts, isolate mRNA from total RNA using oligo(dT) beads or columns that bind the poly-A tail of messenger RNA [60].
- Gel Electrophoresis: Separate 5-20 μg of total RNA or 0.5-2 μg of mRNA on a denaturing formaldehyde agarose gel. Formaldehyde prevents secondary structure formation, ensuring separation by molecular weight. Include an RNA ladder as a size marker. Visualize the gel with ethidium bromide or a fluorescent nucleic acid stain under UV light to confirm RNA integrity and equal loading [15] [60].
- Blotting and Immobilization: Transfer the RNA from the gel to a positively charged nylon membrane using capillary or vacuum blotting. Vacuum blotting is faster (1-2 hours) and more reproducible. After transfer, immobilize the RNA onto the membrane by crosslinking using UV light or baking at ~80°C [15] [60].
- Pre-hybridization and Hybridization: Incubate the membrane in a pre-hybridization buffer containing a blocking agent (e.g., denatured salmon sperm DNA) to block non-specific binding sites. Subsequently, hybridize the membrane with a labeled (radioactive or chemiluminescent) DNA or RNA probe that is complementary to your target sequence. Incubate overnight at a temperature optimized for your probe [15] [60].
- Washing and Detection: Wash the membrane stringently to remove any non-specifically bound probe. The stringency is controlled by the salt concentration and temperature of the wash buffer. Detect the hybridized probe using autoradiography (for radioactive probes) or chemiluminescent imaging. The signal intensity corresponds to the abundance of the target RNA [15] [60].
Diagram 1: Northern blotting workflow.
Detailed Protocol: RNA-Seq for Transcriptome Analysis
This protocol outlines a standard bulk RNA-Seq workflow for differential gene expression analysis, from raw data to visualization.
- Quality Control and Trimming: Begin with raw sequencing data in FASTQ format. Use FastQC to assess read quality. Then, use a tool like Trimmomatic to trim adapter sequences and remove low-quality bases. This step is critical for reducing false positives in downstream analysis [63].
- Alignment to Reference Genome: Map the quality-filtered reads to a reference genome (e.g., GRCh38) using a splice-aware aligner such as STAR or HISAT2. These tools are essential for accurately mapping reads that span exon-exon junctions [62] [63].
- Quantification of Gene Expression: Using the aligned reads (BAM file), count the number of reads mapping to each gene or transcript. Tools like featureCounts (from the Subread package) are commonly used for this step, generating a count matrix where rows represent genes and columns represent samples [63].
- Differential Expression Analysis: Import the count matrix into R and use the DESeq2 package (available through Bioconductor) to perform statistical analysis and identify differentially expressed genes (DEGs). DESeq2 models the count data and corrects for library size differences and variance [63].
- Data Visualization: Create visualizations to interpret the results. Generate a heatmap of significant DEGs using the pheatmap package to show expression patterns across samples. Create a volcano plot using ggplot2 and ggrepel to visualize the relationship between statistical significance (p-value) and magnitude of change (fold-change) for all genes [63].
Diagram 2: RNA-Seq analysis workflow.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Tools for Gene Expression Analysis
| Item | Function/Application |
|---|---|
| TRIzol Reagent | Monophasic solution of phenol and guanidine isothiocyanate for effective total RNA isolation from cells and tissues [60]. |
| Oligo(dT) Beads | For isolation of polyadenylated mRNA from total RNA, enriching for coding transcripts and increasing assay sensitivity [60]. |
| Formaldehyde | Denaturing agent used in agarose gel electrophoresis to break down RNA secondary structures, ensuring separation by size [15]. |
| Positively Charged Nylon Membrane | Robust membrane for immobilizing negatively charged RNA via crosslinking for Northern blotting [15] [60]. |
| STAR Aligner | Spliced Transcripts Alignment to a Reference, a widely used aligner for RNA-Seq data that accurately handles splice junctions [62] [63]. |
| DESeq2 R Package | A statistical software package for assessing differential expression in RNA-Seq count data, modeling biological variability [63]. |
| VarRNA | A specialized computational tool that uses machine learning to call and classify germline and somatic variants from tumor RNA-Seq data [62]. |
In contemporary research, Northern blotting and RNA-Seq are not mutually exclusive but are complementary technologies. RNA-Seq provides a powerful, sensitive, and comprehensive discovery platform for identifying novel transcripts and global expression changes. Meanwhile, Northern blotting retains its critical role as a highly specific orthagonal method for validating key findings, particularly in confirming transcript size and integrity. A strategic approach that leverages the high sensitivity of RNA-Seq for discovery alongside the high specificity of Northern blotting for targeted confirmation offers a robust framework for generating reliable and actionable gene expression data, thereby strengthening research outcomes and drug development pipelines.
