Next-Generation Sequencing for DNA and RNA Analysis: A Comprehensive Guide for Biomedical Research and Drug Development
This article provides a comprehensive overview of next-generation sequencing (NGS) technologies and their transformative impact on genomic research and drug development.
Next-Generation Sequencing for DNA and RNA Analysis: A Comprehensive Guide for Biomedical Research and Drug Development
Abstract
This article provides a comprehensive overview of next-generation sequencing (NGS) technologies and their transformative impact on genomic research and drug development. It covers foundational principles, from first-generation Sanger sequencing to current short- and long-read platforms like Illumina, PacBio, and Nanopore. The scope extends to methodological applications in oncology, rare diseases, and infectious diseases, alongside detailed protocols for library preparation, data analysis, and bioinformatics. The content also addresses critical troubleshooting, optimization strategies for common challenges, and rigorous validation frameworks required for clinical implementation. Designed for researchers, scientists, and drug development professionals, this guide synthesizes current trends, practical applications, and future directions to empower the effective use of NGS in advancing precision medicine.
The NGS Revolution: From Sanger Sequencing to Modern Genomics
The ability to decipher the genetic code has fundamentally transformed biological research and clinical diagnostics. The evolution of DNA sequencing technology, from its inception to the modern era, represents a journey of remarkable scientific innovation, characterized by exponential increases in speed and throughput and precipitous drops in cost [1] [2]. These advances have propelled diverse fields, from personalized medicine to drug discovery, by providing an unparalleled view into the blueprints of life [3] [4]. This article traces the development of sequencing technologies through three distinct generations, providing a structured comparison of their characteristics and detailing foundational protocols that underpin contemporary next-generation sequencing (NGS) workflows for DNA and RNA analysis.
The Generations of Sequencing Technology
First-Generation Sequencing: The Foundations
The first major breakthrough in DNA sequencing occurred in 1977 with the introduction of two methods: the chemical degradation method by Alan Maxam and Walter Gilbert and the chain-termination method by Frederick Sanger and colleagues [1] [5]. The Maxam-Gilbert method used chemicals to cleave DNA at specific bases (A, G, C, or T/C), followed by separation of the resulting fragments via gel electrophoresis and detection by autoradiography [1] [2]. While groundbreaking, this method was technically challenging and used hazardous chemicals.
The Sanger method, which became the dominant first-generation technology, uses a different approach. It relies on the random incorporation of dideoxynucleoside triphosphates (ddNTPs) during in vitro DNA replication [1]. These ddNTPs lack a 3'-hydroxyl group, which prevents the formation of a phosphodiester bond with the next incoming nucleotide, thereby terminating DNA strand elongation [1]. In its original form, four separate reactions—each containing one of the four ddNTPs—were run. The resulting fragments of varying lengths were separated by gel electrophoresis, and the sequence was determined based on their migration [5].
A critical advancement came with the automation of Sanger sequencing. The development of fluorescently labeled ddNTPs and the replacement of slab gels with capillary electrophoresis enabled the creation of automated DNA sequencers [1] [2]. The first commercial automated sequencer, the ABI 370 from Applied Biosystems, significantly increased throughput and accuracy and became the primary workhorse for the landmark Human Genome Project [1] [5]. Despite its accuracy, automated Sanger sequencing remained relatively low-throughput and expensive for sequencing large genomes, highlighting the need for new technologies [2].
Second-Generation Sequencing: The High-Throughput Revolution
Second-generation sequencing, or next-generation sequencing (NGS), is defined by its ability to perform massively parallel sequencing, enabling the simultaneous analysis of millions to billions of DNA fragments [6] [4]. This paradigm shift dramatically reduced the cost and time required for sequencing, making large-scale projects like whole-genome sequencing accessible to individual labs [4].
A key differentiator of most second-generation platforms is their reliance on a template amplification step prior to sequencing. Three primary amplification methods are used:
- Emulsion PCR: Used by 454 pyrosequencing and Ion Torrent platforms, where DNA fragments are amplified on beads in water-in-oil emulsions [6].
- Bridge Amplification: Used by Illumina platforms, where DNA fragments are amplified on a solid surface (a flow cell) to form clonal clusters [6] [2].
- DNA Nanoball Generation: Used by BGI/MGI, where rolling circle amplification creates DNA nanoballs that are deposited on a patterned flow cell [6] [7].
Table 1: Comparison of Major Second-Generation Sequencing Platforms
| Platform (Company) | Sequencing Chemistry | Amplification Method | Key Principle | Typical Read Length | Key Limitations |
|---|---|---|---|---|---|
| 454 GS FLX (Roche) [7] [2] | Pyrosequencing | Emulsion PCR | Detection of pyrophosphate (PPi) release, converted to light via luciferase | 400-1000 bp | Difficulty with homopolymer regions, leading to insertion/deletion errors |
| Ion Torrent (Thermo Fisher) [7] | Sequencing by Synthesis | Emulsion PCR | Semiconductor detection of hydrogen ions (H+) released upon nucleotide incorporation | 200-400 bp | Signal degradation in homopolymer regions affects accuracy |
| Illumina (e.g., MiSeq, NovaSeq) [6] [7] [4] | Sequencing by Synthesis (SBS) | Bridge PCR | Fluorescently-labeled, reversible terminator nucleotides; imaging after each base incorporation | 36-300 bp | Signal decay and dephasing over many cycles can lead to errors |
| SOLiD (Applied Biosystems) [7] | Sequencing by Ligation | Emulsion PCR | Ligation of fluorescently-labeled di-base probes | 75 bp | Short reads and complex data analysis |
The massive data output of these platforms necessitated parallel advances in bioinformatics for data assembly, alignment, and variant calling [7]. While second-generation sequencing provides high accuracy and low cost per base, its primary limitation is short read length, which complicates the assembly of complex genomic regions and the detection of large structural variations [6].
Third-Generation Sequencing: The Long-Read Era
Third-generation sequencing technologies emerged to address the limitations of short reads. Their defining characteristic is the ability to sequence single DNA molecules in real time, without the need for a prior amplification step, producing long reads that can span thousands to tens of thousands of bases [8] [9]. This is particularly valuable for de novo genome assembly, resolving repetitive regions, identifying large structural variants, and detecting epigenetic modifications directly [6] [9].
Table 2: Comparison of Major Third-Generation Sequencing Platforms
| Platform (Company) | Sequencing Chemistry | Template Preparation | Key Principle | Typical Read Length | Accuracy & Key Features |
|---|---|---|---|---|---|
| PacBio SMRT Sequencing [6] [8] [9] | Single-Molecule Real-Time (SMRT) | SMRTbell library (circulared DNA) | Real-time detection of fluorescent nucleotide incorporation by polymerase immobilized in a zero-mode waveguide (ZMW) | >15,000 bp (average) | HiFi Reads: >99.9% accuracy via circular consensus sequencing (CCS) [6] [8] |
| Oxford Nanopore Technologies (ONT) [6] [7] [8] | Nanopore Sequencing | Ligation of adapters to native DNA | Measurement of changes in ionic current as DNA strand passes through a protein nanopore | 10,000-30,000 bp (average) | ~99% raw accuracy (Q20) with latest kits; real-time, portable sequencing (MinION) [8] |
Initially, third-generation technologies were characterized by higher per-base error rates compared to second-generation platforms. However, continuous improvements have substantially increased their accuracy. PacBio's HiFi (High-Fidelity) reads achieve high accuracy by repeatedly sequencing the same circularized DNA molecule to generate a consensus sequence [8]. Oxford Nanopore's accuracy has been improved through updated chemistries (e.g., Kit 14) and advanced base-calling algorithms like Dorado, with duplex reads now exceeding Q30 (>99.9% accuracy) [8] [9].
Experimental Protocol: A Standard Illumina NGS Workflow for Whole Transcriptome RNA Sequencing (RNA-Seq)
The following protocol outlines a standard workflow for preparing RNA-Seq libraries for sequencing on Illumina's short-read platforms, a cornerstone of gene expression analysis [4].
Principle
RNA Sequencing (RNA-Seq) utilizes NGS to determine the sequence and abundance of RNA molecules in a biological sample. It allows for the discovery of novel transcripts, quantification of gene expression levels, and analysis of alternative splicing events [4].
Reagents and Equipment
- Total RNA or mRNA sample (high quality, RIN > 8)
- Oligo(dT) magnetic beads or rRNA depletion kits for enrichment
- Fragmentation buffer
- Reverse Transcriptase and random hexamer/oligo(dT) primers
- DNA Polymerase I, RNase H, and dNTPs
- NGS Library Preparation Kit (e.g., Illumina TruSeq)
- SPRIselect beads or other solid-phase reversible immobilization (SPRI) beads for cleanup
- Indexing Adapters (Illumina) for sample multiplexing
- Quantification instruments (Qubit, Bioanalyzer/Tapestation)
- Illumina Sequencing Platform (e.g., MiSeq, NovaSeq)
Step-by-Step Procedure
- RNA Extraction and Quality Control: Isolate total RNA from your sample (e.g., cells or tissue) using a validated method. Assess RNA integrity and concentration using an instrument like the Agilent Bioanalyzer.
- RNA Enrichment:
- For mRNA Sequencing: Use oligo(dT) magnetic beads to selectively enrich for poly-adenylated mRNA.
- For Total RNA Sequencing: Use commercial kits to deplete abundant ribosomal RNA (rRNA).
- RNA Fragmentation and Priming: Fragment the purified RNA using divalent cations at elevated temperature (e.g., Mg²⁺ at 94°C for several minutes) to generate fragments of a desired size (e.g., 200-300 nucleotides). Prime the fragmented RNA using random hexamer primers.
- First-Strand cDNA Synthesis: Synthesize complementary DNA (cDNA) using Reverse Transcriptase and dNTPs. This creates an RNA-DNA hybrid.
- Second-Strand cDNA Synthesis: Remove the RNA template using RNase H and synthesize the second cDNA strand using DNA Polymerase I and dNTPs, resulting in double-stranded cDNA (ds-cDNA).
- Library End-Repair and A-Tailing: The ds-cDNA fragments are blunt-ended, and a single 'A' nucleotide is added to the 3' ends to facilitate ligation with the indexing adapters, which have a complementary 'T' overhang.
- Adapter Ligation: Ligate the unique dual-indexed adapters provided in the kit to the A-tailed cDNA fragments. These adapters contain the sequences required for binding to the Illumina flow cell and for priming the sequencing reactions.
- Library Amplification and Cleanup: Perform a limited-cycle PCR (e.g., 10-15 cycles) to enrich for adapter-ligated fragments and amplify the library. Purify the final library using SPRI beads to remove excess primers and adapter dimers.
- Library Quality Control and Quantification: Validate the library's size distribution using the Bioanalyzer and accurately quantify the concentration using a fluorescence-based method (Qubit).
- Pooling and Sequencing: Normalize and pool multiple, uniquely indexed libraries into a single tube for multiplexed sequencing. Load the pool onto the Illumina flow cell for cluster generation and sequencing-by-synthesis.
The following workflow diagram illustrates the key steps in this protocol:
Diagram: RNA-Seq Library Preparation Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Their Functions in NGS Workflows
| Research Reagent / Solution | Function in NGS Workflow | Key Characteristics |
|---|---|---|
| Magnetic SPRI Beads [4] | Size-selective cleanup and purification of DNA fragments at various steps (post-fragmentation, post-ligation, post-PCR). | Solid-phase reversible immobilization; allow for buffer-based size selection and removal of enzymes, salts, and short fragments. |
| Fragmentation Enzymes/Buffers [4] | Controlled digestion of DNA or RNA into fragments of optimal size for sequencing. | Enable reproducible and tunable fragmentation (e.g., via acoustic shearing or enzymatic digestion). |
| Indexing Adapters [4] | Unique oligonucleotide sequences ligated to DNA fragments to allow multiplexing of multiple samples in a single sequencing run. | Contain flow cell binding sequences, priming sites, and a unique dual index for sample identification post-sequencing. |
| Polymerases for Library Amplification [4] | High-fidelity PCR amplification of the adapter-ligated library to generate sufficient material for sequencing. | Exhibit high processivity and fidelity to minimize amplification biases and errors. |
| Flow Cells [6] [4] | The solid surface where clonal clusters are generated and the sequencing reaction occurs. | Coated with oligonucleotides complementary to the adapters; patterned flow cells (Illumina) increase density and data output. |
| SBS Chemistries (e.g., XLEAP-SBS) [4] | The chemical cocktail of fluorescently-labeled, reversible terminator nucleotides and enzymes used for sequencing-by-synthesis. | Determine the speed, accuracy, and read length of the sequencing run. |
The evolution from first- to third-generation sequencing technologies has been nothing short of revolutionary, each generation building upon the limitations of the last. The field continues to advance rapidly, with emerging trends focusing on multi-omic integration, spatial transcriptomics, and even higher throughput at lower costs [6] [8]. As these technologies mature and become more integrated into research and clinical pipelines, they promise to further deepen our understanding of genome biology and accelerate the development of novel diagnostics and therapeutics in the era of precision medicine.
Next-generation sequencing (NGS) is a transformative technology that enables the ultra-high-throughput, parallel sequencing of millions of DNA fragments simultaneously [10] [4]. This approach has revolutionized genomics by making large-scale sequencing dramatically faster and more cost-effective than traditional methods, facilitating a wide range of applications from basic research to clinical diagnostics [7].
Table of Contents
- Introduction to Core Principles
- NGS Workflow and Technologies
- Applications and Experimental Protocols
- The Scientist's Toolkit
The core principles of NGS represent a fundamental shift from first-generation sequencing. The key differentiator is massively parallel sequencing, which allows for the concurrent reading of billions of DNA fragments, as opposed to the one-fragment-at-a-time approach of Sanger sequencing [10]. This parallelism directly enables the other two principles: high-throughput data generation and significant cost-effectiveness.
The impact of these principles is profound. The Human Genome Project, which relied on Sanger sequencing, took 13 years and cost nearly $3 billion [10] [4]. In stark contrast, modern NGS platforms can sequence an entire human genome in hours for under $1,000 [10]. This democratization of sequencing has opened up possibilities for population-scale studies and routine clinical application.
Comparative Analysis of Sequencing Generations
Table 1: Key characteristics of different sequencing generations.
| Feature | First-Generation (Sanger) | Second-Generation (NGS) | Third-Generation (Long-Read) |
|---|---|---|---|
| Sequencing Principle | Chain-termination | Massively parallel sequencing by synthesis or ligation [4] [7] | Real-time, single-molecule sequencing [10] [7] |
| Throughput | Low (single fragment per run) | Very High (millions to billions of fragments per run) [4] | High (hundreds of thousands of long fragments) |
| Read Length | Long (500-1000 base pairs) [10] | Short (50-600 base pairs, typically) [10] | Very Long (10,000 - 30,000+ base pairs on average) [7] |
| Cost per Genome | ~$3 billion [10] | <$1,000 [10] | Higher than NGS |
| Primary Applications | Targeted sequencing, validation | Whole-genome sequencing, transcriptomics, targeted panels [4] | De novo genome assembly, resolving complex regions, epigenetic modification detection [10] |
NGS Workflow and Technologies
The standard NGS workflow is a multi-stage process that converts a purified nucleic acid sample into actionable digital sequence data. The following diagram illustrates the primary workflow and the underlying technology that enables parallel sequencing.
NGS Workflow and Sequencing Principle
Detailed Workflow Breakdown
Library Preparation
The process begins with library preparation, where input DNA or RNA is fragmented into appropriate sizes, and platform-specific adapter sequences are ligated to the ends of these fragments [10]. These adapters are essential for the subsequent steps of amplification and sequencing.
Protocol: Standard DNA Library Prep (Illumina)
- Input: 100-1000 ng of high-quality genomic DNA.
- Fragmentation: Use a Covaris sonicator or enzymatic fragmentation (e.g., Nextera tagmentation) to shear DNA to a target size of 200-500 bp.
- End-Repair & A-Tailing: Convert fragmented DNA into blunt-ended, 5'-phosphorylated fragments, then add a single 'A' nucleotide to the 3' ends.
- Adapter Ligation: Ligate indexed adapters containing the 'T' overhang to the 'A'-tailed fragments using T4 DNA ligase. Purify the ligated product with SPRI beads.
- Library QC: Validate the final library size distribution using an Agilent Bioanalyzer or TapeStation and quantify using qPCR.
Cluster Amplification
In this step, the adapter-ligated library is loaded onto a flow cell, a glass surface containing immobilized oligonucleotides complementary to the adapters. Each DNA fragment binds to the flow cell and is amplified locally in a process called bridge amplification, generating millions of clonal clusters [10]. Each cluster ultimately produces a strong enough signal to be detected by the sequencer's camera.
Parallel Sequencing
Sequencing by Synthesis (SBS) is the most common chemistry (used by Illumina). The flow cell is cyclically flooded with fluorescently tagged nucleotides. As each nucleotide is incorporated into the growing DNA chain by a polymerase, its fluorescent dye is imaged, revealing the sequence of each cluster. This process happens for hundreds of millions of clusters in parallel, creating an enormous throughput [4]. Recent advancements like XLEAP-SBS chemistry have further increased the speed and fidelity of this process [4].
Comparison of NGS Platform Technologies
Table 2: Overview of major short- and long-read NGS platform technologies.
| Platform | Technology | Amplification Method | Read Length | Key Limitation |
|---|---|---|---|---|
| Illumina | Sequencing by Synthesis | Bridge PCR [7] | 36-300 bp [7] | Short reads complicate assembly of repetitive regions [10] |
| Ion Torrent | Semiconductor (H+ detection) | Emulsion PCR [7] | 200-400 bp [7] | Signal degradation in homopolymer regions [7] |
| PacBio SMRT | Real-time sequencing (fluorescence) | None (Single Molecule) [7] | Average 10,000-25,000 bp [7] | Higher cost per run compared to short-read platforms [7] |
| Oxford Nanopore | Electrical signal detection (Nanopore) | None (Single Molecule) [7] | Average 10,000-30,000 bp [7] | Raw read error rate can be higher than other technologies [7] |
Applications and Experimental Protocols
The principles of NGS enable its application across diverse fields. The following diagram outlines the primary clinical and research applications.
Primary NGS Application Areas
Protocol: Somatic Variant Detection in Cancer
This protocol identifies tumor-specific mutations for therapy selection and resistance monitoring [11].
- Objective: To detect somatic single nucleotide variants (SNVs), insertions/deletions (indels), and copy number variations (CNVs) from matched tumor-normal sample pairs.
- Sample Requirements: 50-100 ng of DNA from both FFPE tumor tissue and matched normal (e.g., blood or saliva). Input can be lowered to 10 ng using specialized library prep kits.
- Library Preparation: Use a hybrid-capture-based targeted panel (e.g., a comprehensive cancer gene panel) or perform whole-exome sequencing. Follow manufacturer's protocol for end-repair, A-tailing, adapter ligation, and PCR amplification.
- Sequencing: Sequence on an Illumina platform to a minimum depth of 250x for tumor and 100x for normal samples for WES; deeper coverage (500-1000x) is required for targeted panels to detect low-frequency variants.
- Bioinformatic Analysis:
- Quality Control: Use FastQC to assess read quality. Trim adapters and low-quality bases with Trimmomatic [11].
- Alignment: Map reads to a reference genome (e.g., GRCh38) using a short-read aligner like BWA-MEM [11].
- Variant Calling: Use MuTect2 for SNVs/indels and Control-FREEC or GATK for CNVs. Filter variants against population databases (e.g., gnomAD, dbSNP) to remove common polymorphisms.
- Annotation & Reporting: Annotate variants with ANNOVAR or SnpEff [11]. Interpret clinically actionable variants using resources like CIViC, OncoKB, and COSMIC [11].
Protocol: Liquid Biopsy for Therapy Monitoring
Liquid biopsies analyze circulating tumor DNA (ctDNA) from blood plasma, offering a non-invasive method to track tumor dynamics [10].
- Objective: To quantify allele frequency of known tumor mutations in plasma ctDNA for monitoring treatment response and emergent resistance.
- Sample Collection & Processing: Collect 10 mL of blood in cell-free DNA collection tubes (e.g., Streck). Process within 6 hours: double centrifugation (1600xg then 16,000xg) to isolate plasma. Extract cell-free DNA using a commercial kit (e.g., QIAamp Circulating Nucleic Acid Kit).
- Library Prep & Sequencing: Prepare libraries from 10-50 ng of cell-free DNA. Use unique molecular identifiers (UMIs) to correct for PCR amplification errors and sequencing artifacts. Sequence with a targeted panel covering known resistance mutations to very high depth (>10,000x).
- Data Analysis:
- Process data through a standard alignment and variant calling pipeline (as above).
- Use UMI-based error suppression tools (e.g., fgbio) to distinguish true low-frequency variants from technical noise.
- Track the variant allele fraction (VAF) of key mutations over time. A decreasing VAF indicates treatment response, while an emerging or increasing VAF suggests resistance.
The Scientist's Toolkit
Successful NGS experimentation relies on a suite of specialized reagents, instruments, and computational tools.
Research Reagent Solutions
Table 3: Essential reagents and materials for NGS workflows.
| Item | Function | Example Kits/Platforms |
|---|---|---|
| Library Prep Kit | Prepares DNA/RNA fragments for sequencing by adding platform-specific adapters [10]. | Illumina Nextera, NEBNext Ultra II |
| Hybrid-Capture Probes | Biotinylated oligonucleotides used to enrich for specific genomic regions of interest in targeted sequencing [11]. | IDT xGen Lockdown Probes, Twist Core Exome |
| Cluster Generation Reagents | Enzymes and nucleotides for the bridge amplification on the flow cell, creating clonal clusters for sequencing [10]. | Illumina Exclusion Amplification reagents |
| SBS Chemistry Kit | Fluorescently labeled nucleotides and enzymes for the cyclic sequencing-by-synthesis reaction during the run [4]. | Illumina XLEAP-SBS Chemistry |
| Quality Control Kits | For assessing the quality, quantity, and fragment size of input DNA and final libraries pre-sequencing. | Agilent Bioanalyzer DNA High Sensitivity Kit, Qubit dsDNA HS Assay |
Essential Bioinformatics Tools and Databases
The massive data output of NGS requires robust bioinformatics pipelines for analysis and interpretation [11].
- Primary Analysis Tools:
- Key Databases for Interpretation:
- dbSNP: Catalog of common human genetic variations [11].
- COSMIC (Catalogue of Somatic Mutations in Cancer): Curated database of somatic mutations in cancer [11].
- The Cancer Genome Atlas (TCGA): Repository of cancer genomic and clinical data [11].
- Genome Aggregation Database (gnomAD): Population database of genetic variation used for filtering common variants [11].
- Cloud Computing Platforms: To handle the computational and data storage demands, cloud-based solutions like Illumina Connected Analytics and DRAGEN pipelines are widely used [11] [4].
Next-generation sequencing (NGS) technologies have revolutionized genomic research by enabling the high-throughput analysis of DNA and RNA. This section details the core principles and comparative specifications of the three major platforms: Illumina, Pacific Biosciences (PacBio), and Oxford Nanopore Technologies (ONT).
Illumina utilizes sequencing-by-synthesis (SBS) chemistry. This technology employs fluorescently labeled nucleotides that serve as reversible terminators. During each cycle, a single nucleotide is incorporated, its fluorescence is imaged for base identification, and the terminator is cleaved to allow the next cycle. This process generates massive volumes of short reads (typically 50-300 bp) with high per-base accuracy, often exceeding Q30 (99.9%) [4].
PacBio Single Molecule, Real-Time (SMRT) Sequencing is based on the real-time observation of DNA synthesis. A single DNA polymerase molecule is anchored to the bottom of a zero-mode waveguide (ZMW). As nucleotides are incorporated, each base-specific fluorescent label is briefly illuminated and detected. The key feature is the Circular Consensus Sequencing (CCS) mode, where a single DNA molecule is sequenced repeatedly in a loop. This produces HiFi (High-Fidelity) reads that combine long read lengths (typically 10-25 kb) with very high accuracy (exceeding 99.9%) [12] [13].
Oxford Nanopore Technologies (ONT) sequencing is based on the translocation of nucleic acids through protein nanopores. As DNA or RNA passes through a nanopore embedded in an electrically resistant membrane, it causes characteristic disruptions in an ionic current. These current changes are decoded in real-time to determine the nucleotide sequence. A primary advantage of this technology is its ability to sequence long fragments (from 50 bp to over 4 Mb) directly from native DNA/RNA, thereby preserving base modification information like methylation as a standard feature [14] [15].
Table 1: Comparative Specifications of Major NGS Platforms
| Feature | Illumina | PacBio (SMRT Sequencing) | Oxford Nanopore (ONT) |
|---|---|---|---|
| Core Technology | Sequencing-by-Synthesis (SBS) [4] | Single Molecule, Real-Time (SMRT) in Zero-Mode Waveguides (ZMWs) [13] | Nanopore-based current sensing [15] |
| Read Length | Short reads (up to 300 bp, paired-end) [4] | Long reads (average 10-25 kb) [13] | Ultra-long reads (50 bp to >4 Mb) [15] |
| Typical Accuracy | >Q30 (99.9%) [4] | >Q27 (99.9%) for HiFi reads [12] | ~Q20 (99%) with latest chemistries [12] |
| Primary Strengths | High throughput, low per-base cost, well-established workflows [4] | High accuracy long reads, direct methylation detection [16] [13] | Ultra-long reads, real-time analysis, portability [14] [15] |
| Key Limitations | Short reads limit resolution in complex regions, amplification bias [15] | Lower throughput per instrument compared to Illumina, higher DNA input requirements [13] | Higher raw error rate than competitors, though improving [17] |
| Methylation Detection | Requires specialized prep (bisulfite sequencing) [18] | Built-in (kinetics-based) for 6mA and more [13] | Built-in (signal-based) for 5mC, 6mA, and more [14] |
Performance Comparison in Microbial Community Profiling
Amplicon sequencing of the 16S rRNA gene is a foundational method for profiling microbial communities. The choice of sequencing platform significantly impacts taxonomic resolution and perceived community composition, as demonstrated by recent comparative studies.
A 2025 study on rabbit gut microbiota directly compared Illumina (V3-V4 region), PacBio (full-length 16S), and ONT (full-length 16S). The results highlighted a clear advantage for long-read platforms in species-level resolution. ONT classified 76% of sequences to the species level, PacBio classified 63%, while Illumina classified only 48%. However, a critical limitation was noted across all platforms: a majority of sequences classified at the species level were assigned ambiguous names like "uncultured_bacterium," indicating persistent gaps in reference databases rather than a failure of the technology itself [12].
Furthermore, the same study found significant differences in beta diversity analysis, showing that the taxonomic compositions derived from the three platforms were not directly interchangeable. This underscores that the sequencing platform and the choice of primers are significant variables in experimental design [12]. A separate 2025 study on soil microbiomes concluded that PacBio and ONT provided comparable assessments of bacterial diversity, with PacBio showing a slight edge in detecting low-abundance taxa. Importantly, despite ONT's inherent higher error rate, its results closely matched PacBio's for well-represented taxa, suggesting that the errors do not critically impact the broader interpretation of community structure [19].
Table 2: 16S rRNA Sequencing Performance Across Platforms
| Metric | Illumina (V3-V4) | PacBio (Full-Length) | ONT (Full-Length) |
|---|---|---|---|
| Species-Level Classification Rate | 48% [12] | 63% [12] | 76% [12] |
| Genus-Level Classification Rate | 80% [12] | 85% [12] | 91% [12] |
| Representative Read Length | 442 bp [12] | 1,453 bp [12] | 1,412 bp [12] |
| Impact on Beta Diversity | Significant differences observed compared to long-read platforms [12] | Significant differences observed compared to other platforms [12] | Significant differences observed compared to other platforms [12] |
| Reported Community Richness | Higher (in respiratory microbiome study) [17] | Comparable to ONT in soil study [19] | Slightly lower than Illumina in respiratory study [17] |
Detailed Experimental Protocol: Full-Length 16S rRNA Amplicon Sequencing
The following protocol details a standardized workflow for full-length 16S rRNA gene sequencing, adapted for both PacBio and Oxford Nanopore platforms, based on recently published methods [12] [19].
Sample Preparation and DNA Extraction
- Sample Collection: For gut microbiota studies, collect soft feces or intestinal content samples and immediately freeze them at -72°C until DNA extraction [12]. For soil microbiomes, homogenize soil samples and pass through a 1 mm sieve under sterile conditions before storage [19].
- DNA Extraction: Isolate bacterial genomic DNA using a dedicated kit, such as the DNeasy PowerSoil Kit (QIAGEN) or the Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research), following the manufacturer's protocol [12] [19].
- Quality Control: Quantify the extracted DNA using a fluorescence-based method (e.g., Qubit Fluorometer). Assess DNA quality and integrity via agarose gel electrophoresis or a Fragment Analyzer [19].
PCR Amplification and Library Preparation
The steps diverge based on the target platform.
A. PacBio HiFi Library Preparation
- PCR Amplification: Amplify the full-length 16S rRNA gene using universal primers 27F and 1492R, tailed with PacBio barcode sequences. Use a high-fidelity DNA polymerase (e.g., KAPA HiFi HotStart) and run the reaction for 27-30 cycles [12] [19].
- Amplicon Pooling: Verify the PCR product size and quantity using a Fragment Analyzer. Pool amplicons from different samples in equimolar concentrations.
- SMRTbell Library Construction: Prepare the library from the pooled amplicons using the SMRTbell Express Template Prep Kit 2.0 or 3.0. This creates a circular DNA template suitable for SMRT sequencing [12] [19].
- Sequencing: Sequence the library on a PacBio Sequel II or Revio system using a sequencing kit such as the Sequel II Sequencing Kit 2.0, typically with a 10-hour movie time [12] [19].
B. Oxford Nanopore Library Preparation
- PCR Amplification: Amplify the full-length 16S rRNA gene (V1-V9 regions) using the primers from the 16S Barcoding Kit (e.g., SQK-16S024). Perform PCR amplification for 40 cycles [12].
- Amplicon Purification and Pooling: Purify the PCR product using magnetic beads (e.g., KAPA HyperPure Beads). Quantify the purified amplicons and pool them in equimolar amounts [19].
- Sequencing: Load the pooled library onto a MinION flow cell (e.g., R10.4.1) and sequence on a MinION device. Sequencing can be run for a predetermined time (e.g., 24-72 hours) or until the flow cell is exhausted [12] [17].
Bioinformatic Analysis
- PacBio Data Processing: Process the raw subreads to generate Circular Consensus Sequences (CCS) reads, which are high-fidelity HiFi reads. Demultiplex the reads by sample barcodes. Use the DADA2 pipeline in QIIME2 for denoising, error-correction, and generation of Amplicon Sequence Variants (ASVs) [12].
- ONT Data Processing: Basecall and demultiplex raw FAST5 files using Dorado with a high-accuracy (HAC) model. Due to the higher error profile, ONT reads are often processed using specialized pipelines like Spaghetti or the EPI2ME 16S Workflow, which employ an Operational Taxonomic Unit (OTU) clustering approach for denoising [12] [17].
- Taxonomic Assignment: For both platforms, import high-quality sequences into QIIME2. A Naïve Bayes classifier, trained on a reference database (e.g., SILVA) and customized for the specific primers and read length, is typically used for taxonomic annotation from phylum to species level [12].
- Downstream Analysis: Perform diversity analysis (alpha and beta diversity) using tools like phyloseq in R. Filter out sequences classified as Archaea, Eukaryota, or unassigned, and remove low-abundance ASVs/OTUs (e.g., those with a relative abundance below 0.01%) to minimize artifacts [12].
Diagram 1: Full-length 16S rRNA amplicon sequencing workflow for PacBio and ONT platforms.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Reagents and Kits for NGS Workflows
| Item | Function | Example Products |
|---|---|---|
| DNA Extraction Kit | Isolation of high-quality, inhibitor-free genomic DNA from complex samples. | DNeasy PowerSoil Kit (QIAGEN) [12], Quick-DNA Fecal/Soil Microbe Microprep Kit (Zymo Research) [19] |
| High-Fidelity DNA Polymerase | Accurate amplification of the target 16S rRNA gene with low error rates during PCR. | KAPA HiFi HotStart DNA Polymerase [12] |
| Full-Length 16S Primers | Amplification of the ~1,500 bp full-length 16S rRNA gene. | Universal primers 27F / 1492R [12] [19] |
| Multiplexing Barcodes | Sample-specific nucleotide tags allowing pooled sequencing of multiple libraries. | PacBio Barcoded Primers [12], ONT Native Barcoding Kit 96 [19] |
| SMRTbell Prep Kit | Construction of SMRTbell libraries for PacBio circular consensus sequencing. | SMRTbell Express Template Prep Kit 2.0/3.0 [12] [19] |
| ONT 16S Barcoding Kit | An all-in-one kit for amplification, barcoding, and library prep for Nanopore 16S sequencing. | SQK-16S024 [12] |
| Sequencing Kit & Flow Cell | Platform-specific reagents and consumables for the sequencing run. | Sequel II Sequencing Kit 2.0, SMRT Cell [12], MinION Flow Cell (R10.4.1) [17] |
Next-generation sequencing (NGS) has revolutionized genomic research and clinical diagnostics by enabling high-throughput, cost-effective analysis of DNA and RNA. This guide details the experimental protocols and key applications of four major sequencing methodologies.
Whole Genome Sequencing (WGS)
Whole Genome Sequencing provides a comprehensive view of an organism's complete genetic code, enabling the discovery of variants across coding, non-coding, and structural regions.
WGS identifies single nucleotide polymorphisms (SNPs), insertions/deletions (indels), structural variations (SVs), and copy number variations (CNVs) across the entire genome. The UK Biobank project demonstrated its power by sequencing 490,640 participants, identifying approximately 1.5 billion genetic variants—42 times more than what was detectable through whole-exome sequencing [20]. This comprehensive approach is invaluable for population genetics, rare disease research, and characterizing the non-coding genome.
Experimental Protocol: Illumina Short-Read WGS
Library Preparation
- DNA Extraction: Obtain high-quality, high-molecular-weight DNA from source material (e.g., blood, tissue, cells) using systems like Autopure LS (Qiagen) or GENE PREP STAR NA-480 (Kurabo). Assess DNA quality and quantity with fluorescence-based methods (e.g., Quant-iT PicoGreen dsDNA kit) [21].
- Fragmentation: Fragment genomic DNA to an average target size of 550 bp using a focused-ultrasonicator (e.g., Covaris LE220) [21].
- Library Construction: Use a PCR-free library prep kit (e.g., TruSeq DNA PCR-free HT for Illumina or MGIEasy PCR-Free DNA Library Prep Set for MGI platforms) to prevent amplification bias. Ligate platform-specific adapters, which often include unique dual indexes for multiplexing [21].
- Automation: Employ automated liquid handling systems (e.g., Agilent Bravo, Biomek NXp, or MGI SP-960) for high-throughput, reproducible library preparation [21].
Sequencing & Data Analysis
- Sequencing: Load libraries onto a high-throughput platform (e.g., Illumina NovaSeq X Plus, NovaSeq 6000, or DNBSEQ-T7). Sequence to a minimum coverage of 30x for human genomes; the UK Biobank achieved an average coverage of 32.5x [20].
- Primary Analysis: Convert raw signal data (e.g., base calls) into FASTQ files containing sequence reads and quality scores.
- Secondary Analysis:
- Alignment: Map reads to a reference genome (e.g., GRCh38) using aligners like BWA or BWA-mem2 [21].
- Variant Calling: Identify SNPs and indels using GATK HaplotypeCaller. Perform joint genotyping across multiple samples with GATK GnarlyGenotyper, followed by variant quality score recalibration (VQSR) [21].
- SV Calling: Detect larger structural variants (>50 bp) using specialized callers like DRAGEN SV [20].
WGS Research Reagent Solutions
| Item | Function | Example Products/Kits |
|---|---|---|
| DNA Extraction Kit | Iserts high-quality, high-molecular-weight DNA for accurate long-range analysis. | Autopure LS (Qiagen), GENE PREP STAR NA-480 (Kurabo) [21] |
| DNA Quantitation Kit | Precisely measures DNA concentration using fluorescent dye binding, critical for optimal library preparation. | Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen) [21] |
| PCR-free Library Prep Kit | Prepares sequencing libraries without PCR amplification to prevent associated biases and errors. | TruSeq DNA PCR-free HT (Illumina), MGIEasy PCR-Free Set (MGI) [21] |
| Unique Dual Indexes | Allows multiplexing of numerous samples by tagging each with unique barcodes, enabling sample pooling and post-sequencing demultiplexing. | IDT for Illumina TruSeq DNA UD Indexes [21] |
| Sequencing Reagent Kit | Provides enzymes, buffers, and nucleotides required for the sequencing-by-synthesis chemistry on the platform. | NovaSeq X Plus 10B/25B Reagent Kit (Illumina) [21] |
Whole Exome Sequencing (WES)
Whole Exome Sequencing targets the protein-coding regions of the genome (the exome), which constitutes about 1-2% of the genome but harbors the majority of known disease-causing variants.
WES provides a cost-effective method for identifying variants in exonic regions, making it highly efficient for diagnosing rare Mendelian disorders and other conditions linked to coding sequences. It is considered medically necessary for specific clinical presentations, such as multiple anomalies not suggestive of a specific condition, developmental delay, or congenital epilepsy of unknown cause [22].
WES vs. WGS Quantitative Comparison
| Parameter | Whole Exome Sequencing (WES) | Whole Genome Sequencing (WGS) |
|---|---|---|
| Target Region | ~1.5% of genome (exonic regions) [22] | 100% of genome [20] |
| Typical Coverage | 100x - 150x | 30x - 40x |
| Variant Discovery (in UK Biobank) | ~25 million variants [20] | ~1.5 billion variants (60x more) [20] |
| 5' and 3' UTR Variant Capture | Low (e.g., ~10-30% of variants) [20] | ~99% of variants [20] |
| Primary Clinical Use | Diagnosis of rare genetic disorders, idiopathic developmental delay [22] | Comprehensive variant discovery, non-coding region analysis, structural variation [20] |
Experimental Protocol: Illumina Short-Read WES
Library Preparation & Target Enrichment
- Library Construction: Follow a similar initial protocol as WGS: fragment DNA and ligate sequencing adapters.
- Target Capture: The critical differentiating step for WES. Hybridize the library to biotinylated probes (e.g., Illumina Exome Panel, IDT xGen Exome Research Panel) that are complementary to the exonic regions. These probes "pull down" the target fragments.
- Capture Enrichment: Use streptavidin-coated magnetic beads to bind the biotinylated probe-DNA hybrids. Wash away non-hybridized, off-target fragments, leaving an enriched library of exonic sequences.
Sequencing & Data Analysis
- Sequencing: Sequence the enriched library on a short-read platform (e.g., Illumina NovaSeq) to a high depth of coverage (>100x is standard) to ensure confidence in variant calls.
- Data Analysis: The bioinformatics pipeline is similar to WGS (alignment, variant calling), but analysis is focused on the exonic regions. Annotation and prioritization of variants within coding sequences and splice sites are paramount.
Transcriptome Sequencing (RNA-seq)
Transcriptome sequencing analyzes the complete set of RNA transcripts in a cell at a specific point in time, enabling the study of gene expression, alternative splicing, and gene fusions.
RNA-seq provides a quantitative and qualitative profile of the transcriptome. It is pivotal for understanding cellular responses, disease mechanisms, and identifying biomarkers. Single-cell RNA-sequencing (scRNA-seq) has revolutionized this field by resolving cellular heterogeneity within complex tissues, uncovering novel and rare cell types, and mapping gene expression in the context of tissue structure (spatial transcriptomics) [23] [24]. Key applications include tumor microenvironment dissection, drug discovery, and developmental biology [23].
Experimental Protocol: Bulk RNA-seq
Library Preparation
- RNA Extraction: Isolve total RNA or mRNA from samples (tissue, cells) using kits that preserve RNA integrity (e.g., Qiagen RNeasy). Assess RNA Quality (e.g., RIN score via Bioanalyzer).
- rRNA Depletion or Poly-A Selection: Enrich for messenger RNA (mRNA) by either:
- Poly-A Selection: Use oligo(dT) beads to capture mRNA molecules with poly-A tails.
- rRNA Depletion: Use probes to remove abundant ribosomal RNA (rRNA).
- cDNA Synthesis: Convert RNA into double-stranded cDNA using reverse transcriptase and DNA polymerase.
- Library Construction: Fragment the cDNA, ligate sequencing adapters, and amplify the final library via PCR.
Sequencing & Data Analysis
- Sequencing: Sequence on an Illumina platform (e.g., NovaSeq) with paired-end reads (e.g., 2x150 bp) recommended for splicing analysis.
- Data Analysis:
- Quality Control: Assess raw read quality with FastQC.
- Alignment: Map reads to a reference genome/transcriptome using splice-aware aligners (e.g., STAR, HISAT2).
- Quantification: Count reads aligned to genes/transcripts to create an expression matrix.