Enduring Role in Cell Line Characterization and Biologics QC
Within the rigorous demands of modern biologics quality control (QC) and cell line characterization, Northern blotting maintains an enduring role as a robust and reliable method for target gene expression monitoring. As the biopharmaceutical industry increasingly relies on complex protein-based therapeutics, the need for sensitive, precise, and accessible analytical techniques has never been greater [65]. Biologics, including monoclonal antibodies, are characterized by significant molecular complexity, where small variations in transcription and translation—including misincorporation at the DNA, RNA, or amino acid level—can alter a biologic's charge, stability, target affinity, and even lead to unintended aggregation with significant clinical consequences [65]. While high-throughput genomic technologies provide broad screening capabilities, Northern blot analysis is widely recognized as a highly valid and robust analytical approach for gene expression confirmation, offering direct visualization of transcript size and the ability to detect alternatively spliced variants [53] [2]. Its role in validating data from other platforms, such as microarrays, makes it an indispensable tool for ensuring the accuracy and reliability of gene expression data throughout the drug development pipeline, from discovery and clinical trials to large-scale production [53] [65].
Table: Key Advantages of Northern Blotting in Biologics Development
| Feature | Advantage for Biologics QC & Cell Line Characterization |
|---|---|
| Direct Size Determination | Confirms expected transcript size and detects splice variants, verifying genetic integrity of production cell lines [2]. |
| High Specificity & Validity | Provides robust, reliable data considered highly valid for confirming gene expression patterns identified by other methods [53]. |
| Quantitative Capability | Allows direct relative comparison of mRNA abundance between samples on a single membrane [2]. |
| Data Versatility | Can use DNA, RNA, or oligonucleotide probes, and sequences with partial homology (e.g., from different species) [2]. |
Application Note: Monitoring Critical Quality Attributes in a Model System
This application note illustrates a protocol for utilizing Northern blotting to monitor the expression of stress-responsive genes (ATF3, ATF4, GADD153) in Vero and H1299 cell lines following infection with the infectious bronchitis virus (IBV), serving as a model for cellular stress during bioproduction [7]. Such models are crucial for assessing how production cell lines respond to process-related stresses, ensuring consistent product quality and lot-to-lot purity.
Experimental Workflow
The following diagram outlines the complete Northern blotting procedure from sample preparation through to detection:
Research Reagent Solutions
The following table details key reagents and their critical functions in the Northern blotting protocol for ensuring reliable gene expression data.
Table: Essential Reagents for Northern Blot Analysis
| Reagent/Kit | Function in Protocol |
|---|---|
| TRIzol Reagent | A monophasic solution of phenol and guanidine isothiocyanate designed for effective RNA isolation from cells and tissues by disrupting cells and denaturing proteins while inhibiting RNases [7]. |
| DIG Labeling Kit | Utilizes digoxigenin, a plant steroid, to non-radioactively label DNA, RNA, or oligonucleotide probes via PCR or in vitro transcription for highly sensitive detection [7]. |
| NorthernMax Kits | Provide a complete set of RNase-free, optimized reagents (including buffers, agarose, and ULTRAhyb hybridization buffer) for either formaldehyde or glyoxal-based denaturing gels, increasing sensitivity and reliability [2]. |
| DIG Easy Hyb Buffer | A ready-to-use hybridization solution that minimizes background and maximizes specific signal during the membrane hybridization step with DIG-labeled probes [7]. |
| Anti-digoxigenin-AP Fab fragments | Antibody fragments conjugated to alkaline phosphatase that bind specifically to the DIG label on the hybridized probe, enabling subsequent chemiluminescent detection [7]. |
| CDP-Star | A highly sensitive chemiluminescent substrate for alkaline phosphatase. Upon dephosphorylation, it emits light, which is captured on X-ray film or a digital imager to visualize target RNA bands [7]. |
| BrightStar-Plus Membrane | A positively charged nylon membrane that optimally binds negatively charged nucleic acids after transfer, crucial for high sensitivity and low background [2]. |
Detailed Protocol: High-Resolution Northern Blotting
RNA Isolation and Integrity Assessment
Procedure:
- Cell Lysis: Culture cells in 100 mm dishes. After experimental treatment (e.g., infection, stress induction), rinse cells with PBS and lyse directly in the culture dish using 1 ml of TRIzol reagent. Incubate for 5 minutes at room temperature [7].
- Phase Separation: Transfer the lysate to a microcentrifuge tube. Add 0.2 ml of chloroform per 1 ml of TRIzol used. Shake tubes vigorously by hand for 15 seconds and incubate for 3 minutes at room temperature. Centrifuge at 12,000 × g for 15 minutes at 4°C [7].
- RNA Precipitation: Carefully transfer the upper, aqueous phase to a new tube. Mix with an equal volume of 100% isopropanol. Incubate for 10 minutes at room temperature and precipitate the RNA by centrifugation at 12,000 × g for 10 minutes at 4°C [7].
- RNA Wash: Wash the resulting RNA pellet with 1 ml of 70% RNase-free ethanol. Centrifuge at 7,500 × g for 5 minutes. Air-dry the pellet briefly and dissolve in 100 µl of RNase-free H₂O by incubating at 65°C for 15 minutes [7].
- Quality Control: Determine the concentration and purity (A260/A280 ratio) using a spectrophotometer like NanoDrop. Assess RNA integrity by running a small aliquot on a denaturing agarose gel; sharp ribosomal RNA bands indicate intact RNA, which is critical for Northern success [2].
Probe Generation by RT-PCR and DIG Labeling
Procedure:
- cDNA Synthesis: Combine 2 µg of total RNA with 10 pmoles of oligo(dT) primer in a 10.5 µl volume. Denature at 65°C for 10 minutes and immediately place on ice. Add reagents to a final 20 µl reaction containing 10 mM dNTPs, 20 units of RNasin, 1x reverse transcriptase buffer, and 50 units of reverse transcriptase. Synthesize first-strand cDNA at 43°C for 1 hour [7].
- DIG-Labeled PCR: In a 0.2 ml PCR tube, assemble the following on ice: 32.25 µl nuclease-free H₂O, 5 µl 10x PCR buffer, 5 µl PCR DIG Labeling Mix, 5 µl forward primer (10 µM), 5 µl reverse primer (10 µM), 0.75 µl enzyme mix, and 2 µl of the cDNA template. Vortex and centrifuge briefly [7].
- PCR Amplification: Place the sample in a thermal cycler and run with the following conditions: initial denaturation at 95°C for 2 minutes; followed by 30-40 cycles of denaturation at 95°C for 10 seconds, annealing at 60°C for 30 seconds, and elongation at 72°C for 30 seconds; final elongation at 72°C for 5 minutes [7]. The resulting PCR product is a DIG-labeled DNA probe ready for hybridization.
Denaturing Gel Electrophoresis and Transfer
Procedure:
- Gel Preparation: Prepare a 1.3% denaturing agarose gel by dissolving agarose in 1x MOPS buffer, then adding formaldehyde (for a formaldehyde gel) or using a glyoxal/DMSO system (NorthernMax-Gly Kit) for superior safety and potentially sharper bands [2] [7].
- Sample Denaturation & Loading: Mix RNA samples (typically 5-30 µg total RNA) with an appropriate volume of RNA loading buffer containing denaturants (e.g., formaldehyde or glyoxal). Denature the samples at 65°C for 10-15 minutes, then cool on ice. Load the denatured samples onto the gel alongside an appropriate RNA molecular weight ladder [2] [7].
- Electrophoresis: Run the gel in 1x MOPS or glyoxal running buffer at 5-6 V/cm until the dye front has migrated sufficiently. Do not recirculate the buffer for glyoxal gels [2].