- Differential Expression: Use tools like DESeq2 or edgeR to identify significantly differentially expressed genes between conditions.
scRNA-seq Research Reagent Solutions
| Item | Function | Example Products/Kits |
|---|---|---|
| Single-Cell Isolation System | Gently dissociates tissue and isolates individual live cells for downstream processing. | 10x Genomics Chromium Controller, Fluorescent-Activated Cell Sorter (FACS) [23] |
| Single-Cell Library Prep Kit | Creates barcoded sequencing libraries from single cells, enabling thousands of cells to be processed in one experiment. | 10x Genomics Single Cell Gene Expression kits [23] |
| Cell Lysis Buffer | Breaks open individual cells to release RNA while preserving RNA integrity. | Component of commercial scRNA-seq kits [23] |
| Reverse Transcriptase Master Mix | Converts the RNA from each single cell into stable, barcoded cDNA during the GEM (Gel Bead-in-Emulsion) step. | Component of commercial scRNA-seq kits [23] |
| Barcoded Beads | Microbeads containing cell- and molecule-specific barcodes that uniquely tag all cDNA from a single cell. | 10x Genomics Barcoded Gel Beads [23] |
Epigenomics
Epigenomics involves the genome-wide study of epigenetic modifications—heritable changes in gene expression that do not involve changes to the underlying DNA sequence. Key modifications include DNA methylation, histone modifications, and non-coding RNA-associated silencing.
Epigenetic aberrations are crucial in tumor diseases, cardiovascular disease, diabetes, and neurological disorders [25]. Clinical sampling for epigenetics can involve tissue biopsies, blood (for cell-free DNA analysis), saliva, and isolated specific cell types (e.g., circulating tumor cells) [25]. HiFi long-read sequencing (PacBio) can now detect base modifications like 5mC methylation simultaneously with standard sequencing, providing phased haplotyping and methylation profiles from a single experiment [26].
Experimental Protocol: DNA Methylation Analysis (Bisulfite Sequencing)
Bisulfite Conversion & Library Prep
- Bisulfite Treatment: Treat DNA with sodium bisulfite, which converts unmethylated cytosines to uracils (which are read as thymines in sequencing), while methylated cytosines (5mC) remain unchanged. This creates sequence differences based on methylation status.
- Library Preparation: Prepare a sequencing library from the bisulfite-converted DNA. Specialized kits are available that account for the DNA damage caused by bisulfite treatment.
- Note on Long-Read Methods: With PacBio HiFi sequencing, 5mC detection in CpG contexts is achieved natively without bisulfite conversion, as the polymerase kinetics are sensitive to the modification [26].
Sequencing & Data Analysis
- Sequencing: Sequence the library on an appropriate platform. For whole-genome bisulfite sequencing (WGBS), this is typically done on an Illumina system.
- Data Analysis:
- Alignment: Use bisulfite-aware aligners (e.g., Bismark, BSMAP) that account for the C-to-T conversion in the reads.
- Methylation Calling: Calculate the methylation ratio at each cytosine position by comparing the number of reads reporting a 'C' (methylated) versus a 'T' (unmethylated).
The Impact of NGS on Genomics Research and Precision Medicine
Next-generation sequencing (NGS) has fundamentally transformed genomics research and clinical diagnostics by enabling the rapid, high-throughput sequencing of DNA and RNA molecules [11]. This technology allows researchers to sequence millions to billions of DNA fragments simultaneously, providing unprecedented insights into genome structure, genetic variations, gene expression profiles, and epigenetic modifications [7]. The versatility of NGS platforms has expanded the scope of genomics research, facilitating studies on rare genetic diseases, cancer genomics, microbiome analysis, infectious diseases, and population genetics [27]. NGS has progressed through multiple generations of technological advancement, beginning with first-generation Sanger sequencing, evolving to dominant second-generation short-read platforms, and expanding to include third-generation long-read and real-time sequencing technologies [28] [7]. The continuous innovation in NGS technologies has driven down costs while dramatically increasing speed and accessibility, making large-scale genomic studies and routine clinical applications feasible [24].
NGS Technologies and Platforms
Platform Diversity and Specifications
The NGS landscape features multiple platforms employing distinct sequencing chemistries, each with unique advantages and limitations. Key players include Illumina's sequencing-by-synthesis, Thermo Fisher's Ion Torrent semiconductor sequencing, Pacific Biosciences' single-molecule real-time (SMRT) sequencing, and Oxford Nanopore's nanopore-based sequencing [7]. These platforms differ significantly in their throughput, read length, accuracy, cost, and application suitability, requiring researchers to carefully match platform capabilities to their specific research goals [28].
Table 1: Comparison of Major NGS Platforms and Their Capabilities
| Platform | Sequencing Technology | Read Length | Key Advantages | Primary Applications |
|---|---|---|---|---|
| Illumina | Sequencing-by-Synthesis (SBS) | 75-300 bp (short) | High accuracy (99.9%), high throughput, low cost per base | Whole genome sequencing, targeted sequencing, gene expression, epigenetics [27] [7] |
| PacBio SMRT | Single-molecule real-time | Average 10,000-25,000 bp (long) | Long reads, detects epigenetic modifications | De novo genome assembly, structural variant detection, full-length transcript sequencing [7] |
| Oxford Nanopore | Nanopore detection | Average 10,000-30,000 bp (long) | Real-time sequencing, portable options, longest read lengths | Real-time surveillance, field sequencing, structural variation [24] [7] |
| Ion Torrent | Semiconductor sequencing | 200-400 bp (short) | Fast run times, simple workflow | Targeted sequencing, small genome sequencing [7] |
| Roche SBX | Sequencing by Expansion | Information Missing | Ultra-rapid, high signal-to-noise, scalable | Whole genome, exome, and RNA sequencing (promised for future applications) [29] |
Emerging Innovations in NGS Technology
The NGS technology landscape continues to evolve with emerging innovations that address limitations of current platforms. In 2025, Roche unveiled its novel Sequencing by Expansion (SBX) technology, which represents a new approach to NGS [29]. This method uses a sophisticated biochemical process to encode the sequence of a target nucleic acid molecule into a measurable surrogate polymer called an Xpandomer, which is fifty times longer than the original molecule [29]. These Xpandomers encode sequence information into high signal-to-noise reporters, enabling highly accurate single-molecule nanopore sequencing using a Complementary Metal Oxide Semiconductor (CMOS)-based sensor module [29]. This technology promises to reduce the time from sample to genome from days to hours, potentially significantly speeding up genomic research and clinical applications [29]. Additionally, advances in long-read sequencing technologies from PacBio and Oxford Nanopore are continuously improving accuracy and read length while reducing costs, enabling more comprehensive genome analysis and closing gaps in genomic coverage [30] [7].
NGS Workflow: From Sample to Insight
Standardized Experimental Protocol
The NGS workflow consists of three major stages: (1) template preparation, (2) sequencing and imaging, and (3) data analysis [28]. The following protocol details the critical steps for DNA whole-genome sequencing using Illumina platforms, which can be adapted for other applications and platforms with appropriate modifications.
Template Preparation
Day 1: Nucleic Acid Extraction and Quality Control (2-4 hours)
- Extract DNA from biological samples using validated extraction kits suitable for your sample type (e.g., blood, tissue, cells).
- Quantify DNA using fluorometric methods (e.g., Qubit) to ensure accurate concentration measurement.
- Assess DNA quality through agarose gel electrophoresis or automated electrophoresis systems (e.g., TapeStation, Bioanalyzer). High-quality DNA should show minimal degradation with a DNA Integrity Number (DIN) >7.0.
- Dilute DNA to working concentration in low-EDTA TE buffer or nuclease-free water based on library preparation requirements.
Day 1-2: Library Preparation (6-8 hours)
- Fragment DNA to desired size (typically 200-500bp) using enzymatic fragmentation (e.g., tagmentation) or acoustic shearing (e.g., Covaris).
- Perform end-repair and A-tailing to create blunt-ended, 5'-phosphorylated fragments with a single 3'A overhang.
- Ligate adapters containing platform-specific sequences, sample indexes (barcodes), and sequencing primer binding sites using DNA ligase.
- Clean up ligation reaction using solid-phase reversible immobilization (SPRI) beads to remove excess adapters and reaction components.
- Amplify library via limited-cycle PCR (typically 4-10 cycles) to enrich for properly ligated fragments and incorporate complete adapter sequences.
- Perform final library cleanup using SPRI beads to remove PCR reagents and select for appropriate fragment sizes.
- Validate library quality using automated electrophoresis systems to confirm expected size distribution and absence of adapter dimers.
- Quantify final library using fluorometric methods for accurate concentration measurement.
Sequencing and Imaging
Day 2-3: Cluster Generation and Sequencing (1-3 days depending on platform)
- Dilute libraries to appropriate concentration for loading onto flow cell.
- Denature library to create single-stranded DNA templates.
- Load library onto flow cell for cluster generation via bridge amplification.
- Perform sequencing using the appropriate sequencing kit and cycle number for your desired read length.
- For paired-end sequencing: Sequence from both ends of the fragments by performing an additional round of sequencing after fragment reversal.
Data Analysis
Day 3-5: Bioinformatics Processing (Timing varies with computational resources)
- Convert base calls from binary format to FASTQ sequence files.
- Perform quality control on raw reads using FastQC or similar tools.
- Trim adapters and low-quality bases using tools like Trimmomatic or Cutadapt [11].
- Align reads to reference genome using aligners such as BWA (Burrows-Wheeler Aligner) or STAR (for RNA-Seq) [11].
- Process aligned reads in BAM format, including sorting, duplicate marking, and local realignment around indels.
- Call variants using appropriate tools (e.g., GATK for SNPs and indels, Manta for structural variants) [11].
- Annotate variants using databases such as dbSNP, gnomAD, ClinVar, and COSMIC to determine functional and clinical significance [11].
- Interpret results in biological and clinical context.
Research Reagent Solutions
Successful NGS experiments require high-quality reagents and materials throughout the workflow. The following table details essential research reagent solutions for NGS library preparation and sequencing.
Table 2: Essential Research Reagents for NGS Workflows
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Nucleic Acid Extraction Kits | QIAamp DNA/RNA Blood Mini Kit, DNeasy Blood & Tissue Kit | Isolate high-quality DNA/RNA from various sample types | Critical for obtaining high-quality input material; choice depends on sample source and yield requirements |
| Library Preparation Kits | KAPA HyperPrep Kit, Illumina DNA Prep | Fragment DNA, repair ends, add adapters, and amplify library | Kit selection depends on application (WGS, targeted, RNA-Seq) and input DNA quantity/quality |
| Target Enrichment Kits | Illumina Nextera Flex for Enrichment, Twist Target Enrichment | Enrich specific genomic regions of interest | Essential for targeted sequencing; uses hybridization capture or amplicon-based approaches |
| Sequencing Kits | Illumina SBS Kits, PacBio SMRTbell Prep Kit | Provide enzymes, nucleotides, and buffers for sequencing reaction | Platform-specific; determine read length, quality, and output |
| Quality Control Reagents | Qubit dsDNA HS/BR Assay Kits, Agilent High Sensitivity DNA Kit | Quantify and qualify nucleic acids at various workflow stages | Essential for ensuring library quality before sequencing; prevents failed runs |
| Cleanup Reagents | AMPure XP Beads, ProNex Size-Selective Purification System | Remove enzymes, nucleotides, and short fragments; size selection | SPRI bead-based methods are standard for most cleanup and size selection steps |
| Barcodes/Adapters | Illumina TruSeq DNA UD Indexes, IDT for Illumina Nextera DNA UD Indexes | Enable sample multiplexing and platform compatibility | Unique dual indexes (UDIs) enable higher-plex multiplexing and reduce index hopping |
Clinical Applications and Impact
Precision Oncology Applications
NGS has revolutionized cancer diagnostics and treatment selection through comprehensive genomic profiling of tumors [27]. By identifying somatic mutations, gene fusions, copy number alterations, and biomarkers of therapy response, NGS enables molecularly guided treatment strategies in precision oncology [27]. Key applications in oncology include:
1. Comprehensive Genomic Profiling (CGP) CGP utilizes large NGS panels (typically 300-500 genes) to simultaneously identify multiple classes of genomic alterations in tumor tissue [27]. This approach replaces sequential single-gene testing, conserving valuable tissue and providing a more complete molecular portrait of the tumor. CGP can identify actionable mutations in genes such as EGFR, ALK, ROS1, BRAF, and KRAS, guiding selection of targeted therapies [27]. The FDA-approved AVENIO Tumor Tissue CGP Automated Kit (Roche/Foundation Medicine collaboration) exemplifies the translation of NGS into validated clinical diagnostics [29].
2. Liquid Biopsy and Minimal Residual Disease (MRD) Monitoring Liquid biopsy analyzes circulating tumor DNA (ctDNA) from blood samples, providing a non-invasive method for cancer detection, therapy selection, and monitoring [27] [31]. These tests can detect tiny fragments of tumor DNA floating in someone's bloodstream, with minimal residual disease (MRD) tests capable of detecting cancer recurrence months before traditional scans would show evidence [31]. Liquid biopsies are particularly valuable when tumor tissue is unavailable or for monitoring treatment response and resistance in real-time [27].
3. Immunotherapy Biomarker Discovery NGS enables identification of biomarkers that predict response to immune checkpoint inhibitors, including tumor mutational burden (TMB), microsatellite instability (MSI), and PD-L1 expression [27]. High TMB (typically ≥10 mutations/megabase) correlates with improved response to immunotherapy across multiple cancer types, and NGS panels can simultaneously assess TMB while detecting other actionable alterations [27].
Rare Disease Diagnostics
NGS has dramatically improved the diagnostic yield for rare genetic disorders, which often involve heterogeneous mutations across hundreds of genes [24]. Whole exome sequencing (WES) and whole genome sequencing (WGS) can identify pathogenic variants in previously undiagnosed cases, ending diagnostic odysseys for patients and families [24]. Rapid whole-genome sequencing (rWGS) has shown particular utility in neonatal and pediatric intensive care settings, where rapid diagnosis can directly impact acute management decisions [24]. The ability to simultaneously analyze trio samples (proband and parents) enhances variant interpretation by establishing inheritance patterns and de novo mutation rates [11].
Pharmacogenomics and Personalized Therapeutics
NGS facilitates personalized drug therapy by identifying genetic variants that influence drug metabolism, efficacy, and toxicity [24] [31]. Pharmacogenomic profiling using targeted NGS panels can detect clinically relevant variants in genes such as CYP2C9, CYP2C19, CYP2D6, VKORC1, and TPMT, guiding medication selection and dosing [31]. This approach minimizes adverse drug reactions and improves therapeutic outcomes by aligning treatment with individual genetic profiles [24].
Future Directions and Challenges
Emerging Trends in NGS Technology
The NGS field continues to evolve with several emerging trends shaping its future development and application. A significant shift is occurring toward multi-omics integration, combining genomic data with transcriptomic, epigenomic, proteomic, and metabolomic information from the same sample [24] [30] [31]. This approach provides a more comprehensive view of biological systems, linking genetic information with molecular function and phenotypic outcomes [24]. The multi-omics market is projected to grow substantially from USD 3.10 billion in 2025 to USD 12.65 billion by 2035, reflecting increased adoption [31].
Spatial transcriptomics and single-cell sequencing represent another frontier, enabling researchers to explore cellular heterogeneity and gene expression patterns within tissue architecture [24] [30]. These technologies allow direct sequencing of genomic variations such as cancer mutations and immune receptor sequences in single cells within their native spatial context in tissue, empowering exploration of complex cellular interactions and disease mechanisms with unprecedented precision [30]. The year 2025 is expected to see increased routine 3D spatial studies to comprehensively assess cellular interactions in the tissue microenvironment [30].
Artificial intelligence and machine learning are increasingly integrated into NGS data analysis, enhancing variant detection, interpretation, and biological insight extraction [24] [30]. AI-powered tools like Google's DeepVariant utilize deep learning to identify genetic variants with greater accuracy than traditional methods [24]. Machine learning models analyze polygenic risk scores to predict individual susceptibility to complex diseases and accelerate drug discovery by identifying novel therapeutic targets [24].
Addressing Implementation Challenges
Despite rapid advancements, several challenges remain for widespread NGS implementation in research and clinical settings. Data management and analysis present significant hurdles, with each human genome generating approximately 100 gigabytes of raw data [11] [31]. The massive scale and complexity of genomic datasets demand advanced computational tools, cloud computing infrastructure, and bioinformatics expertise [11] [24]. Cloud platforms such as Amazon Web Services (AWS) and Google Cloud Genomics provide scalable solutions for storing, processing, and analyzing large genomic datasets while ensuring compliance with regulatory frameworks like HIPAA and GDPR [24].
Cost and accessibility issues persist, particularly for comprehensive genomic tests and in resource-limited settings [27] [32]. While the cost of whole-genome sequencing has dropped dramatically to approximately $200 per genome on platforms like Illumina's NovaSeq X Plus, the total cost of ownership including instrumentation, reagents, and computational infrastructure remains substantial [32]. Efforts to democratize access through streamlined workflows, automated library preparation (e.g., Roche's AVENIO Edge system), and decentralized manufacturing for cell and gene therapies are helping to address these barriers [24] [29].
Interpretation of variants of uncertain significance (VUS) and standardization of clinical reporting continue to challenge the field [27]. As more genes are sequenced across diverse populations, the number of identified VUS increases, creating uncertainty for clinicians and patients [27]. Developing more sophisticated functional annotation tools, aggregating data across institutions, and implementing AI-driven interpretation platforms will be essential for improving variant classification [27] [30].
The United States NGS market reflects these dynamic trends, with projections indicating substantial growth from US$3.88 billion in 2024 to US$16.57 billion by 2033, driven by personalized medicine applications, research expansion into agriculture and environmental sciences, and continued technological advancements in automation and data analysis [32]. This growth trajectory underscores the transformative impact NGS continues to have across biomedical research and clinical practice.
NGS in Action: Techniques and Transformative Applications in Research and Drug Discovery
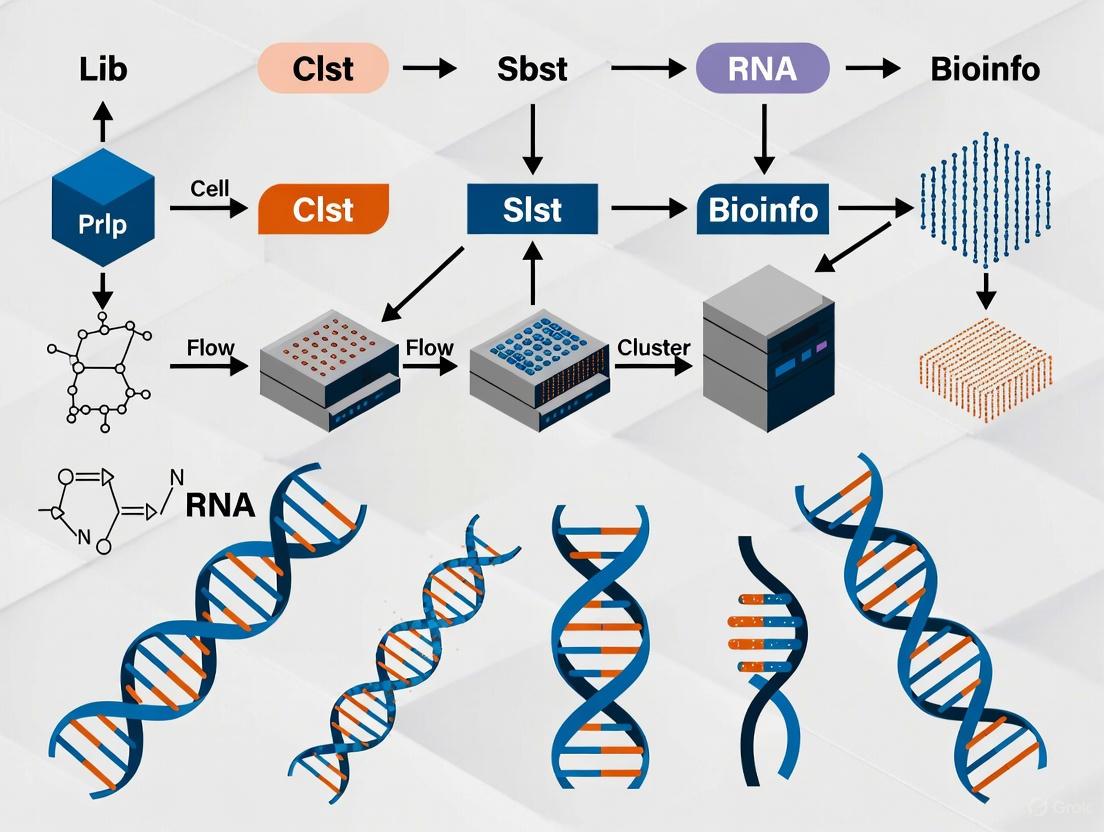
Next-generation sequencing (NGS) has revolutionized genomics research by enabling the massively parallel sequencing of millions of DNA fragments, providing ultra-high throughput, scalability, and speed at a fraction of the cost and time of traditional methods [10] [4]. This transformative technology has made large-scale whole-genome sequencing accessible and practical for average researchers, shifting from the decades-old Sanger sequencing method that could only read one DNA fragment at a time [10] [28]. The NGS workflow encompasses all steps from biological sample acquisition through computational data analysis, with each phase being critical for generating accurate, reliable genetic information [33] [28]. This application note provides a comprehensive overview of the end-to-end NGS workflow, detailed protocols, and current technological applications framed within the context of DNA and RNA analysis for research and drug development.