- Capillary Transfer: Set up a passive downward alkaline transfer. Place the gel on a platform over a reservoir of transfer buffer. Place the membrane on top of the gel, followed by stack of absorbent paper (e.g., tissue paper) and a weight on top. Transfer for 2-4 hours. This method is faster and produces tighter bands than traditional overnight upward capillary transfer [2].
- Immobilization: After transfer, crosslink the RNA to the positively charged nylon membrane using ultraviolet light (UV crosslinker) following the manufacturer's instructions. This step is preferred over baking for optimal RNA retention [2].
Pre-hybridization, Hybridization, and Detection
Procedure:
- Pre-hybridization: Place the membrane in a hybridization tube with a sufficient volume of ULTRAhyb Ultrasensitive Hybridization Buffer. Pre-hybridize for 1-2 hours at the hybridization temperature (e.g., 42-68°C, depending on probe type and buffer) in a hybridization oven [2] [7].
- Hybridization: Denature the DIG-labeled DNA probe by heating at 95°C for 5 minutes, then immediately cooling on ice. Add the denatured probe directly to the pre-hybridization buffer in the tube. Hybridize for a minimum of 2 hours or overnight for low-abundance targets [2] [7].
- Stringency Washes: Discard the hybridization solution and perform a series of washes to remove unbound probe. Typically, start with a low-stringency wash (e.g., 2x SSC, 0.1% SDS) at room temperature, followed by one or more high-stringency washes (e.g., 0.1x SSC, 0.1% SDS) at the hybridization temperature or higher [2] [7].
- Immunodetection: Wash the membrane briefly in a washing buffer. Incubate the membrane with a blocking solution, followed by incubation with Anti-digoxigenin-AP Fab fragments (typically diluted 1:10,000) for 30 minutes. Wash the membrane thoroughly to remove unbound antibody [7].
- Chemiluminescent Visualization: Equilibrate the membrane in a detection buffer. Place the membrane in a transparent plastic sheet protector, add the chemiluminescent substrate (CDP-Star) evenly over the surface, and incubate for 5 minutes. Drain excess substrate, seal the bag, and expose to X-ray film or a digital imaging system in a cassette for various time periods (seconds to hours) to achieve an optimal signal [7].
Data Presentation and Analysis
Quantitative Data from Northern Analysis
Northern blotting generates quantitative data on transcript abundance, which can be summarized for comparison across different experimental conditions, such as cell lines or treatments.
Table: Example Quantitative Summary from a Northern Blot Experiment
| Cell Line / Treatment Condition | Target mRNA Level (Relative Units) | mRNA Size (kb) | Internal Control (e.g., GAPDH) | Normalized Expression |
|---|---|---|---|---|
| Vero Cells - Control | 1.0 | 1.5 | 1.0 | 1.00 |
| Vero Cells - IBV Infected (8h) | 5.2 | 1.5 | 1.1 | 4.73 |
| H1299 Cells - Control | 0.8 | 1.5 | 0.9 | 0.89 |
| H1299 Cells - IBV Infected (8h) | 8.5 | 1.5 | 1.0 | 8.50 |
Advanced Application: High-Resolution Analysis
High-resolution Northern blotting, utilizing optimized protocols and high-percentage gels, can provide single-nucleotide resolution. This is particularly valuable for characterizing microRNA (miRNA) biogenesis and the length heterogeneity of short interfering RNAs (siRNAs) or miRNA mimetics used in RNAi technology [54]. This application allows researchers to monitor the precision of Drosha and Dicer cleavages and the distribution of different length variants of these small RNAs, which is critical for understanding their function and potential off-target effects [54]. The data from such high-resolution analyses have been shown to correlate well with deep sequencing results for abundant miRNAs, providing a reliable and quantitative method for characterizing the most common and functionally important variants [54].
Northern blotting remains a foundational technique in the molecular biologist's toolkit, providing an unmatched combination of specificity, direct size information, and quantitative robustness for gene expression analysis. Its enduring role in the critical fields of cell line characterization and biologics quality control is secured by its ability to deliver highly reliable data that validates and refines findings from newer, high-throughput technologies. By following the detailed protocols and utilizing the optimized reagents outlined in this document, researchers can ensure the generation of high-quality, reproducible data essential for rigorous scientific discovery and the development of safe, effective biologic therapeutics.