The versatility of NGS platforms has expanded the scope of genomics research across diverse domains including clinical diagnostics, cancer genomics, rare genetic diseases, microbiome analysis, infectious diseases, and population genetics [7]. By allowing researchers to rapidly sequence entire genomes, deeply sequence target regions, analyze epigenetic factors, and quantify gene expression, NGS has become an indispensable tool for precision medicine approaches and targeted therapy development [24] [4]. The continuous evolution of NGS technologies has driven consistent improvements in sequencing accuracy, read length, and cost-effectiveness, supporting increasingly sophisticated applications in both basic research and clinical settings [7].
The fundamental NGS workflow consists of three primary stages: (1) sample preparation and library construction, (2) sequencing, and (3) data analysis [28] [4]. This process transforms biological samples into interpretable genetic information through a coordinated series of molecular and computational steps. The following diagram illustrates the complete end-to-end workflow:
Figure 1: Comprehensive overview of the end-to-end NGS workflow from sample acquisition to data analysis, highlighting the four major stages with their key components.
Sample Preparation and Library Construction
Nucleic Acid Extraction
The initial step in every NGS protocol involves extracting high-quality nucleic acids (DNA or RNA) from biological samples [33]. The quality of extracted nucleic acids directly depends on the quality of the starting material and appropriate sample storage, typically involving freezing at specific temperatures [33].
Protocol: Nucleic Acid Extraction from Blood Samples
- Sample Lysis: Add 20 mL of blood to 30 mL of red blood cell lysis buffer, mix by inversion, and incubate for 10 minutes at room temperature. Centrifuge at 2,000 × g for 10 minutes and discard supernatant [33].
- Cell Lysis: Resuspend pellet in 10 mL of cell lysis buffer with proteinase K, mix thoroughly, and incubate at 56°C for 60 minutes with occasional mixing [33].
- RNA Separation: For RNA extraction, add RNA-specific binding buffers and purify using spin-column technology. For DNA, proceed with organic extraction [33].
- Precipitation and Washing: Add equal volume of isopropanol to precipitate nucleic acids. Centrifuge at 5,000 × g for 10 minutes, wash pellet with 70% ethanol, and air dry for 10 minutes [33].
- Elution: Resuspend nucleic acid pellet in 50-100 μL of TE buffer or nuclease-free water. Quantify using fluorometric methods and assess quality by agarose gel electrophoresis or bioanalyzer [33].
The success of downstream sequencing applications critically depends on optimal nucleic acid quality. Common challenges include sample degradation, contamination, and insufficient quantity, which can be mitigated through proper handling, use of nuclease-free reagents, and working in dedicated pre-amplification areas [33].
Library Preparation
Library preparation converts the extracted nucleic acids into a format compatible with sequencing platforms through fragmentation, adapter ligation, and amplification [33] [28]. Different applications require specific library preparation methods:
Protocol: DNA Library Preparation for Illumina Platforms
- Fragmentation: Fragment 100-500 ng of genomic DNA to 200-500 bp fragments using enzymatic digestion (e.g., fragmentase) or acoustic shearing. Verify fragment size using agarose gel electrophoresis or bioanalyzer [28].
- End Repair and A-Tailing: Repair fragment ends using a combination of T4 DNA polymerase and Klenow fragment to create blunt ends. Add single A-overhangs using Klenow exo- polymerase to facilitate adapter ligation [33] [28].
- Adapter Ligation: Ligate platform-specific adapters containing sequencing primer binding sites and sample barcodes using T4 DNA ligase. Purify ligated products using magnetic beads to remove unincorporated adapters [28].
- Library Amplification: Amplify adapter-ligated fragments using 4-12 cycles of PCR with high-fidelity DNA polymerase. Incorporate complete adapter sequences and sample indexes during amplification [33].
- Library Quantification and Normalization: Quantify final library using qPCR and determine size distribution using bioanalyzer. Normalize libraries to 4 nM and pool if multiplexing [33].
For RNA sequencing, the workflow includes additional steps such as mRNA enrichment using poly-A selection or rRNA depletion, followed by reverse transcription to cDNA before library construction [33]. The introduction of tagmentation reactions, which combine fragmentation and adapter attachment into a single step, has significantly reduced library preparation time and costs [33].
Table 1: Comparison of NGS Library Preparation Methods for Different Applications
| Application | Starting Material | Key Preparation Steps | Special Considerations |
|---|---|---|---|
| Whole Genome Sequencing | Genomic DNA | Fragmentation, adapter ligation, PCR amplification | High DNA integrity crucial; avoid amplification bias |
| Whole Exome Sequencing | Genomic DNA | Fragmentation, hybrid capture with exome probes, PCR | Efficient target enrichment critical for coverage uniformity |
| RNA Sequencing | Total RNA or mRNA | Poly-A selection/rRNA depletion, reverse transcription, cDNA synthesis | Strand-specific protocols preserve transcript orientation |
| Targeted Sequencing | Genomic DNA | Fragmentation, hybrid capture or amplicon generation, PCR | High coverage depth required for rare variant detection |
| Single-Cell Sequencing | Single cells | Cell lysis, reverse transcription, whole transcriptome amplification | Address amplification bias from minimal starting material |
Sequencing Platforms and Technologies
Sequencing Technologies
NGS platforms utilize different biochemical principles for sequencing, with the most common being sequencing by synthesis (SBS) [28] [4]. The key technologies include:
Sequencing by Synthesis (SBS): This method, employed by Illumina platforms, uses fluorescently-labeled reversible terminator nucleotides that are added sequentially to growing DNA chains [28] [4]. Each nucleotide incorporation event is detected through fluorescence imaging, with the terminator cleavage allowing the next cycle to begin. Recent advances like XLEAP-SBS chemistry have increased speed and fidelity compared to standard SBS chemistry [4].
Semiconductor Sequencing: Used by Ion Torrent platforms, this technology detects hydrogen ions released during DNA polymerization rather than using optical detection [28]. When a nucleotide is incorporated into a growing DNA strand, a hydrogen ion is released, changing the local pH that is detected by a semiconductor sensor [28].
Single-Molecule Real-Time (SMRT) Sequencing: Developed by Pacific Biosciences, this third-generation technology observes DNA synthesis in real-time using zero-mode waveguides (ZMWs) [7]. The technology provides exceptionally long read lengths (average 10,000-25,000 bp) that are valuable for resolving complex genomic regions, though with higher per-base error rates than short-read technologies [7].
Nanopore Sequencing: Oxford Nanopore Technologies' method involves measuring changes in electrical current as DNA molecules pass through protein nanopores [7]. This technology can produce ultra-long reads (average 10,000-30,000 bp) and enables real-time data analysis, though it historically has higher error rates (up to 15%) [7].
Table 2: Comparison of Major NGS Platforms and Technologies (2025)
| Platform | Technology | Read Length | Accuracy | Throughput per Run | Run Time | Key Applications |
|---|---|---|---|---|---|---|
| Illumina NovaSeq X | Sequencing by Synthesis | 50-300 bp | >99.9% | Up to 16 Tb | 13-44 hours | Large WGS, population studies |
| PacBio Revio | SMRT Sequencing | 10,000-25,000 bp | >99.9% (after correction) | 360-1080 Gb | 0.5-30 hours | De novo assembly, structural variants |
| Oxford Nanopore | Nanopore Sequencing | 10,000-30,000+ bp | ~99% (after correction) | 10-320 Gb | 0.5-72 hours | Real-time sequencing, metagenomics |
| Ion Torrent Genexus | Semiconductor Sequencing | 200-400 bp | >99.5% | 50 Mb-1.2 Gb | 8-24 hours | Targeted sequencing, rapid diagnostics |
| Element AVITI | Sequencing by Synthesis | 50-300 bp | >99.9% | 20 Gb-1.2 Tb | 12-40 hours | RNA-seq, exome sequencing |
Platform Selection Considerations
Choosing the appropriate NGS platform depends on multiple factors including research objectives, required throughput, read length, accuracy needs, and budget constraints [28]. Benchtop sequencers are ideal for small-scale studies and targeted panels, while production-scale systems are designed for large genome projects and high-volume clinical testing [28].
Recent advancements include the launch of Illumina's NovaSeq X Series, which provides extraordinary sequencing power with increased speed and sustainability, capable of producing over 20,000 whole genomes annually [4]. The ongoing innovation in sequencing chemistry, such as the development of XLEAP-SBS with twice the speed and three times the accuracy of previous methods, continues to push the boundaries of what's possible with NGS technology [4].
Data Analysis Workflow
Primary and Secondary Analysis
The NGS data analysis workflow consists of multiple stages that transform raw sequencing data into biological insights [28]. The massive volume of data generated by NGS platforms (often terabytes per project) requires sophisticated computational infrastructure and bioinformatics expertise [24] [28].
Primary Analysis involves base calling, demultiplexing, and quality control. Raw signal data (images or electrical measurements) are converted into sequence reads (FASTQ files) with associated quality scores [28]. Quality control metrics include per-base sequence quality, adapter contamination, and overall read quality, with tools like FastQC commonly used for this purpose.
Protocol: Primary Data Analysis and QC
- Base Calling: Convert raw signal data to nucleotide sequences using platform-specific algorithms. Generate FASTQ files containing sequences and quality scores [28].
- Demultiplexing: Separate pooled samples using barcode sequences assigned during library preparation. Assign reads to individual samples based on unique barcodes [28].
- Quality Control: Assess read quality using FastQC or similar tools. Trim adapter sequences and low-quality bases using Trimmomatic or Cutadapt. Remove poor-quality reads (Q-score < 20) [28].
Secondary Analysis encompasses read alignment and variant calling. Processed reads are aligned to a reference genome (BAM files), followed by identification of genetic variants (VCF files) [28].
Protocol: Secondary Analysis - Alignment and Variant Calling
- Read Alignment: Map quality-filtered reads to reference genome using aligners like BWA-MEM or Bowtie2. For RNA-seq data, use splice-aware aligners such as STAR [34] [28].
- Post-Alignment Processing: Sort and index aligned BAM files. Mark PCR duplicates using tools like Picard MarkDuplicates to minimize amplification bias [33] [28].
- Variant Calling: Identify genetic variants (SNPs, indels) using callers like GATK HaplotypeCaller or DeepVariant. For tumor samples, use MuTect2 for somatic variant detection [24] [34].
- Variant Filtering: Apply quality filters based on read depth, mapping quality, and variant frequency. Remove low-confidence calls to minimize false positives [34].
Tertiary Analysis and Interpretation
Tertiary analysis focuses on biological interpretation through variant annotation, pathway analysis, and data visualization [28]. This stage extracts meaningful biological insights from variant data by connecting genetic changes to functional consequences.
Protocol: Tertiary Analysis and Biological Interpretation
- Variant Annotation: Annotate variants using databases like dbSNP, ClinVar, COSMIC, and gnomAD. Predict functional impact with tools like SnpEff and ANNOVAR [34].
- Pathway Analysis: Identify enriched biological pathways using tools like GSEA or Enrichr. Connect genetic variants to molecular pathways and disease mechanisms [34].
- Data Visualization: Create visual representations of results using genome browsers (IGV), Circos plots, and custom visualizations in R or Python [28].
The integration of artificial intelligence and machine learning has significantly enhanced NGS data analysis, with tools like Google's DeepVariant utilizing deep learning to identify genetic variants with greater accuracy than traditional methods [24]. Cloud computing platforms such as Amazon Web Services (AWS) and Google Cloud Genomics provide scalable infrastructure to store, process, and analyze massive NGS datasets efficiently [24].
Applications in Research and Drug Development
Clinical Genomics and Personalized Medicine
NGS has transformed clinical genomics by enabling comprehensive genetic analysis for disease diagnosis and treatment selection [34] [7]. Key applications include:
Rare Genetic Disorders: Whole exome and genome sequencing have dramatically reduced the diagnostic odyssey for patients with rare genetic conditions, particularly in neonatal care [24] [10]. The Deciphering Developmental Disorders project has diagnosed thousands of children's conditions by identifying causative mutations in genes previously unassociated with disease [10].
Cancer Genomics: Comprehensive tumor profiling through NGS facilitates personalized oncology by identifying driver mutations, gene fusions, and biomarkers that guide targeted therapies [24] [34]. Liquid biopsies using circulating tumor DNA (ctDNA) enable non-invasive cancer detection, monitoring of treatment response, and early identification of resistance mechanisms [10].
Pharmacogenomics: NGS-based approaches predict how genetic variations influence drug metabolism and response, allowing clinicians to optimize drug selection and dosage while minimizing adverse effects [24] [10]. This has proven particularly valuable in fields like psychiatry and cardiology where drug response variability is significant [10].
Drug Discovery and Development
NGS technologies have accelerated multiple stages of the drug discovery pipeline through target identification, mechanism of action studies, and biomarker development [24] [4].
Target Identification: Whole-genome and exome sequencing of large patient cohorts identifies novel disease-associated genes and pathways as potential therapeutic targets [24]. Integration of multi-omics data (genomics, transcriptomics, proteomics) provides a comprehensive view of biological systems, linking genetic information to molecular function and phenotypic outcomes [24].
Mechanism of Action Studies: RNA sequencing reveals how drug treatments alter gene expression patterns, cellular pathways, and regulatory networks [4]. Single-cell RNA sequencing provides unprecedented resolution to study heterogeneous responses to therapeutic compounds across different cell types within complex tissues [24].
Biomarker Development: NGS enables the discovery of genomic, transcriptomic, and epigenetic biomarkers for patient stratification, treatment selection, and monitoring therapeutic efficacy [4]. Cell-free RNA sequencing has emerged as a promising approach for non-invasive biomarker discovery and health monitoring [4].
Essential Research Reagent Solutions
Successful NGS experiments require high-quality reagents and materials at each workflow stage. The following table details essential research reagent solutions and their functions:
Table 3: Essential Research Reagent Solutions for NGS Workflows
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Nucleic Acid Extraction Kits | Qiagen DNeasy Blood & Tissue Kit, Zymo Research Quick-DNA/RNA kits | Isolate high-quality DNA/RNA from various sample types | Select kits based on sample source (blood, tissue, cells) and required yield |
| Library Preparation Kits | Illumina DNA Prep, KAPA HyperPrep, NEB Next Ultra II | Fragment DNA, add adapters, amplify libraries | Consider insert size, yield, and bias characteristics for specific applications |
| Target Enrichment Kits | IDT xGEN, Twist Bioscience Panels, Agilent SureSelect | Capture specific genomic regions of interest | Evaluate based on target size, uniformity, and off-target rates |
| Sequencing Reagents | Illumina SBS chemistry, PacBio SMRTbell, Nanopore R9/R10 | Enable nucleotide incorporation and detection | Platform-specific reagents with defined read lengths and run parameters |
| Quality Control Tools | Agilent Bioanalyzer/TapeStation, Qubit fluorometer, qPCR kits | Assess nucleic acid quality, quantity, and library integrity | Implement at multiple workflow stages to ensure success |
| Enzymes | High-fidelity PCR polymerases, T4 DNA ligase, fragmentase | Amplify, ligate, and fragment nucleic acids | Select enzymes with minimal bias for accurate representation |
| Purification Materials | SPRI beads, silica membrane columns, magnetic stands | Purify nucleic acids between reaction steps | Bead-based methods preferred for automation compatibility |
| NGS Validation Tools | Orthogonal PCR assays, Sanger sequencing, digital PCR | Confirm key NGS findings | Essential for clinical applications and variant verification |
Challenges and Future Directions
Despite significant advancements, NGS workflows still face several challenges including managing massive datasets, ensuring data privacy, standardizing analytical protocols, and reducing costs further [24] [7]. Sample preparation remains a critical bottleneck, with issues such as PCR amplification bias, inefficient library construction, and sample contamination affecting data quality [33].
Emerging trends focus on increasing automation, improving long-read technologies, reducing sample input requirements, and integrating multi-omics approaches [24] [7]. The adoption of AI and machine learning in data analysis continues to grow, enabling more accurate variant calling and biological interpretation [24]. Companies like Volta Labs are addressing workflow challenges through automated sample prep systems that support DNA, RNA, and single-cell workflows across multiple sequencing platforms [35].
The NGS data analysis market is projected to grow from $3.43 billion in 2024 to $8.24 billion by 2029, reflecting a compound annual growth rate of 18.8% [36]. This growth is driven by increasing adoption of liquid biopsy methodologies, incorporation of artificial intelligence into analytical processes, expansion of precision medicine programs, and development of innovative sequencing technologies [36].
As NGS technologies continue to evolve, they will further transform biological research and clinical diagnostics, enabling increasingly sophisticated applications and deeper insights into human health and disease mechanisms.
Next-generation sequencing (NGS) has revolutionized biological research by enabling high-throughput, parallel sequencing of nucleic acids. A critical first step in any NGS workflow is library preparation, which involves converting nucleic acid samples (gDNA or cDNA) into a library of uniformly sized, adapter-ligated DNA fragments that can be sequenced [37]. For many applications, targeted sequencing offers significant advantages over whole-genome sequencing by enabling focused, in-depth analysis of specific genomic regions while reducing costs, time, and data management challenges [38] [39]. Targeted sequencing identifies both known and novel variants within regions of interest and generally requires less sample input [38]. The two predominant methods for target enrichment are hybridization capture and amplification-based approaches (commonly called amplicon sequencing), each with distinct characteristics, applications, and methodological considerations [38] [39]. This application note provides a detailed comparison of these methods, along with experimental protocols, to guide researchers in selecting and implementing the optimal approach for their NGS research.
Key Comparison and Method Selection
Technical Comparison of Hybrid-Capture and Amplicon Methods
The choice between hybridization capture and amplicon sequencing involves multiple technical considerations, from workflow complexity to genomic coverage capabilities.
Table 1: Technical Comparison of Hybrid-Capture and Amplicon Sequencing Methods
| Feature | Hybridization Capture | Amplicon Sequencing |
|---|---|---|
| Number of Steps | More extensive workflow [38] | Fewer steps, streamlined process [38] |
| Number of Targets per Panel | Virtually unlimited [38] | Usually fewer than 10,000 amplicons [38] [39] |
| Input DNA Requirement | 1-250 ng for library prep + 500 ng for capture [39] | 10-100 ng [39] |
| Sensitivity | <1% variant detection [39] | <5% variant detection [39] |
| Total Time | More time required [38] | Less time [38] |
| Cost per Sample | Varies | Generally lower [38] |
| On-target Rate | Lower than amplicon [38] | Naturally higher [38] |
| Coverage Uniformity | Greater uniformity [38] | Less uniform due to amplification bias [38] |
| False Positives/Noise | Lower levels [38] | Higher potential for false positives [38] |
Application-Based Selection Guide
Selecting the appropriate targeted sequencing method depends heavily on the specific research goals, sample characteristics, and practical constraints.