Integrating Northern Blotting with Omics Technologies
In the contemporary landscape of gene expression analysis, high-throughput omics technologies provide powerful, genome-wide screening capabilities. However, their quantitative data requires rigorous validation using established, targeted methods. Northern blotting remains a cornerstone technique for this purpose, offering direct, reliable quantification of specific transcripts that is essential for confirming results from genomics, transcriptomics, and multi-omics studies [59] [16]. Its unique ability to provide information about transcript size and integrity makes it an indispensable gold standard for verifying gene expression changes discovered via microarrays or RNA-Seq, ensuring that findings related to target gene expression are both accurate and biologically meaningful [2].
Quantitative Comparison of Gene Expression Techniques
The integration of any technique into a modern workflow requires an understanding of its performance relative to other available methods. The table below summarizes key characteristics of common gene expression techniques, highlighting the niche that Northern blotting occupies.
Table 1: Comparison of Gene Expression Analysis Techniques
| Technique | Throughput | Sensitivity | Quantitative Nature | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|
| DNA Microarray | High | Slightly inferior to Northern blot [59] | Provides quantitative data comparable to Northern blot [59] | Expression profile of thousands of genes simultaneously [59] | Can fail to detect expression changes caught by Northern analysis [59] |
| Northern Blot | Low | Standard sensitivity is lower than RT-PCR [16] | Direct relative comparison of message abundance on a single membrane; true quantitation [16] [2] | Information on transcript size, integrity, and alternative splicing; highly valid data [16] [2] | Labor-intensive; requires higher quality RNA [16] |
| RT-qPCR | Medium | Extremely high [16] | Extrapolated value influenced by RT and amplification efficiency [16] | High throughput, speed, and specificity [16] | Risk of inconsistent/misleading data due to normalization issues [16] |
Modified Northern Blot Protocol for Enhanced Sensitivity and Reliability
This protocol incorporates modifications designed to improve sensitivity, particularly for low-abundance transcripts, and to make the procedure more reliable and controllable for the researcher [16].
RNA Isolation and Integrity Check
- RNA Isolation: Obtain high-quality, intact total RNA or poly(A) RNA using a method that ensures cellular lysis, effective ribonuclease inhibition, deproteinization, and recovery of intact RNA. Degraded RNA severely compromises data quality and quantitation [2].
- Integrity Check (EtBr Pre-staining): Before electrophoresis, pre-stain the RNA sample with ethidium bromide (EtBr). This allows for direct visualization of RNA bands at any time during or immediately after electrophoresis, enabling real-time evaluation of RNA integrity and gel loading uniformity [16].
Denaturing Agarose Gel Electrophoresis
- Gel Preparation: Prepare a 1% agarose gel containing 12% formaldehyde as the denaturant. This moderate formaldehyde concentration adequately maintains RNA in a denatured state while also helping to denature any contaminated exogenous RNases [16].
- Electrophoresis: Load 15-20 µg of total RNA per lane. Electrophorese until sufficient separation of ribosomal RNA bands is achieved. The pre-stained rRNA bands (18S and 28S) should appear sharp, indicating good RNA integrity.
Transfer to Membrane
- Capillary Transfer: Use a passive, downward capillary transfer system for 2 hours. This method is faster than traditional upward transfer and results in tighter bands and more signal [2]. The transfer buffer is slightly alkaline [2].
- Membrane and Immobilization: Use a positively charged nylon membrane. After transfer, crosslink the RNA to the membrane using ultraviolet light (preferred) or baking [2].
Probe Generation and Hybridization
- Probe Selection: For maximum sensitivity, use in vitro transcribed RNA probes. RNA probes can be 10-fold more sensitive than random-primed DNA probes and allow for more stringent washing conditions [2]. Alternatively, for a quicker method, use asymmetric PCR-generated DNA probes [2].
- Hybridization: Hybridize the membrane using a specialized ultrasensitive hybridization buffer (e.g., ULTRAhyb). Such buffers can increase sensitivity up to 100-fold compared to standard solutions and allow for hybridization times as short as 2 hours for many messages [2].
Post-Hybridization Washes and Detection
- Modified Washes (Key Step for Sensitivity): Do not follow traditional sequential low- and high-stringency washes. Instead, perform post-hybridization washes only under moderate-stringency conditions. Monitor the level of radioactivity on the filter (e.g., using a Geiger counter) and continue washing only until the background level drops to 20–50 counts per second. This quantitatively controlled approach maximizes the retention of specifically bound probes, thereby improving detection sensitivity for low-expression genes [16].
- Detection: Detect the specifically bound probes using an appropriate method (e.g., autoradiography for radiolabeled probes). The membrane can be subsequently stripped and re-probed up to eight times without significant signal loss [16].
Workflow: Integrating Northern Blotting with Multi-Omics Research
The following diagram illustrates the synergistic role of Northern blotting within a multi-omics research framework aimed at understanding complex biological traits, such as disease resistance in plants [66].
Essential Research Reagent Solutions
The table below lists key reagents and their optimized functions for a successful Northern blotting experiment, drawing from both standard and modified protocols.
Table 2: Key Reagents for Northern Blot Analysis
| Reagent / Kit | Function / Purpose | Key Feature / Benefit |
|---|---|---|
| ULTRAhyb Hybridization Buffer | Prehybridization and hybridization solution [2] | Increases sensitivity up to 100-fold; allows for 2-hour hybridizations for many messages [2] |
| BrightStar-Plus Membranes | Positively charged nylon membrane for RNA immobilization [2] | Minimizes background and maximizes signal transfer efficiency [2] |
| MAXIscript Kit | In vitro transcription for RNA probe synthesis [2] | Produces highly sensitive RNA probes; allows for incorporation of isotopic/nonisotopic labels [2] |
| DECAprime II Kit | Random-primed DNA probe synthesis [2] | Quick (10-minute) reaction to obtain high specific activity DNA probes [2] |
| NorthernMax-Gly Kit | Complete system for Northern analysis using glyoxal/DMSO [2] | Avoids safety issues of formaldehyde; no fume hood needed; sharper bands [2] |
| Formaldehyde (12%) Gel | Denaturing gel electrophoresis [16] | Adequate denaturation of RNA and contaminating RNases; balances safety and performance [16] |
| Ethidium Bromide | Pre-staining of RNA samples [16] | Enables direct visualization of RNA integrity during electrophoresis [16] |
Northern blotting maintains its critical role in the validation pipeline of modern molecular biology. By integrating the sensitivity enhancements from modified protocols—such as quantitatively controlled washes and EtBr pre-staining—with the robust, quantitative data inherent to the method, researchers can confidently bridge the gap between high-throughput omics discovery and functional analysis of specific target genes. This ensures that the conclusions drawn from large-scale datasets are grounded by a reliable and definitive measure of gene expression.
Conclusion
Northern blotting remains an indispensable tool in the molecular biology arsenal, offering unparalleled specificity and the unique ability to provide direct information on transcript size and integrity. While high-throughput methods excel in discovery, Northern blotting's role as a gold standard for validation is secure, especially in critical applications like cell bank characterization for therapeutic development. Future directions point not to replacement, but to integration—where Northern blotting works in concert with RNA-Seq and qPCR, providing orthogonal validation and tackling specific questions about RNA biology that other methods cannot. Continued innovation in non-radioactive detection and protocol streamlining will further solidify its value in both basic research and clinical diagnostics, ensuring its relevance for years to come.