Table 2: Application-Based Method Selection Guide
| Application | Recommended Method | Rationale |
|---|---|---|
| Exome Sequencing | Hybridization Capture [38] [40] | Suitable for large target sizes (megabases) [41] |
| Oncology Research | Hybridization Capture [38] [39] | Better for detecting low-frequency somatic variants [39] |
| Rare Variant Identification | Hybridization Capture [38] [39] | Higher sensitivity (<1%) [39] |
| Genotyping, Germline SNPs/Indels | Amplicon Sequencing [38] [39] | Sufficient sensitivity (≤5%) for germline variants [39] |
| CRISPR Edit Validation | Amplicon Sequencing [38] [39] | Ideal for specific, small target verification [38] |
| Small Target Panels (<50 genes) | Amplicon Sequencing [41] | More affordable and simpler workflow [41] |
| Large Target Panels (>50 genes) | Hybridization Capture [41] | More comprehensive for profiling all variant types [41] |
Diagram 1: Method Selection Decision Tree
Hybridization Capture Protocol
Hybridization capture utilizes biotinylated oligonucleotide baits complementary to genomic regions of interest to enrich target sequences from fragmented DNA libraries [39] [42]. This method involves several key steps: library preparation, hybridization with target-specific probes, capture using streptavidin-coated magnetic beads, washing to remove non-specifically bound fragments, and amplification of captured targets [42]. The approach is particularly valuable for applications requiring comprehensive coverage of large genomic regions, such as exome sequencing or large gene panels [38] [41]. A significant advantage of hybridization capture is its minimal sequence bias compared to PCR-based methods, as it does not rely on primer-based amplification of targets [39]. Recent advancements have simplified traditional hybridization workflows, reducing processing time while maintaining or improving capture specificity and library complexity [42].
Detailed Experimental Protocol
Library Preparation
- DNA Fragmentation: Fragment genomic DNA to 200-300bp using enzymatic digestion (e.g., fragmentase) or mechanical shearing (e.g., sonication, nebulization) [37]. Enzymatic fragmentation is faster but may introduce biases, while physical methods provide more random fragmentation [37].
- End Repair: Convert fragmented DNA to blunt-ended, 5'-phosphorylated fragments using T4 DNA polymerase (5'→3' polymerase and 3'→5' exonuclease activities) and T4 Polynucleotide Kinase [37].
- A-Tailing: Add single adenosine overhangs to 3' ends using Klenow Fragment (exo-) or other polymerases with terminal transferase activity to facilitate ligation of thymine-overhang adapters [37] [43].
- Adapter Ligation: Ligate platform-specific adapters containing sample barcodes to DNA fragments using T4 DNA ligase [37] [43]. Adapters typically include:
- Sequencing adapters: For flow cell binding
- Barcodes/Indices: Unique nucleotide sequences for sample multiplexing [37]
- Library Cleanup and Size Selection: Remove excess adapters and adapter dimers using magnetic bead-based cleanups (e.g., SPRI beads) or gel electrophoresis [37]. Perform quality control assessment using fluorometric methods (e.g., Qubit) and fragment analyzers [37].
Target Enrichment by Hybridization Capture
- Hybridization: Combine prepared libraries with biotinylated oligonucleotide baits (typically 50-100bp in length) in hybridization buffer. Include blockers (e.g., Cot-1 DNA, adapter-specific blockers) to prevent non-specific binding [42]. Incubate at 65°C for 16-24 hours to allow specific hybridization of baits to target sequences [42].
- Capture: Add streptavidin-coated magnetic beads to the hybridization mixture and incubate to allow binding of biotinylated baits (with hybridized targets) to beads [42].
- Washing: Perform a series of temperature-controlled washes (typically at 65°C) to remove non-specifically bound fragments while retaining target-bound complexes [42]. Washes typically include:
- Low-stringency wash (2× SSC, SDS) to remove non-specific hybrids
- High-stringency wash (0.1× SSC, SDS) to eliminate weakly bound fragments
- Elution: Release captured DNA from beads using alkaline elution or nuclease-free water at elevated temperature [42].
- Amplification: Amplify captured libraries using high-fidelity DNA polymerase (typically 10-14 PCR cycles) to generate sufficient material for sequencing [42]. Alternatively, for a PCR-free workflow, use circularization and amplification approaches on specialized flow cells [42].
Simplified Hybrid Capture Protocol (Trinity Method)
Recent developments have introduced streamlined hybrid capture workflows that eliminate multiple steps:
- Library Preparation: Prepare libraries using standard or PCR-free methods [42].
- Fast Hybridization: Hybridize libraries with biotinylated probes for 1-2 hours using fast hybridization protocols [42].
- Direct Loading: Instead of bead-based capture and post-hybridization PCR, directly load hybridization products onto streptavidin-functionalized flow cells [42].
- On-Flow Cell Amplification: Circularize and amplify captured targets directly on the flow cell surface [42].
This simplified approach reduces total processing time by over 50% while maintaining high on-target rates and improving indel calling accuracy [42].
Amplification-Based (Amplicon) Protocol
Amplicon sequencing employs polymerase chain reaction (PCR) with target-specific primers to amplify genomic regions of interest prior to sequencing [39]. This method creates DNA fragments (amplicons) that are subsequently converted into sequencing libraries [39]. The approach is characterized by its high specificity, relatively simple workflow, and cost-effectiveness for studying limited numbers of targets [38] [41]. A key advantage of amplicon sequencing is its capacity for highly multiplexed PCR, where hundreds to thousands of primer pairs simultaneously amplify different genomic regions in a single reaction [39]. However, primer design challenges and amplification biases can lead to uneven coverage or failure to amplify certain targets, particularly those with high GC content or sequence variations at primer binding sites [41]. Amplicon sequencing is particularly well-suited for applications requiring deep coverage of small genomic regions, such as variant validation, CRISPR edit confirmation, and pathogen detection [38] [39].
Detailed Experimental Protocol
Primer Design and Validation
- Target Selection: Define specific genomic regions of interest for amplification.
- Primer Design: Design target-specific primers with the following considerations:
- Amplicon size: 150-300bp for optimal NGS sequencing
- Tm: 58-62°C with minimal variation across all primers in multiplex
- Avoid primer-dimer formation and secondary structures
- Ensure specificity to target regions
- For highly multiplexed panels, use specialized software for primer design
- Primer Validation: Test individual primer pairs for amplification efficiency and specificity before multiplexing.
Library Preparation via Multiplex PCR
- Multiplex PCR Amplification:
- Set up PCR reactions containing:
- 10-100ng genomic DNA [39]
- Multiplex primer pool (hundreds to thousands of primer pairs)
- High-fidelity DNA polymerase with strong processivity
- Use thermocycling conditions optimized for multiplex amplification:
- Initial denaturation: 95°C for 2 minutes
- 25-35 cycles of: 95°C for 30s, 60-65°C for 30s, 72°C for 1 minute
- Final extension: 72°C for 5 minutes
- Set up PCR reactions containing:
- Amplicon Purification: Clean up PCR products using magnetic beads or columns to remove primers, enzymes, and salts.
- Indexing PCR: Add platform-specific adapters and sample barcodes via a limited-cycle PCR (typically 8-10 cycles) using primers containing:
- Adapter sequences: For flow cell binding
- Dual indices: Unique combinatorial barcodes for sample multiplexing [41]
- Library Normalization and Pooling: Quantify individual libraries, normalize concentrations, and pool samples based on desired sequencing depth.
- Final Cleanup and QC: Purify pooled libraries and assess quality using capillary electrophoresis (e.g., Bioanalyzer, Fragment Analyzer) and quantitation (e.g., qPCR).
Streamlined Amplicon Library Preparation
Recent innovations have simplified amplicon library preparation through transposase-based methods:
- PCR Amplification: Generate amplicons using target-specific primers as described above [41].
- Transposase-Mediated Tagmentation: Use optimized Tn5 transposase to simultaneously fragment amplicons and tag them with sequencing adapters in a single reaction [41].
- Limited-Cycle PCR: Add complete adapter sequences and sample barcodes via 8-12 cycles of PCR [41].
- Pooling and Cleanup: Normalize and pool libraries before sequencing [41].
This streamlined approach reduces hands-on time and improves library complexity compared to traditional methods [41].
Diagram 2: Comparative Workflows of Hybridization Capture and Amplicon Sequencing
Research Reagent Solutions
Successful implementation of hybrid-capture or amplicon-based NGS requires carefully selected reagents and materials at each workflow stage.
Table 3: Essential Research Reagents for Targeted NGS Library Preparation
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Fragmentation Enzymes | Fragmentase, DNase I, Transposases | Fragment DNA to optimal size for sequencing | Physical methods (sonication) provide more random fragmentation than enzymatic approaches [37] |
| End-Repair Enzymes | T4 DNA Polymerase, T4 PNK | Create blunt-ended, 5'-phosphorylated DNA fragments | Essential for efficient adapter ligation [37] |
| A-Tailing Enzymes | Klenow Fragment (exo-), Taq Polymerase | Add 3'A-overhangs for TA-ligation | Required for specific adapter types [37] |
| Ligation Enzymes | T4 DNA Ligase | Covalently attach adapters to DNA fragments | High-efficiency ligation critical for library complexity [37] |
| High-Fidelity Polymerases | Q5, KAPA HiFi, Phusion | Amplify libraries with minimal errors | Essential for both target amplification and library PCR [37] |
| Hybridization Baits | xGen Exome Panels, Twist Panels | Biotinylated oligonucleotides for target capture | Panel size and design impact coverage uniformity [42] |
| Capture Beads | Streptavidin Magnetic Beads | Bind biotinylated baits for target selection | Bead quality affects capture efficiency and specificity [42] |
| Target-Specific Primers | Custom amplicon panels | Amplify genomic regions of interest | Multiplexing capability depends on primer design [41] |
| NGS Adapters | Illumina-compatible adapters | Enable sequencing and sample multiplexing | May include unique dual indices for improved demultiplexing [43] [41] |
| Cleanup Reagents | SPRI beads, Column purifications | Remove enzymes, primers, and unwanted fragments | Bead-based methods enable precise size selection [37] |
Hybridization capture and amplification-based approaches represent two fundamentally different strategies for targeted NGS library preparation, each with distinct advantages and limitations. Hybridization capture excels in applications requiring comprehensive coverage of large genomic regions (e.g., exome sequencing), detection of low-frequency variants, and minimal amplification bias [38] [42]. In contrast, amplicon sequencing offers a simpler, more cost-effective solution for focused studies of limited genomic regions, with higher on-target rates and faster turnaround times [38] [39]. Method selection should be guided by specific research objectives, including the number of targets, required sensitivity, sample quality and quantity, and available resources [38] [41]. Recent methodological advances, such as simplified hybrid capture workflows [42] and transposase-based amplicon library preparation [41], continue to enhance the efficiency, accuracy, and accessibility of both approaches. As targeted NGS technologies evolve, they will undoubtedly expand their transformative impact on genomics research and clinical applications.
Application Note: NGS in Clinical Oncology
Comprehensive Genomic Profiling for Targeted Therapy
Next-generation sequencing (NGS) has become a cornerstone of precision oncology, enabling comprehensive genomic profiling of tumors to identify actionable alterations that inform targeted treatment strategies. By simultaneously interrogating hundreds of cancer-related genes, NGS panels provide a molecular compass for therapeutic decision-making in advanced cancers, substantially improving clinical outcomes compared to traditional single-gene tests [44].
Current National Comprehensive Cancer Network (NCCN), European Society for Medical Oncology, and American Society of Clinical Oncology (ASCO) guidelines recommend NGS in patients with advanced lung, breast, colorectal, prostate, and ovarian cancer [44]. The paradigm has shifted from organ-based cancer classification to molecularly-driven diagnosis, as exemplified by the 2021 EANO Guidelines for gliomas, which mandate broad molecular profiling for accurate diagnosis and treatment selection [44].
Table 1: Clinically Validated NGS Panels in Oncology
| NGS Test | Technology Used | Sample Type | Key Genes Detected | Turnaround Time |
|---|---|---|---|---|
| FoundationOne CDx | Hybrid Capture NGS | Tumor Tissue | EGFR, ALK, BRAF, BRCA1/2, MET, NTRK, ROS1, PIK3CA, RET, ERBB2 (HER2), KRAS | 14 days |
| FoundationOne Liquid CDx | cfDNA Sequencing | Blood (Liquid Biopsy) | EGFR, ALK, BRAF, BRCA1/2, MET, RET, ERBB2 (HER2), PIK3CA, KRAS | 10-14 days |
| Guardant360 CDx | cfDNA Sequencing | Blood (Liquid Biopsy) | EGFR, ALK, BRAF, BRCA1/2, MET, RET, ERBB2 (HER2), PIK3CA, KRAS | 7 days |
| MSK-IMPACT | Hybrid Capture NGS | Tumor Tissue | 468 cancer-relevant genes including EGFR, ALK, BRAF, BRCA1/2, MET, NTRK, ROS1 | 21 days |
| Tempus xT | Hybrid Capture NGS | Tumor Tissue | >600 genes; includes TMB and MSI analysis | 10-14 days |
Protocol: Hybrid Capture-Based Tumor Sequencing
Principle: This protocol uses biotinylated probes to enrich for specific genomic regions of interest from tumor DNA, enabling high-sensitivity detection of mutations, copy number alterations, and structural variants.
Materials:
- Tumor tissue (FFPE or fresh frozen) or blood-derived cfDNA
- QIAamp DNA FFPE Tissue Kit or similar for DNA extraction
- KAPA HyperPrep Kit or similar for library preparation
- IDT xGen Lockdown Panels or similar hybridization probes
- Streptavidin-coated magnetic beads
- Illumina sequencing platform (MiSeq, NextSeq, or NovaSeq)
Procedure:
Nucleic Acid Extraction
- Extract genomic DNA from tumor tissue using commercial kits with a minimum input of 50-200ng.
- For liquid biopsy, isolate cell-free DNA from 4-10mL plasma using circulating nucleic acid kits.
- Assess DNA quality and quantity using fluorometry (Qubit) and fragment analysis (TapeStation).
Library Preparation
- Fragment DNA to 300bp using acoustic shearing (Covaris).
- Repair DNA ends and add 'A' bases to 3' ends.
- Ligate Illumina adapters with unique dual indices to enable sample multiplexing.
- Clean up libraries using SPRIselect beads and amplify with 4-6 PCR cycles.
Hybrid Capture Enrichment
- Pool libraries in equimolar ratios (up to 96 samples per capture reaction).
- Hybridize with biotinylated probes for 4-16 hours at 65°C.
- Capture probe-bound fragments using streptavidin magnetic beads.
- Wash to remove non-specific binding and amplify captured libraries (10-14 cycles).
Sequencing and Data Analysis
- Pool final libraries and quantify by qPCR.
- Sequence on appropriate Illumina platform to achieve minimum 500x coverage for tissue and 10,000x for liquid biopsy.
- Align reads to reference genome (hg38) using BWA-MEM or similar.
- Call variants using specialized algorithms (MuTect2 for SNVs, Control-FREEC for CNVs).
- Annotate variants and interpret clinical significance using databases like OncoKB.
Quality Control:
- Minimum coverage: 500x for tissue, 10,000x for liquid biopsy
- Minimum DNA input: 50ng for FFPE, 30ng for liquid biopsy
- Include positive and negative controls in each run
- Validate variants with allele frequency >5% for tissue, >0.5% for liquid biopsy
Application Note: NGS in Rare Genetic Diseases
Ending the Diagnostic Odyssey
Next-generation sequencing has dramatically reduced the diagnostic odyssey for patients with rare genetic diseases, with exome and genome sequencing providing molecular diagnoses in cases that previously remained unsolved after extensive testing. A user-based valuation study found that at least half of potential users consider costs of up to CAD$10,000 acceptable if the chance of obtaining a diagnosis through exome sequencing is at least 50% [45].
Population-based genome projects, such as the Hong Kong Genome Project (HKGP), demonstrate the real-world implementation of genome sequencing for rare disease diagnosis. These initiatives utilize both short-read and long-read genome sequencing technologies to comprehensively detect single nucleotide variants, copy number variants, and structural variants across the genome [46].
Table 2: NGS Approaches for Rare Disease Diagnosis
| Sequencing Approach | Target Region | Variant Types Detected | Diagnostic Yield | Key Applications |
|---|---|---|---|---|
| Whole Genome Sequencing (WGS) | Entire genome (coding and non-coding) | SNVs, CNVs, SVs, repeat expansions, non-coding variants | 40-60% | Unexplained genetic disorders, complex phenotypes |
| Whole Exome Sequencing (WES) | Protein-coding regions (1-2% of genome) | SNVs, small indels in exons | 30-40% | Mendelian disorders, neurodevelopmental conditions |
| Targeted Gene Panels | Pre-defined gene sets | SNVs, indels, specific CNVs | 10-30% (depending on phenotype) | Disorder-specific testing (e.g., cardiomyopathies, epilepsies) |
Protocol: Whole Genome Sequencing for Rare Diseases
Principle: This protocol provides comprehensive sequencing of the entire genome to identify pathogenic variants across all genomic regions, including coding, non-coding, and structural variants that may be missed by targeted approaches.
Materials:
- Peripheral blood DNA (minimum 1μg) or saliva-derived DNA
- Illumina DNA PCR-Free Library Prep Kit or similar
- Illumina sequencing platforms (NovaSeq 6000)
- Oxford Nanopore or PacBio systems for long-read sequencing (optional)
- Bioinformatic tools for variant calling (GATK, DeepVariant, Manta)
Procedure:
Sample Preparation and Quality Control
- Extract high-molecular-weight DNA from blood or saliva using automated systems.
- Assess DNA quality by agarose gel electrophoresis or Fragment Analyzer.
- Quantify DNA using fluorometric methods (Qubit dsDNA HS Assay).
- Ensure DNA integrity number (DIN) >7.0 for optimal library preparation.
PCR-Free Library Preparation
- Fragment DNA to 350bp using Covaris ultrasonication.
- Perform end repair, A-tailing, and adapter ligation using Illumina DNA Prep reagents.
- Clean up libraries using SPRIselect beads without PCR amplification.
- Quantify libraries using Qubit and qPCR (KAPA Library Quantification Kit).
Sequencing
- Pool libraries in equimolar ratios (up to 32 samples per NovaSeq S4 flow cell).
- Sequence on Illumina NovaSeq 6000 using 2x150bp paired-end chemistry.
- Target coverage: 30-40x for short-read WGS.
- For complex cases, supplement with long-read sequencing (PacBio HiFi or ONT) for structural variant detection.
Bioinformatic Analysis
- Align reads to reference genome (GRCh38) using BWA-MEM or DRAGEN.
- Perform base quality recalibration and variant calling using GATK best practices.
- Annotate variants using Ensembl VEP or similar tools.
- Prioritize variants based on population frequency (<1% in gnomAD), predicted pathogenicity, and phenotypic match (HPO terms).
- Report variants according to ACMG/AMP guidelines.
Quality Control:
- Minimum sequencing depth: 30x coverage for >95% of genome
- Mean insert size: 300-500bp
- >Q30 bases: >85% of reads
- Contamination estimate: <3%
- Sex concordance check between reported and genetic sex
Application Note: NGS in Infectious Disease Surveillance
Pathogen Genomics for Public Health
Next-generation sequencing has revolutionized infectious disease surveillance by enabling high-resolution pathogen typing, outbreak investigation, and antimicrobial resistance detection. The US Department of Defense's Global Emerging Infections Surveillance (GEIS) program has established a Next-Generation Sequencing and Bioinformatics Consortium (NGSBC) with a three-tiered framework for building and maintaining genomic surveillance capabilities [47].
NGS methods provide unbiased detection of pathogens from various sample types without prior knowledge of the organism, making them particularly valuable for identifying novel or unexpected pathogens. During the COVID-19 pandemic, this capability proved critical for tracking SARS-CoV-2 evolution and monitoring the emergence of variants of concern [48].
Table 3: NGS Methods for Pathogen Genomic Surveillance
| Sequencing Method | Primary Use | Advantages | Limitations | Example Applications |
|---|---|---|---|---|
| Whole Genome Sequencing of Isolates | Complete genome sequencing of cultured isolates | High-quality complete genomes, detection of low-frequency variants | Requires culture, longer turnaround time | Reference genome generation, transmission tracking |
| Amplicon Sequencing | Targeted sequencing of known pathogens | High sensitivity, low input requirements, rapid | Limited to known targets, primer sensitivity to mutations | SARS-CoV-2 variant monitoring, viral outbreak investigation |
| Hybrid Capture | Detection and characterization of multiple known pathogens | High sensitivity, tolerant to sequence variations | Requires probe design, higher cost | Respiratory virus panels, antimicrobial resistance detection |
| Shotgun Metagenomics | Comprehensive pathogen detection without prior knowledge | Unbiased detection of all microorganisms, pathogen discovery | Lower sensitivity, high host contamination, complex data analysis | Investigation of unknown etiology, microbiome studies |
Protocol: Amplicon Sequencing for Viral Pathogen Surveillance
Principle: This protocol uses multiplex PCR to amplify specific regions of a viral genome directly from clinical specimens, enabling rapid sequencing and characterization of known pathogens for outbreak investigation and variant monitoring.
Materials:
- Clinical specimens (nasopharyngeal swabs, wastewater, tissue samples)
- Nucleic acid extraction kits (QIAamp Viral RNA Mini Kit)
- Reverse transcription reagents (SuperScript IV Reverse Transcriptase)
- ARTIC Network primers or similar tiled amplicon schemes
- Q5 Hot Start High-Fidelity DNA Polymerase
- Illumina DNA Prep Kit or Nextera XT Library Prep Kit
- MiniSeq, MiSeq, or iSeq sequencing systems
Procedure:
Nucleic Acid Extraction and Reverse Transcription
- Extract viral RNA from 200μL clinical specimen using commercial kits.
- Synthesize cDNA using random hexamers and reverse transcriptase.
- Assess RNA quality by RT-qPCR (Ct value <32 for optimal sequencing).
Multiplex PCR Amplification
- Design tiled amplicons (~400bp) covering the entire viral genome with ~50bp overlaps.
- Perform two pooled multiplex PCR reactions using tiled primer pools.
- Use high-fidelity polymerase to minimize amplification errors.
- Cycle conditions: 98°C for 30s; 35 cycles of 98°C for 15s, 60°C for 5m; 72°C for 5m.
Library Preparation and Sequencing
- Quantify amplicons using Fragment Analyzer or TapeStation.
- Tagment amplicons using Illumina DNA Prep or Nextera XT.
- Attach dual indices via limited-cycle PCR (8-10 cycles).
- Normalize and pool libraries equimolarly.
- Sequence on MiSeq (2x150bp) or iSeq (2x150bp) systems.
Bioinformatic Analysis
- Demultiplex reads and trim primers using iVar or similar tools.
- Map reads to reference genome using BWA-MEM or minimap2.
- Call consensus sequence using ≥10x coverage threshold.
- Perform phylogenetic analysis with Nextstrain or USHER pipeline.
- Identify variants of concern by comparison to reference sequences.
Quality Control:
- Minimum Ct value: <32 for reliable sequencing
- Minimum coverage: 100x for >90% of genome
- Amplicon balance: even coverage across all amplicons
- Include positive and negative extraction controls
- Monitor for cross-contamination between samples
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents for NGS Applications
| Category | Specific Products | Function | Key Applications |
|---|---|---|---|
| Library Preparation | Illumina DNA Prep, KAPA HyperPrep, NEBNext Ultra II | Convert nucleic acids to sequencing-compatible libraries | All NGS applications |
| Target Enrichment | IDT xGen Lockdown Probes, Illumina Respiratory Virus Enrichment Kit, Twist Pan-Cancer Panel | Enrich for specific genomic regions of interest | Targeted sequencing, hybrid capture |
| Sequencing Platforms | Illumina NovaSeq, MiSeq, iSeq; Oxford Nanopore MinION; PacBio Sequel | Generate sequence data from prepared libraries | All NGS applications, varying by scale and read length needs |
| Bioinformatics Tools | BWA-MEM, GATK, SAMtools, DRAGEN, CLC Genomics Server | Process, analyze, and interpret sequencing data | All NGS applications |
| Quality Control | Agilent TapeStation, Fragment Analyzer, Qubit Fluorometer | Assess nucleic acid quality, quantity, and fragment size | Critical QC step for all NGS workflows |
Workflow Visualization
NGS Biomedical Applications Workflow: This diagram illustrates the complete workflow from sample collection through data analysis, highlighting the convergence of different sample types and processing methods toward three key biomedical applications: oncology, rare genetic diseases, and infectious disease surveillance.
Tiered Genomic Surveillance Network: This diagram illustrates the three-tiered framework for genomic surveillance capabilities as implemented by the GEIS Next-Generation Sequencing and Bioinformatics Consortium, showing how cases move through point-of-care, regional, and national laboratories to inform public health action.
Next-Generation Sequencing (NGS) has revolutionized pharmaceutical research by providing high-throughput, cost-effective genomic analysis capabilities that permeate every stage of drug discovery and development [7]. The global NGS in drug discovery market, valued at $1.45 billion in 2024, is projected to reach $4.27 billion by 2034, growing at a compound annual growth rate (CAGR) of 18.3% [49]. This growth is fueled by NGS's ability to accelerate target identification, biomarker discovery, and personalized medicine development while addressing the critical challenge of drug resistance across therapeutic areas [49].
NGS technologies enable comprehensive analysis of DNA and RNA molecules, providing unprecedented insights into genome structure, genetic variations, gene expression profiles, and epigenetic modifications [7]. The versatility of NGS platforms has expanded the scope of genomics research, facilitating studies on rare genetic diseases, cancer genomics, microbiome analysis, infectious diseases, and population genetics [7]. This review details specific applications and protocols for employing NGS in target identification, biomarker discovery, and combating drug resistance, providing researchers with practical experimental frameworks.
NGS Technologies and Platforms
Technology Comparison
NGS platforms offer complementary strengths for different applications in drug discovery. Short-read technologies like Illumina provide high accuracy for variant calling, while long-read technologies from Pacific Biosciences and Oxford Nanopore enable resolution of complex genomic regions and structural variations [7].
Table 1: Comparison of Major NGS Platforms and Their Applications in Drug Discovery
| Platform | Technology | Read Length | Key Strengths | Primary Drug Discovery Applications |
|---|---|---|---|---|
| Illumina | Sequencing-by-synthesis | 36-300 bp | High accuracy, high throughput | Variant calling, expression profiling, target identification [7] |
| Pacific Biosciences (SMRT) | Single-molecule real-time sequencing | 10,000-25,000 bp | Long reads, epigenetic detection | Structural variation, complex genome assembly, haplotype phasing [7] |
| Oxford Nanopore | Nanopore sensing | 10,000-30,000 bp | Ultra-long reads, real-time analysis | Microbial resistance genotyping, transcriptomics [50] [7] |
| Ion Torrent | Semiconductor sequencing | 200-400 bp | Rapid turnaround, simple workflow | Oncology panels, infectious disease resistance [7] |
The Researcher's Toolkit: Essential Reagents and Solutions
Table 2: Key Research Reagent Solutions for NGS in Drug Discovery
| Reagent/Solution | Function | Application Examples |
|---|---|---|
| DeepChek Assays (ABL Diagnostics) | Target amplification for resistance-associated genomic regions | HIV protease/RT genotyping, HBV RT genotyping, SARS-CoV-2 whole genome [50] |
| TruSight Oncology 500 (Illumina) | Comprehensive genomic profiling for solid tumors | Detection of gene amplifications, fusions, deletions in DNA and RNA [51] |
| Pillar Biosciences Assays | Targeted NGS for oncology | Solid tumor, liquid biopsy, and haematology testing in single-tube workflow [51] |
| Watchmaker Genomics Kits | Library preparation for DNA and RNA sequencing | Automated library preparation systems for enhanced sequencing performance [51] |
| 10x Genomics Single-Cell Assays | Single-cell RNA sequencing workflow | Processing up to 96 samples concurrently for tumor heterogeneity studies [51] |
NGS in Target Identification
Protocol: Genome-Wide Association Study for Novel Target Discovery
Purpose: Identify genetic variants associated with disease susceptibility for novel therapeutic target identification [49].
Materials and Equipment:
- Illumina NovaSeq X or similar high-throughput sequencer
- Whole genome sequencing library preparation kit
- Cloud computing access (AWS, Google Cloud Genomics)
- AI-based variant calling tools (e.g., DeepVariant)
- Population-scale genomic datasets (UK Biobank, 1000 Genomes)
Procedure:
- Cohort Selection: Recruit 1000 cases (disease-positive) and 1000 controls (disease-negative) with appropriate power calculation
- DNA Extraction: Isolate high-molecular-weight DNA from blood or tissue samples using standardized protocols
- Library Preparation: Fragment DNA and attach platform-specific adapters following manufacturer instructions
- Sequencing: Perform whole genome sequencing at minimum 30x coverage using paired-end reads
- Variant Calling: Process raw sequencing data through bioinformatic pipeline (quality control, alignment, variant identification)
- Association Analysis: Conduct statistical analysis to identify variants enriched in case population
- Functional Validation: Prioritize hits based on functional impact scores and pathway enrichment
Data Analysis:
- Identify non-synonymous variants, regulatory elements, and structural variants associated with disease
- Calculate odds ratios and p-values with multiple testing correction
- Integrate with gene expression data (e.g., GTEx) to prioritize targetable pathways
Case Study: KRAS G12C Inhibitor Development
The discovery and development of KRAS G12C inhibitors exemplifies successful NGS-driven target identification. KRAS mutations occur in approximately 20% of solid tumors, with particularly high prevalence in pancreatic (>90%), colorectal (30-50%), and non-small cell lung cancers (20-30%) [52]. NGS-based tumor profiling identified the specific G12C mutation as a druggable target, leading to the development of covalent inhibitors such as sotorasib and adagrasib that selectively bind to the GDP-bound state of mutant KRAS [52].
NGS in Biomarker Discovery
Protocol: Multi-Omics Approach for Predictive Biomarker Identification
Purpose: Identify composite biomarkers predictive of therapeutic response through integrated genomic, transcriptomic, and epigenomic analysis [53] [24].
Materials and Equipment:
- Paired tumor-normal tissue samples from clinical trial participants
- NGS platforms for WGS, RNA-seq, and bisulfite sequencing
- Single-cell sequencing equipment (10x Genomics)
- Cloud-based bioinformatics infrastructure
- Multi-omics integration software
Procedure:
- Sample Collection: Obtain pre-treatment tumor biopsies and matched normal tissue
- Nucleic Acid Extraction: Isolate DNA, RNA, and perform quality assessment
- Multi-Omics Profiling:
- Perform whole genome sequencing (100x coverage)
- Conduct total RNA sequencing (50 million reads/sample)
- Execute whole genome bisulfite sequencing for epigenomics
- Optional: single-cell RNA-seq for heterogeneity assessment
- Clinical Data Integration: Correlate molecular profiles with treatment response data
- Biomarker Identification: Use machine learning approaches to identify response-associated features
Data Analysis:
- Identify mutational signatures, expression subtypes, and methylation patterns
- Develop predictive models using random forest or neural network algorithms
- Validate biomarkers in independent cohort using targeted NGS approaches
Application: Biomarkers in Acute Myeloid Leukemia
In Acute Myeloid Leukemia (AML), NGS-based biomarker discovery has enabled precise classification and treatment selection. Common AML biomarkers include FLT3-ITD (poor prognosis), NPM1 mutations (favorable prognosis when FLT3-wildtype), and IDH1/2 mutations (targetable with IDH inhibitors) [54]. These biomarkers are detected using targeted NGS panels that enable simultaneous assessment of multiple genetic alterations from minimal specimen material, guiding therapeutic decisions and monitoring treatment response through measurable residual disease (MRD) detection [54].
Table 3: Clinically Actionable Biomarkers in AML and Their Therapeutic Implications
| Biomarker | Frequency | Prognostic Impact | Targeted Therapies | NGS Detection Method |
|---|---|---|---|---|
| FLT3-ITD | ~30% | Poor | FLT3 inhibitors (gilteritinib) | Amplification-based NGS [54] |
| NPM1 | ~30% | Favorable (if FLT3-wt) | Indirect impact | Targeted gene panels [54] |
| IDH1/IDH2 | ~20% | Intermediate | IDH inhibitors (ivosidenib, enasidenib) | DNA sequencing [54] |
| TP53 | 5-10% | Very poor | Emerging therapies | Whole exome or targeted sequencing [54] |
NGS in Combating Drug Resistance
Protocol: Comprehensive Resistance Mechanism Analysis
Purpose: Identify genomic, transcriptomic, and epigenetic mechanisms underlying drug resistance to inform combination therapy strategies [52] [53].
Materials and Equipment:
- Cell line models or patient-derived xenografts with acquired resistance
- Short-read and long-read NGS platforms
- DeepChek software or similar bioinformatic tools
- Automation systems for high-throughput screening (e.g., Biomek i7)
- Access to resistance databases (CARD, COSMIC)
Procedure:
- Resistance Model Development: Generate resistant models through prolonged drug exposure
- Multi-Modal Sequencing:
- Whole genome sequencing to identify secondary mutations
- RNA sequencing for pathway reactivation analysis
- Single-cell sequencing for heterogeneous resistance mechanisms
- Epigenetic profiling for chromatin accessibility changes
- Functional Validation: Use CRISPR screening to confirm resistance mechanisms
- Therapeutic Strategy Design: Develop combination approaches to overcome identified resistance
Data Analysis:
- Compare pre- and post-resistance genomic profiles
- Identify significantly altered pathways and networks
- Prioritize clinically actionable resistance mechanisms
Case Study: KRAS G12C Inhibitor Resistance
Despite initial efficacy, KRAS G12C inhibitors face resistance challenges that NGS has helped elucidate. Multiple studies utilizing whole exome and transcriptome sequencing have identified diverse resistance mechanisms including secondary KRAS mutations (e.g., R68S, Y96D), bypass signaling activation (RTK upstream activation, MET amplification), and phenotypic transformation (epithelial-to-mesenchymal transition, histological transformation) [52]. These findings have informed multiple combination strategies currently in clinical trials, including KRAS G12C inhibitors with SHP2 inhibitors, EGFR inhibitors, and CDK4/6 inhibitors [52].
Application: Antimicrobial Resistance Detection
Purpose: Rapid detection of antimicrobial resistance genes in bacterial pathogens to guide appropriate antibiotic therapy [50] [55].
Materials and Equipment:
- Clinical isolates from infected patients
- DNA extraction kits for microbial cells
- Targeted NGS panels for resistance genes
- Oxford Nanopore MinION for rapid turnaround
- DeepChek or similar interpretation software
Procedure:
- Sample Processing: Isolate DNA from bacterial cultures or directly from clinical specimens
- Library Preparation: Amplify resistance gene targets using pathogen-specific primers
- Sequencing: Perform targeted NGS using Illumina or Nanopore platforms
- Bioinformatic Analysis: Align sequences to resistance gene databases
- Report Generation: Interpret variants and provide therapeutic recommendations
Data Analysis:
- Identify single nucleotide polymorphisms in resistance genes
- Detect gene amplifications and horizontal transfer events
- Correlate genotypic findings with established resistance phenotypes
Table 4: NGS-Based Detection of Drug Resistance Across Pathogens
| Pathogen | Primary Resistance Mechanisms | NGS Detection Method | Clinical Impact |
|---|---|---|---|
| Mycobacterium tuberculosis | katG, rpoB, rpsL mutations | Whole genome sequencing | Guides MDR-TB treatment [55] |
| HIV-1 | Reverse transcriptase, protease mutations | Targeted deep sequencing | Optimizes ART regimens [50] |
| Hepatitis B/C | Polymerase, NS5A, NS3 mutations | Amplicon-based deep sequencing | Informs DAA selection [50] |
| Klebsiella pneumoniae | Carbapenemase genes (KPC, NDM) | Metagenomic NGS | Directs carbapenem therapy [55] |
Integrated Data Analysis and Future Directions
Protocol: AI-Enhanced Multi-Omics Data Integration
Purpose: Leverage artificial intelligence and cloud computing to integrate diverse NGS datasets for enhanced biomarker and target discovery [49] [24].
Materials and Equipment:
- Cloud computing platforms (AWS, Google Cloud, Azure)
- AI/ML frameworks (TensorFlow, PyTorch)
- Multi-omics data integration tools
- Secure data storage compliant with regulatory standards
Procedure:
- Data Collection: Aggregate NGS data from multiple sources (genomic, transcriptomic, epigenomic)
- Data Preprocessing: Normalize and harmonize diverse data types
- Feature Selection: Identify most informative features using dimensionality reduction
- Model Training: Develop predictive models using machine learning algorithms
- Validation: Test model performance on independent datasets
- Deployment: Implement models in clinical trial design or diagnostic development
Data Analysis:
- Use deep learning for variant calling (e.g., Google's DeepVariant)
- Apply neural networks for drug response prediction
- Implement natural language processing for literature-based validation
The integration of AI with NGS data has demonstrated significant improvements in interpretation efficiency, with one CDC implementation reporting a 40% increase in interpretation efficiency through machine learning algorithms [49]. Furthermore, cloud-based NGS data analysis has reduced development cycles by up to 20% in pharmaceutical applications [49].
Emerging Trends
The field of NGS in drug discovery continues to evolve with several emerging trends:
- Real-time NGS monitoring in clinical trials: Using circulating tumor DNA sequencing to monitor treatment effectiveness and identify resistance mutations earlier than traditional methods [49]
- Automated NGS workflows: Integration of robotic systems that reduce hands-on time from 23 hours to 6 hours per run while improving data consistency [51]
- Portable sequencing technologies: Oxford Nanopore platforms enabling field-based sequencing for rapid outbreak investigation and resistance monitoring [7]
- Spatial transcriptomics: Mapping gene expression within tissue architecture to understand microenvironmental contributions to drug response [24]
NGS technologies have become indispensable tools throughout the drug discovery and development pipeline, from initial target identification to combating drug resistance. The protocols and applications detailed herein provide researchers with practical frameworks for implementing NGS approaches in their discovery efforts. As sequencing technologies continue to advance, with improvements in accuracy, throughput, and accessibility, and as analytical methods become increasingly sophisticated through AI integration, the impact of NGS on pharmaceutical research is poised to grow even further. Strategic partnerships between technology developers, pharmaceutical companies, and research institutions will be crucial for realizing the full potential of NGS in delivering novel therapeutics to patients.
Next-generation sequencing (NGS) has revolutionized DNA and RNA analysis, providing unprecedented capabilities for characterizing genetic material with high throughput and precision [56]. These technologies are now being deployed across industrial sectors to address critical challenges in agriculture, food security, and biomanufacturing. The applications range from enhancing crop resilience and monitoring foodborne pathogens to ensuring the safety of biological products, collectively contributing to more sustainable and secure production systems [56] [57] [58]. This application note details specific protocols and methodologies that leverage NGS for these emerging industrial applications, providing researchers with practical frameworks for implementation.
Application Notes
NGS in Agricultural Genomics for Orphan Crops
Background: Orphan crops, also known as neglected and underutilized species (NUS), are vital for enhancing nutritional diversity and strengthening food security but have historically received limited research attention [57]. Genomics enables the rapid genetic improvement of these species, which possess inherent resilience to environmental stresses like drought, heat, salinity, and pests [57].
Key Applications:
- Genetic Diversity Analysis: Unraveling evolutionary histories and domestication processes through high-throughput sequencing.
- Marker-Assisted Selection (MAS): Accelerating breeding programs for traits like disease resistance and nutritional quality.
- Genomic Selection: Enhancing genetic gain for complex traits in breeding populations.
Table 1: Genomic Approaches for Orphan Crop Improvement
| Application | Genomic Tool | Outcome | Example Crop |
|---|---|---|---|
| Deciphering Domestication | High-throughput sequencing, DArTSeq | Identification of wild progenitors & migration history | Teff, Finger Millet [57] |
| Trait Mapping | SNP panels, KASP assays | Identification of genomic regions associated with desirable traits | Finger Millet (iron-rich varieties) [57] |
| Accelerated Breeding | Genomic Selection, Speed Breeding | Reduced breeding cycles & enhanced genetic gain | Various orphan cereals [57] |
| De Novo Domestication | Gene editing (CRISPR/Cas), advanced transformation | Introduction of domestication genes (e.g., shattering resistance) | Model species for future orphan crops [57] |
NGS for Food Safety and Authenticity
Background: NGS provides culture-independent methods for pathogen detection, antimicrobial resistance surveillance, and microbial community profiling in complex food matrices, significantly improving food safety monitoring and outbreak prevention [56].
Key Applications:
- Whole Genome Sequencing (WGS): Enables high-resolution traceability of foodborne pathogen outbreaks [56].
- Metagenomics: Characterizes microbial dynamics during food processing and fermentation [56].
- Metatranscriptomics: Provides insights into flavor formation and metabolic pathways during fermentation [56].
Table 2: NGS Platforms and Applications in Food Science
| NGS Platform | Technology Principle | Key Food Science Applications | References |
|---|---|---|---|
| Illumina | Sequencing by synthesis (SBS) | WGS of foodborne pathogens; Metagenomics of fermentation; Metatranscriptomics for flavor formation | [56] |
| Ion Torrent | SBS with H+ ion detection | Metagenetics for seafood quality; Microbial profiling of dairy products | [56] |
| PacBio | Single-molecule real-time (SMRT) sequencing | Long-read metagenetics for analyzing dairy product quality | [56] |
| Oxford Nanopore | Nanopore electrical signal sequencing | Real-time identification of foodborne pathogens and AMR genes; Spoilage microbe detection | [56] |
NGS in Biomanufacturing for Adventitious Virus Detection
Background: The biomanufacturing of biologics requires rigorous testing for adventitious viruses. NGS is emerging as a powerful, broad-spectrum alternative to conventional in vivo and PCR-based assays, as recognized in the ICH Q5A (R2) guideline and the EDQM general chapter 2.6.41 [58].