References
- 1. Northern Blotting - an overview [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/northern-blotting]
- 2. The Basics: Northern Analysis [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/northern-analysis/general-articles/the-basics-northern-analysis.html]
- 3. A Comprehensive Analysis of Northern versus Liquid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8929067/]
- 4. Northern Blot- Definition, Principle, Steps, Results ... [https://microbenotes.com/northern-blot/]
- 5. An Overview of Northern blot [https://www.goldbio.com/blogs/articles/an-overview-of-northern-blot?srsltid=AfmBOorgvT3U1VU2iGuBpfhc0g2mWXeRvZGRQVH3DqtzaoSIOrxwAvxB]
- 6. Northern blot - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4287216/]
- 7. RNA Isolation and Northern Blot Analysis [https://en.bio-protocol.org/en/bpdetail?id=1077&type=0]
- 8. Northern Blotting: Protocols for Radioactive and ... [https://pubmed.ncbi.nlm.nih.gov/39535701/]
- 9. An Overview of Northern blot [https://www.goldbio.com/blogs/articles/an-overview-of-northern-blot?srsltid=AfmBOorMGKJ7GcCTqagyIrAjDeoe3mgYpCEHsx0beyNy5QUVfuVPVgo6]
- 10. Northern Blotting - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/medicine-and-dentistry/northern-blotting]
- 11. Northern Blotting Market | Global Market Analysis Report [https://www.futuremarketinsights.com/reports/northern-blotting-market]
- 12. Protocol to analyze tRNA and rRNA processing using ... [https://www.sciencedirect.com/science/article/pii/S2666166725002722]
- 13. Northern Blotting [https://azurebiosystems.com/western-blotting-applications/northern-blotting/]
- 14. Sensitive and specific detection of microRNAs by northern blot ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC545470/]
- 15. Northern blot [https://en.wikipedia.org/wiki/Northern_blot]
- 16. Modified Northern blot protocol for easy detection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8772191/]
- 17. Analysis of RNA by northern and slot blot hybridization [https://pubmed.ncbi.nlm.nih.gov/18265351/]
- 18. Practical Tips for Optimal Northern Analysis [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/northern-analysis/tech-notes/practical-tips-for-optimal-northern-analysis.html]
- 19. Northern Blotting Market Size & Share, Growth Analysis 2037 [https://www.researchnester.com/reports/northern-blotting-market/721]
- 20. | MyBioSource Learning Center Northern Blotting [https://www.mybiosource.com/learn/testing-procedures/northern-blotting/]
- 21. tDR-quant: a reliable electroporation-based approach for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12509828/]
- 22. An Overview of Northern blot [https://www.goldbio.com/blogs/articles/an-overview-of-northern-blot?srsltid=AfmBOopq9ryOo4TFjOLgAK_7rzd3UNxv-V_D-xjmID2Ic6-ycCVi8_jq]
- 23. Extraction of high quality and high yield RNA from frozen ... [https://www.nature.com/articles/s41598-024-58576-9]
- 24. Optimized SDS-Based Protocol for High-Quality RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11858800/]
- 25. RNA isolation and quality assessment [https://bio-protocol.org/exchange/minidetail?id=213741&type=30]
- 26. An Overview of Northern blot [https://www.goldbio.com/blogs/articles/an-overview-of-northern-blot?srsltid=AfmBOoousRcN57gltcYbygauUTNecKzyAwwJ5xk3aJOEHbDhta0gcpGf]
- 27. Agarose Gel Electrophoresis of RNA [https://www.thermofisher.com/us/en/home/references/protocols/nucleic-acid-purification-and-analysis/rna-protocol/agarose-gel-electrophoresis-of-rna.html]
- 28. Northern Blot probes (non-radioactive) [https://www.jenabioscience.com/rna-technologies/rna-analysis-detection/northern-blot-probes-non-radioactive]
- 29. More Than a Clever Name: Northern Blots [https://bitesizebio.com/27267/more-than-a-clever-name-northern-blots/]
- 30. Protocol for Small RNA Northern Blots: High-Sensitivity, High ... [https://www.clyte.tech/post/protocol-for-small-rna-northern-blots-high-sensitivity-high-resolution?srsltid=AfmBOorolA4dMCz-vfHaHSnQTfQ8cBsW-JAfd-dCkxn1sPlg8He_Z7V0]
- 31. The Origin and Evolution of Northern Blotting in Molecular ... [https://dev.housing.arizona.edu/northern-blotting]
- 32. An Overview of Northern blot [https://www.goldbio.com/blogs/articles/an-overview-of-northern-blot?srsltid=AfmBOops_4gBNH-YuT3k2_p0Aqn7FPZLiMYl7vQYDUDWpm5SlPtDOHea]