Key Applications:
- Replacing In Vivo Assays: NGS can detect a wide range of viral contaminants without the need for animal testing.
- Supplementing/Replacing In Vitro Assays: Provides a more comprehensive viral safety assessment.
- Regulatory Submissions: A suitable validation package enables the use of NGS in regulatory filings for drug safety [58].
Experimental Protocols
Protocol: Metagenomic Analysis of a Food Microbiome
This protocol details the process for characterizing the microbial community of a fermented food product using shotgun metagenomics.
I. Wet Lab Phase: Sample to Library
Step 1: Sample Collection and Storage
- Material: Sterile swabs or sample bags, cryogenic tubes.
- Procedure: Aseptically collect a representative sample (e.g., 25g of food matrix or surface swabs). For raw materials with variable microbiota, use pooled sampling to improve accuracy [56]. Immediately snap-freeze samples in liquid nitrogen and store at -80°C to prevent nucleic acid degradation and microbial growth changes [56].
Step 2: Nucleic Acid Extraction
- Material: Commercial DNA/RNA extraction kit (e.g., QIAamp circulating nucleic acid kit), lysis buffer, proteinase K, ethanol.
- Procedure: Extract total nucleic acids from the homogenized sample according to the manufacturer's protocol. Include mechanical lysis (e.g., bead beating) for robust cell disruption. Assess DNA/RNA quality and quantity using spectrophotometry (e.g., Nanodrop) and fluorometry (e.g., Qubit).
Step 3: Library Preparation
- Material: Library preparation kit (e.g., Illumina Nextera XT), size selection beads (e.g., AMPure XP), PCR reagents.
- Procedure: For Illumina platforms, fragment DNA and ligate platform-specific adapters. Optional: Amplify the library with indexed primers for multiplexing. Perform a clean-up and size selection step to remove adapter dimers and select the desired insert size. Validate the final library using a bioanalyzer or tapestation.
Step 4: Sequencing Run
- Material: Sequencing cartridge (e.g., Illumina MiSeq Reagent Kit v3), flow cell.
- Procedure: Denature and dilute the library according to platform-specific guidelines. Load onto the sequencer (e.g., Illumina MiSeq or NovaSeq) and initiate the run.
II. Dry Lab Phase: Data Analysis
Step 1: Quality Control and Preprocessing
- Tool: FastQC, Trimmomatic.
- Procedure: Assess raw read quality (Phred scores, GC content, adapter contamination). Trim low-quality bases and remove adapter sequences.
Step 2: Metagenomic Assembly
- Tool: MEGAHIT, metaSPAdes.
- Procedure: De novo assemble the quality-filtered reads into longer contigs. Assess assembly quality using metrics like N50 and contig counts.
Step 3: Taxonomic and Functional Profiling
- Tool: Kraken2, MetaPhlAn, HUMAnN2.
- Procedure: Classify reads or contigs against reference databases (e.g., RefSeq, GTDB) to determine taxonomic abundance. Reconstruct and quantify metabolic pathways from the assembled data.
Protocol: Targeted Sequencing for Circulating DNA/RNA in Liquid Biopsies
This protocol is adapted for quality control in biomanufacturing, such as detecting viral contaminants or host cell DNA/RNA in biological products.
I. Wet Lab Phase
Step 1: Sample Collection
- Material: Blood collection tubes (e.g., Streck cfDNA BCT), serum/ plasma collection tubes.
- Procedure: Collect blood or other bodily fluid samples. For cell-free DNA (cfDNA), process plasma within 2-4 hours to prevent genomic DNA contamination.
Step 2: Circulating Nucleic Acid Extraction
- Material: Specialized cfDNA/RNA extraction kit (e.g., QIAamp Circulating Nucleic Acid Kit) [59].
- Procedure: Isolate cfDNA/RNA from plasma or serum according to the kit's protocol, which typically involves protease digestion, binding to a silica membrane, and washing steps. Elute in a low-volume buffer.
Step 3: Targeted Library Preparation
- Material: Targeted DNA + RNA assay panel (e.g., Mission Bio Tapestri Targeted DNA + RNA Assay), hybrid capture reagents (e.g., IDT xGen Lockdown Probes) [60].
- Procedure: For a multi-omics approach, use a platform that enables targeted genotyping and transcript quantification from the same cell [60]. Alternatively, construct libraries and perform hybrid capture with biotinylated probes targeting specific viral or host sequences. Amplify and index the captured libraries.
Step 4: Sequencing
- Procedure: Sequence on an appropriate platform (e.g., Illumina for high-depth targeted sequencing).
II. Dry Lab Phase
Step 1: Data Preprocessing
- Procedure: Perform standard QC and adapter trimming. For single-cell multi-omics data, process DNA and RNA reads separately.
Step 2: Variant Calling and Expression Analysis
- Tool: GATK, Cell Ranger (for single-cell data).
- Procedure: Align DNA reads to a reference genome and call variants (SNVs, indels). Align RNA reads to quantify gene expression. For single-cell data, link genetic mutations to transcriptional phenotypes within the same cell [60].
Step 3: Pathogen Detection
- Tool: Kraken2, BLAST.
- Procedure: Classify non-host reads against microbial databases to identify adventitious viral sequences, confirming the absence of contaminants in biologics [58].
Workflow and Pathway Diagrams
NGS Analysis Workflow for Food and Environmental Samples
Genomic-Assisted Breeding Pipeline for Orphan Crops
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Kits for NGS Applications
| Product Name / Type | Function | Specific Application Example |
|---|---|---|
| QIAamp Circulating Nucleic Acid Kit | Isolation and purification of cell-free DNA and RNA from plasma or serum. | Liquid biopsy analysis for non-invasive prenatal testing or oncology [59]. |
| Tapestri Single-Cell Targeted DNA + RNA Assay | Simultaneous measurement of genotypic and transcriptional readouts from thousands of individual cells. | Linking genetic mutations to functional consequences in complex cell populations (e.g., cancer research, cell therapy development) [60]. |
| xGen Lockdown Probes | Biotinylated oligonucleotide probes for targeted enrichment of specific genomic regions by hybrid capture. | Sequencing tumor DNA to identify biomarkers for precision oncology [59]. |
| Nextera XT DNA Library Preparation Kit | Rapid preparation of Illumina sequencing libraries from genomic DNA through tagmentation. | Metagenomic sequencing of food or environmental samples for microbiome analysis [56]. |
| DArTSeq Platform | A complexity-reduction method based on sequencing genomic representations for cost-effective SNP discovery and genotyping. | Assessing genetic diversity and population structure in orphan crops without a reference genome [57]. |
Navigating NGS Challenges: A Guide to Troubleshooting and Data Optimization
Within next-generation sequencing (NGS) research for DNA and RNA analysis, the pre-analytical phase represents a critical determinant of experimental success. The integrity and interpretation of genomic data are profoundly influenced by two fundamental factors: the quality of the input nucleic acids and the proportion of malignant cells within a tumor specimen. Sample quality directly impacts sequencing library complexity and data yield, while tumor purity affects mutation detection sensitivity and variant allele frequency quantification. This application note provides detailed protocols and quantitative frameworks to address these common pre-analytical challenges, enabling researchers to generate more reliable and reproducible NGS data for drug development and clinical research.
Sample Quality Challenges in NGS
Critical Quality Parameters for Nucleic Acids
The quality of nucleic acids extracted from biological samples must be rigorously assessed through multiple parameters before proceeding with NGS library preparation. The table below summarizes the key quality metrics and their recommended thresholds for successful sequencing.
Table 1: Quality Control Parameters and Thresholds for NGS Sample Preparation
| Parameter | Assessment Method | Recommended Threshold | Significance of Deviation |
|---|---|---|---|
| DNA/RNA Concentration | Qubit fluorometer [61] [62] | Application-dependent | Overestimation with spectrophotometry due to contaminants [61] [63] |
| DNA Purity | NanoDrop 260/280 ratio [62] | ~1.8 [63] [62] | Ratio <1.8 indicates protein/phenol contamination [62] |
| DNA Purity | NanoDrop 260/230 ratio [63] [62] | 2.0-2.2 [63] [62] | Ratio <2.0 indicates chemical contamination [62] |
| RNA Integrity | RNA Integrity Number (RIN) [63] [64] [65] | >7 for RNA-seq [63] [64] [65] | Degraded RNA alters transcript representation [63] |
| DNA Size Distribution | Bioanalyzer/Femto Pulse [62] | Application-dependent | Fragmentation affects library prep efficiency [62] |
Impact of FFPE Processing on Sample Quality
Formalin-fixed paraffin-embedded (FFPE) tissues remain the standard specimen type in clinical cancer research but present significant challenges for NGS. Formalin fixation induces multiple types of DNA damage including crosslinks, fragmentation, cytosine deamination, and abasic sites [61]. Pre-analytical factors such as ischemic time, fixation duration, and storage conditions dramatically impact FFPE sample quality. Under-fixation can cause nucleic acid degradation in deeper tissue regions, while over-fixation increases crosslinking, making nucleic acid extraction more difficult [61]. Storage of FFPE blocks at 4°C rather than room temperature helps prolong biomolecule integrity [61].
DNA Extraction and Quantification Best Practices
Substantial variability exists in commercial laboratory performance for DNA extraction from FFPE samples, with yields varying by 5-10 times between different labs processing identical samples [61]. For FFPE DNA quantification, the Qubit fluorometer demonstrates superior consistency compared to Nanodrop spectrophotometry, which often overestimates DNA concentration due to residual RNA, single-stranded DNA, and other contaminants [61]. For precious or limited samples, double elution during DNA extraction can improve recovery by up to 42%, and overnight Proteinase K digestion often enhances both DNA yield and purity [61].
Figure 1: Comprehensive Quality Control Workflow for NGS Sample Preparation
Tumor Purity Considerations in Cancer Genomics
Traditional versus Computational Purity Assessment
Tumor purity, defined as the proportion of malignant cells in a specimen, significantly impacts mutation detection sensitivity and clinical interpretation of NGS results. Traditional assessment by pathologist review of hematoxylin and eosin (H&E)-stained slides shows limited reproducibility between observers [66] [67]. Computational approaches leverage genetic features from sequencing data to provide more objective purity estimates, with methods including ABSOLUTE, ASCAT, THetA2, and transcriptomic approaches like PUREE [68] [66].
Real-world data from FoundationOne CDx testing demonstrates that computational tumor purity estimates show stronger correlation with quality check status than pathologist-reviewed tumor nuclei percentage [69]. Tumor purity substantially affects the success rate of comprehensive genomic profiling (CGP) tests, with microsatellite status testing significantly more successful in samples with pass versus qualified QC status [69].
Tumor Purity Estimation Methods
Multiple computational approaches have been developed to estimate tumor purity from different data types, each with distinct strengths and applications.
Table 2: Comparison of Tumor Purity Estimation Methods
| Method | Input Data | Underlying Principle | Performance Notes |
|---|---|---|---|
| Pathologist Estimation [66] | H&E-stained slides | Microscopic examination | Limited reproducibility between pathologists [66] |
| ABSOLUTE [66] | SNP array/NGS | Copy number and allele frequencies | Used in TCGA; fails on some samples [66] |
| ASCAT [66] | SNP array | Allele-specific copy number | Fails on some samples [66] |
| THetA2 [66] | NGS | Copy number changes | Returns warning flags for some samples [66] |
| PUREE [68] | RNA-seq | Weakly supervised learning | Pearson r=0.78 vs genomic consensus [68] |
| ESTIMATE [68] | RNA-seq | Immune/stromal signature | Pearson r=0.63 vs genomic consensus [68] |
The PUREE method employs a weakly supervised learning approach trained on gene expression data and genomic consensus purity estimates from 7,864 solid tumor samples across 20 cancer types [68]. It utilizes a linear regression model with 158 carefully selected features that demonstrate enrichment in cancer-related pathways including epithelial-mesenchymal transition, KRAS signaling, and inflammatory response [68]. In benchmarking studies, PUREE outperformed existing transcriptome-based methods with higher correlation (r=0.78) and lower root mean squared error (0.09) compared to genomic consensus purity estimates [68].
Impact of Tumor Purity on Clinical Genomic Testing
Real-world data from 1,204 F1CDx tests reveals that the percentage of tumor nuclei is the most significant factor influencing quality check status, with pancreatic cancer and long-term FFPE block storage representing additional independent risk factors [69]. Receiver operating characteristic (ROC) analysis supports 35% as an ideal percentage of tumor nuclei for CGP submission, substantially higher than the 30% minimum recommendation [69]. Storage time of FFPE blocks significantly associates with qualified status, with blocks older than three years showing increased qualification rates, though this effect is smaller than tumor purity impact [69].
Figure 2: Tumor Purity Estimation Approaches from Different Data Types
Experimental Protocols
Protocol: DNA QC for FFPE NGS Studies
Principle: Assess DNA quantity, purity, and size distribution from FFPE samples to ensure suitability for NGS library preparation [61] [62].
Materials:
- Qubit fluorometer with dsDNA BR Assay Kit
- NanoDrop spectrophotometer
- Agilent TapeStation or Bioanalyzer
- TE buffer
Procedure:
- DNA Quantification:
- Allow Qubit reagents to reach room temperature
- Prepare working solution by diluting Qubit dsDNA BR reagent 1:200 in buffer
- Add 1μL DNA sample to 199μL working solution, mix thoroughly
- Read concentration using Qubit fluorometer
- Compare with NanoDrop A260 measurement; significant discrepancies indicate contamination
Purity Assessment:
- Apply 1-2μL DNA sample to NanoDrop pedestal
- Measure A260/A280 and A260/A230 ratios
- Acceptable ranges: A260/A280 ~1.8, A260/A230 2.0-2.2 [62]
- If ratios are low, perform additional purification or PCR amplification
Size Distribution Analysis:
- Prepare TapeStation or Bioanalyzer according to manufacturer instructions
- Use genomic DNA assay with ladder and marker
- Load 1μL sample
- Analyze fragment size distribution; verify majority of DNA >200bp for successful NGS
Troubleshooting:
- Low yield: Increase Proteinase K digestion time or try double elution [61]
- Poor purity: Perform additional column purification or ethanol precipitation
- Excessive fragmentation: Optimize FFPE sectioning or DNA extraction method
Protocol: RNA QC for Transcriptome Sequencing
Principle: Evaluate RNA integrity and purity to ensure successful RNA-seq library preparation [63] [64] [65].
Materials:
- Agilent TapeStation with RNA ScreenTape
- NanoDrop spectrophotometer
- RNase-free water and tubes
Procedure:
- RNA Quality Assessment:
Concentration and Purity Measurement:
- Apply 1μL RNA to NanoDrop for A260/A280 and A260/A230 ratios
- Acceptable ranges: A260/A280 ~2.0, A260/A230 >1.8 [63]
- Use Qubit RNA HS Assay for more accurate quantification if needed
Visual Inspection of Electrophoresis:
- For eukaryotic samples, verify sharp 28S and 18S ribosomal RNA bands
- Confirm 28S:18S intensity ratio approximately 2:1
- Reject samples with smeared appearance or low molecular weight bands
Troubleshooting:
- Low RIN: Isolate RNA more quickly after tissue collection; use RNAlater
- DNA contamination: Perform DNase digestion
- Poor yields: Optimize homogenization; use different RNA isolation kit
Protocol: Computational Tumor Purity Estimation with PUREE
Principle: Estimate tumor purity from RNA-seq data using a pre-trained machine learning model [68].
Materials:
- Gene expression matrix (TPM or FPKM) from tumor sample
- PUREE software package (available from original publication)
- R or Python environment
Procedure:
- Data Preprocessing:
- Normalize gene expression data using rank-percentile transformation
- Filter to include only protein-coding autosomal genes
- Select the 158-gene feature set used in PUREE model
Purity Estimation:
- Load pre-trained PUREE linear regression model
- Apply model to transformed gene expression data
- Generate purity estimate between 0 and 1
Result Interpretation:
- Compare estimated purity with histological assessment when available
- Use purity estimate to guide downstream analysis
- Flag samples with purity <30% for potential exclusion or special handling
Validation:
- Benchmark against genomic methods when DNA sequencing available
- Compare with ESTIMATE or other transcriptomic methods
- Correlate with pathological review for concordance
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Pre-Analytical QC
| Category | Specific Product/Kit | Application | Performance Notes |
|---|---|---|---|
| DNA Quantification | Qubit dsDNA BR Assay Kit [61] [62] | Accurate DNA mass measurement | Superior to spectrophotometry for FFPE DNA [61] |
| RNA Quality Assessment | Agilent TapeStation [63] | RNA integrity evaluation | Provides RIN score for objective quality assessment [63] |
| Nucleic Acid Extraction | RNeasy kits [63] | RNA purification from tissues | Produces high-purity RNA preparations [63] |
| FFPE DNA Extraction | Proteinase K digestion [61] | DNA release from FFPE | Overnight digestion improves yield and purity [61] |
| RNA Stabilization | RNALater [63] | Tissue preservation | Prevents RNA degradation when immediate isolation impossible [63] |
| Computational Purity Tools | PUREE package [68] | Tumor purity estimation | 0.78 correlation with genomic consensus; pan-cancer application [68] |
Addressing pre-analytical challenges in NGS research requires rigorous attention to both sample quality metrics and tumor purity considerations. Implementation of standardized QC protocols for nucleic acid assessment, combined with computational approaches for tumor purity estimation, significantly enhances the reliability of downstream genomic analyses. The quantitative thresholds and experimental methodologies outlined in this application note provide researchers with a structured framework to optimize these critical pre-analytical factors, ultimately supporting more robust and reproducible cancer genomics research in both basic science and drug development contexts.
Next-generation sequencing (NGS) has revolutionized genomic research, but the quality of its data is heavily dependent on the initial library preparation steps. A central challenge in this process is the introduction of PCR artifacts and duplicate reads, which can compromise data integrity and lead to erroneous biological conclusions [70] [71]. PCR duplicates arise when multiple identical reads originate from a single original DNA or RNA fragment, artificially inflating coverage estimates and potentially obscuring true genetic variants [70] [72]. Distinguishing technical artifacts from biological duplicates is particularly crucial in RNA-Seq, where highly expressed genes naturally generate many identical reads [70]. This application note provides comprehensive strategies and detailed protocols to minimize these artifacts, ensuring the generation of high-quality, reliable sequencing data for research and drug development applications.
Understanding PCR Artifacts and Duplicate Reads
PCR duplicates originate from multiple sources during library preparation. The primary mechanism involves over-amplification of original DNA fragments during the PCR enrichment step, where insufficient starting material necessitates excessive amplification cycles to generate adequate library mass [73] [72]. This bottleneck effect is particularly pronounced with low-input samples, where a limited diversity of original molecules creates libraries dominated by amplified copies of a small subset of fragments [70]. Additionally, optical duplicates can occur during sequencing on patterned flow cells when the same template molecule binds to adjacent sites and is amplified independently [72].
The impact of these artifacts on downstream analysis is substantial. In variant calling, PCR duplicates can create false positive variant calls when polymerase incorporation errors during amplification are misinterpreted as true genetic variants [72]. For quantitative applications like RNA-Seq or ChIP-Seq, duplicates distort abundance measurements, skewing expression estimates and differential analysis [70]. Furthermore, the computational removal of duplicates, while necessary for some applications, wastes sequencing capacity and increases project costs [72].
Distinguishing Technical vs. Biological Duplicates
A critical consideration in duplicate management is distinguishing technical artifacts from natural biological duplicates, especially in RNA-Seq experiments. Unlike genomic DNA applications, where duplicates are predominantly technical artifacts, RNA-Seq libraries naturally contain many identical reads from highly expressed genes [70]. As the dupRadar package authors explain, "the top 5% of expressed genes often account for more than 50% of all reads in a common RNA-Seq dataset" [70]. Removing these reads as technical duplicates would severely underestimate expression levels for highly abundant transcripts.
Tools like dupRadar help distinguish these sources by modeling the relationship between duplication rate and gene expression level [70]. Natural duplication follows a predictable pattern where duplication rates remain low for genes with low to medium expression, then increase sharply as expression levels approach and exceed one read per base pair of the gene model [70]. Artifactual duplication, in contrast, appears as elevated duplication rates across all expression levels, indicating systematic issues with library complexity [73].
Experimental Design Strategies for Artifact Prevention
Input Material and Amplification Optimization
The foundation for minimizing PCR artifacts begins with appropriate input material and careful control of amplification. Table 1 summarizes key optimization strategies for preventing PCR artifacts and duplicate reads.