- 33. An Overview of Northern blot [https://www.goldbio.com/blogs/articles/an-overview-of-northern-blot?srsltid=AfmBOopPv29Se97kW8YYPvVvbd_YTMgp6QAJNgwBfWn2aSzOVqyNT3_3]
- 34. Researchers Revolutionize RNA Detection - UConn Today [https://today.uconn.edu/2025/10/researchers-revolutionize-rna-detection/]
- 35. Single-MicroRNA Detection on High-Selectivity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12613836/]
- 36. The current and future landscape of RNA-based therapies ... [https://www.nature.com/articles/s43856-025-01166-1]
- 37. An Overview of Northern blot [https://www.goldbio.com/blogs/articles/an-overview-of-northern-blot?srsltid=AfmBOoqXdAnW_QuBrl8m5Mpb5jjEaSH7zCKuGdncjew2gmtWdO-7ajZU]
- 38. Northern blot analysis [https://bio-protocol.org/exchange/minidetail?id=2432107&type=30]
- 39. RNA purification and Northern blotting [https://bio-protocol.org/exchange/minidetail?id=5189884&type=30]
- 40. Northern Blot / RNA Blot [https://www.news-medical.net/life-sciences/Northern-Blot-RNA-Blot.aspx]
- 41. Sample Prep for Single Cell/Nuclei RNA-seq [https://biotech.wisc.edu/sample-prep-for-single-cell-nuclei-rna-seq/]
- 42. Optimizing Stringency in Wash Buffers for Hybridization Assays [https://www.letstalkacademy.com/increasing-stringency-hybridization-buffers/]
- 43. Top Ten Northern Analysis Tips [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/northern-analysis/general-articles/top-ten-northern-analysis-tips.html]
- 44. Near-infrared fluorescent northern blot - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6239192/]
- 45. The Top 10 Western Blotting Mistakes (and Solutions!) [https://bitesizebio.com/19799/the-top-10-western-blotting-mistakes-and-solutions/]
- 46. Common Western Blotting Questions, Answered [https://azurebiosystems.com/blog/what-is-western-blotting/]
- 47. The Basics: Northern Analysis [https://www.thermofisher.com/ca/en/home/references/ambion-tech-support/northern-analysis/general-articles/the-basics-northern-analysis.html]
- 48. The Methodology Used to Measure Differential Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2392989/]
- 49. Quantitative Expression Analysis in Brassica napus by ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0163679]
- 50. Northern Blotting - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/northern-blotting]
- 51. Differential Expression of a Novel Gene in Response to ... [https://www.sciencedirect.com/science/article/pii/S0022202X15417166]
- 52. Northern Blotting - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/northern-blotting]
- 53. BlotBase: A northern blot database [https://www.sciencedirect.com/science/article/abs/pii/S0378111908004228]
- 54. Northern blotting analysis of microRNAs, their precursors and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3080303/]
- 55. Analysis of Differential Gene Expression [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/northern-analysis/general-articles/analysis-of-differential-gene-expression.html]
- 56. A Comprehensive Analysis of Northern versus Liquid ... [https://www.mdpi.com/1467-3045/43/2/36]
- 57. A perspective on microarrays: current applications, pitfalls ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1796898/]
- 58. Comparing microarrays and next-generation sequencing ... [https://www.sciencedirect.com/science/article/pii/S0167779910000491]
- 59. Quantitative assessment of DNA microarrays [https://pubmed.ncbi.nlm.nih.gov/11161795/]
- 60. An Overview of Northern blot [https://www.goldbio.com/blogs/articles/an-overview-of-northern-blot?srsltid=AfmBOor50pv3FdKe4yfOfKIr3CiZROoqrjCA4m1h2Fe3gONjAdKTOsKY]
- 61. RNA Sequencing | RNA-Seq methods & workflows [https://www.illumina.com/techniques/sequencing/rna-sequencing.html]
- 62. Variant calling from RNA-Seq data reveals allele-specific ... [https://www.nature.com/articles/s43856-025-00901-y]
- 63. A Guide to Basic RNA Sequencing Data Processing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12067304/]
- 64. Augmenting precision medicine via targeted RNA-Seq ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12166063/]
- 65. Biologics Quality Control: The Growing Need for ... [https://www.biopharminternational.com/view/biologics-quality-control-the-growing-need-for-accessible-proteomics]
- 66. Harnessing multi-omics approaches to combat Karnal bunt ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1687301/full]