Table 1: Optimization Strategies for Preventing PCR Artifacts and Duplicate Reads
| Factor | Recommendation | Impact on Artifacts |
|---|---|---|
| DNA Input | Use sufficient starting material (>500 ng per library for multiplexed hybridization capture) [72] | Reduces molecular bottleneck and need for excessive amplification |
| PCR Cycles | Minimize amplification cycles; use just enough for adequate library yield [72] | Limits over-amplification of individual fragments |
| Enzyme Selection | Use high-fidelity polymerases with proofreading capability [33] | Reduces nucleotide misincorporation errors during amplification |
| Fragmentation Method | Choose between sonication (random fragmentation) or enzymatic (sequence-specific) based on application [71] | Different methods produce distinct artifact patterns that require specific mitigation |
| Unique Molecular Identifiers (UMIs) | Incorporate UMIs during adapter ligation [74] | Enables bioinformatic discrimination of PCR duplicates from original molecules |
The relationship between input amount and duplication rate is particularly critical in multiplexed experiments. Research demonstrates that using 500 ng of each barcoded library in multiplexed hybridization capture experiments maintains consistently low duplication rates (approximately 2.5%), regardless of multiplexing level [72]. In contrast, pooling the same total mass (500 ng) across increasingly complex libraries dramatically increases duplication rates, reaching 13.5% in 16-plex experiments [72].
Fragmentation Method Considerations
The choice between sonication and enzymatic fragmentation significantly influences the types of artifacts encountered. Sonication shears DNA through physical means, producing relatively random fragments, but can generate chimeric artifacts containing inverted repeat sequences (IVSs) when partial single-stranded DNA molecules from the same original molecule incorrectly reanneal [71]. Enzymatic fragmentation using DNA endonucleases is more convenient and minimizes sample loss but can produce artifacts at palindromic sequences (PS) due to the specific cleavage patterns of the enzymes [71].
Recent research has proposed the Pairing of Partial Single Strands Derived from a Similar Molecule (PDSM) model to explain artifact formation mechanisms common to both fragmentation methods [71]. This model hypothesizes that fragmentation generates partial single-stranded DNA molecules that can incorrectly pair with complementary sequences from other fragments, creating chimeric molecules that are amplified in subsequent PCR steps [71]. Understanding this mechanism informs both experimental and computational mitigation strategies.
Detailed Protocols for Library Preparation
Optimized Adapter Ligation Procedure
Adapter ligation is a critical step where inefficiencies can lead to chimeric fragments and low library complexity [75] [33]. The following protocol ensures efficient ligation:
Adapter Preparation: Use freshly prepared or properly stored adapters to prevent degradation or improper annealing [75]. For dual-indexed adapters, verify the compatibility of index sequences to prevent index hopping.
Ligation Conditions:
Molar Ratios: Maintain correct molar ratios between insert DNA and adapters (typically 1:5 to 1:10) to minimize adapter dimer formation while ensuring efficient ligation [75]. Excess adapter can lead to dimer formation that consumes sequencing capacity.
A-tailing Efficiency: Ensure efficient A-tailing of PCR products before adapter ligation, as this universal procedure prevents chimera formation and improves ligation efficiency [33].
Automated liquid handling systems like the I.DOT Liquid Handler can significantly improve reproducibility in adapter ligation by ensuring consistent reaction setups across samples [75].
Enzymatic Fragmentation Workflow
Enzymatic fragmentation offers advantages for low-input samples but requires careful optimization to minimize sequence-specific biases:
Enzyme Selection: Choose enzyme cocktails designed to minimize sequence-specific cutting biases. Verify lot-to-lot consistency for production-scale studies.
Fragmentation Time and Temperature: Optimize conditions for your specific genomic DNA sample type. Typical conditions include 37°C for 15-30 minutes, but manufacturer recommendations vary [71].
Termination: Use recommended stop solutions or heat inactivation precisely to prevent over-digestion, which can reduce library complexity.
Size Selection: Perform rigorous size selection after fragmentation using magnetic bead-based cleanups (0.6-0.8X bead-to-sample ratio for fragment retention) to remove too small or too large fragments [33].
For sonication-based fragmentation, ensure consistent shearing parameters across samples and minimize DNA sample loss during transfer steps, which is particularly problematic for limited samples like biopsies [71].
PCR Enrichment and Library Normalization
Controlled amplification is essential for maintaining library complexity:
Cycle Determination: Use the minimum number of PCR cycles necessary for adequate library yield. Perform test amplifications with varying cycles (8-15) and quantify results to establish optimal conditions [72].
High-Fidelity Polymerases: Select polymerases with demonstrated low error rates and minimal amplification bias [33]. These enzymes reduce nucleotide misincorporation that can be misinterpreted as variants.
Library Normalization: Accurately normalize libraries before pooling to ensure equal representation. Use quantitative PCR or fluorometric methods rather than spectrophotometry for most accurate quantification [75]. Automated systems like the G.STATION NGS Workstation can normalize libraries using integrated protocols and bead-based cleanup, increasing consistency across pooled samples [75].
The following workflow diagram illustrates the optimized library preparation process with key quality control checkpoints:
Essential Reagents and Research Solutions
The selection of appropriate reagents is fundamental to successful library preparation. Table 2 catalogizes key research reagent solutions and their functions in minimizing artifacts.
Table 2: Research Reagent Solutions for Optimal Library Preparation
| Reagent Category | Specific Examples | Function in Artifact Prevention |
|---|---|---|
| High-Fidelity Polymerases | Q5, KAPA HiFi, Phusion | Reduce nucleotide misincorporation errors during PCR amplification [33] |
| Fragmentation Enzymes | 5× WGS fragmentation mix | Provide consistent fragmentation with minimal sequence bias; alternatives to sonication [71] |
| Adapter Systems | Illumina dual-indexed adapters, IDT unique dual indexes | Enable sample multiplexing while reducing index hopping and cross-contamination [74] [72] |
| Library Prep Kits | Illumina DNA Prep, Illumina RNA Prep | Provide optimized, validated workflows with integrated bead-based cleanups [74] |
| Unique Molecular Identifiers (UMIs) | IDT UMIs, Illumina UMIs | Molecular barcoding of original fragments to distinguish biological from technical duplicates [74] |
| Bead-Based Cleanup | AMPure XP, G.PURE NGS Clean-Up Device | Consistent size selection and purification to remove adapter dimers and primers [75] |
Unique Molecular Identifiers (UMIs) represent a particularly powerful tool for artifact mitigation. These short random nucleotide sequences are incorporated into individual molecules before amplification, creating a unique barcode for each original fragment [74]. During bioinformatic processing, reads sharing identical UMIs and mapping coordinates can be confidently identified as PCR duplicates and collapsed into a single consensus read, significantly improving variant calling accuracy [74].
Quality Control and Validation Methods
Pre-sequencing QC Checkpoints
Implementing rigorous quality control throughout library preparation is essential for detecting potential artifacts early. Key checkpoints include:
Post-ligation QC: Assess ligation efficiency using fragment analysis systems (e.g., Bioanalyzer, TapeStation). Successful ligation should show a size shift corresponding to adapter addition with minimal adapter dimer peaks [75].
Post-amplification QC: Verify library concentration and size distribution using fluorometric quantification (e.g., Qubit) combined with fragment analysis. This confirms adequate yield and appropriate size distribution for sequencing [33].
Post-normalization QC: Validate normalization accuracy before pooling, particularly for multiplexed experiments. qPCR-based methods provide the most accurate quantification for sequencing load calculations [75].
Automated systems like the G.STATION NGS Workstation can integrate these QC steps while maintaining detailed audit trails for regulatory compliance and troubleshooting [75].
Bioinformatic Artifact Detection
Despite optimal wet-lab procedures, some artifacts may persist and require computational detection:
Duplicate Assessment Tools: Packages like dupRadar provide specialized analysis for RNA-Seq data, modeling the relationship between duplication rate and expression level to distinguish technical artifacts from natural duplicates [70]. The tool generates diagnostic plots showing expected duplication patterns, with anomalous profiles indicating library preparation issues.
Artifact Filtering Algorithms: For DNA sequencing, tools like ArtifactsFinder identify chimeric reads resulting from specific genomic structures (inverted repeats, palindromic sequences) that are prone to artifact formation during fragmentation [71]. These tools generate custom "blacklists" of problematic genomic regions to filter from variant calls.
Standard Duplicate Marking: Tools like Picard MarkDuplicates and SAMTools rmdup identify reads with identical mapping coordinates, flagging them for removal in DNA-based assays where biological duplicates are unexpected [72].
The following diagram illustrates the PDSM model mechanism for artifact formation, which informs these bioinformatic detection approaches:
Quantitative Data and Performance Metrics
Establishing benchmark metrics is essential for evaluating library preparation success. Table 3 summarizes expected performance values for key quality metrics under optimized conditions.
Table 3: Expected Performance Metrics for Optimized Library Preparation
| Quality Metric | Optimal Range | Acceptable Range | Measurement Method |
|---|---|---|---|
| DNA Input Amount (multiplexed capture) | 500 ng per library [72] | 200-500 ng | Fluorometric quantification (Qubit) |
| Duplication Rate (DNA sequencing) | <5% [72] | 5-10% | Picard MarkDuplicates, SAMTools |
| Library Complexity (unique fragments) | >80% of non-duplicate reads | 70-80% | Estimation from pre- and post-deduplication counts |
| Coverage Uniformity | >95% of targets at 20X [72] | >90% at 20X | Analysis of per-base coverage distribution |
| Adapter Ligation Efficiency | >90% fragments with adapters | >80% | Fragment analysis (Bioanalyzer) |
Monitoring these metrics across experiments enables rapid detection of protocol deviations and facilitates continuous process improvement. For RNA-Seq applications, the interpretation of duplication rates must be expression-level dependent, with expected increases in duplication for highly expressed genes [70]. The dupRadar package provides specialized diagnostics for this purpose, fitting a logistic model to the relationship between duplication rate and expression level (measured as Reads Per Kilobase) to identify libraries with anomalously high duplication across all expression levels [70].
Optimizing library preparation to minimize PCR artifacts and duplicate reads requires a comprehensive approach addressing experimental design, reagent selection, process control, and quality validation. The strategies outlined in this application note provide a roadmap for generating high-quality NGS data with minimal artifacts, enabling more reliable downstream analysis and more confident biological conclusions. As NGS applications continue to expand into more challenging sample types and lower input requirements, these optimization principles become increasingly critical for research success and drug development progress.
Next-generation sequencing (NGS) has revolutionized genomics research by enabling the high-throughput analysis of DNA and RNA, yet significant technical challenges persist in accurately sequencing and aligning reads from complex genomic regions [7]. Short-read sequencing technologies, while cost-effective and accurate for many applications, struggle with repetitive elements and structural variations due to their limited read length, which complicates unambiguous alignment to reference genomes [76] [7]. These limitations are particularly problematic for studying short tandem repeats (STRs), structural variants (SVs), and other complex genomic architectures that play crucial roles in genetic diversity and disease pathogenesis [76].
The fundamental issue stems from the inherent trade-offs between different sequencing technologies. Short-read platforms (e.g., Illumina) generate reads typically between 75-300 base pairs, which are often insufficient to span repetitive regions, leading to misalignment, ambiguous mapping, and false variant calls [7] [77]. Third-generation long-read sequencing technologies from PacBio and Oxford Nanopore address these limitations by producing reads thousands of base pairs long, enabling the direct sequencing through repetitive elements and complex structural variations [76] [7]. This application note details integrated experimental and computational strategies to overcome challenges associated with multi-reads and repetitive regions in genomic studies.
Technical Challenges in Repetitive Region Analysis
Limitations of Short-Read Technologies
Short-read sequencing technologies face several specific challenges in repetitive regions:
- Mapping Ambiguity: Reads originating from repetitive elements often map equally well to multiple genomic locations, resulting in multi-mapped reads that complicate variant calling and genotyping [76]. This ambiguity leads to reduced sensitivity and specificity in variant detection.
- Inadequate Spanning Length: Short reads cannot span entire repetitive regions, making it impossible to resolve the exact structure and composition of STRs and other repeats [76]. For instance, a 150 bp read cannot characterize a 500 bp tandem repeat expansion.
- False Structural Variant Calls: The inability to unambiguously map reads across repetitive sequences results in high false discovery rates for structural variants, with studies reporting up to 85% false positives and sensitivity as low as 30-70% using short-read technologies [76].
Comparative Performance of Sequencing Technologies
Table 1: Sequencing Technology Comparison for Repetitive Region Analysis
| Technology | Read Length | Error Profile | STR Resolution | SV Detection Sensitivity | Best Application |
|---|---|---|---|---|---|
| Illumina (Short-read) | 75-300 bp | Low substitution errors (<0.1%) | Limited for long repeats | Low (30-70%) | SNP detection, targeted sequencing |
| PacBio SMRT (Long-read) | 10,000-25,000 bp | Random indels (∼5-15%) | Excellent for full-length STR sequencing | High (3-4x more SVs than short-read) | De novo assembly, complex SV detection |
| Oxford Nanopore (Long-read) | 10,000-30,000 bp | Higher indel rate (up to 15%) | Capable of spanning most STRs | High for large SVs | Real-time sequencing, methylation detection |
| Sanger Sequencing | 500-1000 bp | Very low error rate | Limited by fragment size | Limited | Validation of NGS findings |
Experimental Solutions and Protocols
Library Preparation for Repetitive Region Sequencing
Protocol: PCR-Free Library Preparation for Complex Genomic Regions
Objective: To minimize amplification bias in repetitive regions during library preparation [21].
Reagents and Equipment:
- Covaris LE220 focused-ultrasonicator or equivalent acoustic shearing system
- TruSeq DNA PCR-free HT sample prep kit (Illumina) or MGIEasy PCR-Free DNA Library Prep Set (MGI Tech)
- Agilent Bravo automated liquid handling system or equivalent
- Qubit dsDNA HS Assay Kit (Life Technologies) for quantification
- Fragment Analyzer (Advanced Analytical) or TapeStation system (Agilent) for size distribution analysis
Procedure:
- DNA Quality Assessment: Verify DNA integrity using pulse-field gel electrophoresis. High molecular weight DNA (>50 kb) is essential for long-read sequencing applications [21].
- DNA Fragmentation: Fragment genomic DNA using focused ultrasonication to a target size of 550 bp for short-read platforms. For long-read technologies, minimize fragmentation to preserve high molecular weight [21].
- Size Selection: Perform rigorous size selection using magnetic beads or gel electrophoresis to remove fragments <300 bp that may represent artifactual products [77].
- Adapter Ligation: Use unique dual indexes (e.g., IDT for Illumina TruSeq DNA Unique Dual indexes for 96 samples) to enable multiplexing while maintaining sample identity [21].
- Library QC: Quantify libraries using fluorescence-based methods (Qubit) and verify size distribution using fragment analyzers. For Illumina platforms, employ qMiSeq methods to determine relative library concentrations for pooling [21].
Sequencing Platform Selection and Optimization
Protocol: Optimizing Sequencing Parameters for Repetitive Regions
Objective: To maximize data quality and coverage uniformity across repetitive genomic regions.
Table 2: Platform-Specific Optimization Parameters
| Platform | Coverage Depth | Read Configuration | Quality Control Metrics | Special Considerations |
|---|---|---|---|---|
| Illumina NovaSeq | 30-50x for WGS | Paired-end 150 bp | % occupied >80%, Pass filter >75% | Use high diversity libraries to reduce index hopping |
| PacBio SMRT | 15-20x for WGS | Single-molecule continuous long reads | Read length N50 >20 kb, subread length | Circular consensus sequencing for error correction |
| Oxford Nanopore | 20-30x for WGS | Ultra-long read mode | Read length N50 >30 kb, pore integrity | DNA integrity critical for ultra-long reads |
Procedure:
- Coverage Planning: For short-read platforms, increase coverage depth to 50-100x in regions known to contain repeats to improve consensus accuracy [77].
- Read Configuration: Utilize paired-end sequencing with insert sizes optimized for the repetitive element of interest. Longer insert sizes (600-800 bp) can help span shorter repeats [77].
- Loading Optimization: For Illumina platforms, carefully titrate library loading concentration to achieve optimal cluster density. Monitor % occupied and pass filter values in real-time using Sequence Analysis Viewer [21].
- Quality Monitoring: Perform real-time quality control using FastQC to assess base quality distribution, duplication rates, and GC content. For long-read technologies, monitor read length distribution continuously [21].
Bioinformatics Strategies for Multi-Reads and Repetitive Regions
Alignment Algorithms for Ambiguous Regions
Protocol: Specialized Alignment of Repetitive Reads
Objective: To improve mapping accuracy for reads originating from repetitive regions.
Workflow Diagram: Repetitive Region Alignment Strategy
Tools and Parameters:
- Short-read Alignment: Use BWA-MEM or BWA-mem2 with disabled seed option (
-k 1000000) to prevent early termination of alignment in repetitive regions [21]. - Long-read Alignment: Employ Minimap2 with recommended presets for PacBio (
-x map-pb) or Oxford Nanopore (-x map-ont) data [76]. - Multi-mapper Handling: Retain all alignment positions for multi-mapped reads using the
-aflag in BWA, then employ probabilistic assignment methods based on paired-end information [77].
Specialized Variant Calling for Repetitive Regions
Protocol: STR and SV Detection Using Long-Read Data
Objective: To accurately call variants in repetitive regions using specialized tools.
Procedure:
- STR Genotyping: For short tandem repeat analysis, use specialized tools like RepeatHMM or HipSTR that incorporate specific error models for repetitive regions [76].
- Structural Variant Calling: Employ multiple complementary approaches:
- Read-depth Methods: Detect copy number variations from coverage irregularities
- Split-read Methods: Identify breakpoints from partially aligned reads
- Assembly-based Methods: Perform local assembly of discordantly mapped reads
- Variant Filtering: Apply context-specific filters to reduce false positives:
- Remove variants with extreme allele frequencies inconsistent with Mendelian inheritance
- Filter variants supported primarily by multi-mapped reads
- Exclude variants in known problematic regions (e.g., segmental duplications)
Workflow Diagram: Variant Calling in Repetitive Regions
Research Reagent Solutions
Table 3: Essential Reagents for Sequencing Complex Genomic Regions
| Reagent/Category | Specific Product Examples | Function in Workflow | Considerations for Repetitive Regions |
|---|---|---|---|
| DNA Extraction Kits | Autopure LS (Qiagen), GENE PREP STAR NA-480 (Kurabo) | High molecular weight DNA isolation | Preserve long fragments >50 kb for long-read sequencing [21] |
| Library Prep Kits | TruSeq DNA PCR-free HT (Illumina), MGIEasy PCR-Free Set (MGI) | Sequencing library construction | PCR-free protocols reduce amplification bias in repeats [21] |
| Fragmentation Systems | Covaris LE220 focused-ultrasonicator | DNA shearing to desired size | Controlled fragmentation preserves repeat integrity [21] |
| Size Selection Beads | AMPure XP (Beckman Coulter) | Fragment size selection | Remove artifactual short fragments that compete during sequencing [77] |
| Quality Control Tools | Qubit dsDNA HS Assay, Fragment Analyzer, TapeStation | Library quantification and sizing | Accurate quantification prevents overloading and improves coverage uniformity [21] |
| Automation Systems | Agilent Bravo, MGI SP-960 | Liquid handling automation | Improve reproducibility in library preparation [21] |
The accurate sequencing and analysis of repetitive genomic regions remains challenging but is increasingly feasible through integrated experimental and computational approaches. The strategic selection of sequencing technologies, combined with specialized library preparation methods and bioinformatics tools, enables researchers to overcome historical limitations in studying multi-reads and repetitive elements. As sequencing technologies continue to evolve, with improvements in both short-read and long-read platforms, along with enhanced algorithmic approaches incorporating artificial intelligence and machine learning, we anticipate further improvements in the resolution of complex genomic regions [78]. These advances will be particularly important for fully elucidating the role of repetitive elements in human health, disease, and evolutionary biology.
Next-generation sequencing (NGS) has revolutionized genomic research, enabling scientists to investigate DNA and RNA with unprecedented depth and precision. However, this powerful technology generates a monumental data deluge that presents substantial computational challenges. The evolution from Sanger sequencing to modern NGS platforms has reduced the cost of sequencing a human genome from nearly $3 billion during the Human Genome Project to a fraction of that cost today, while simultaneously increasing data output to staggering levels [28]. Modern production-scale sequencers can generate over 16 terabytes of data in a single run, creating immense pressures on bioinformatics infrastructure [28] [30]. This article examines the critical computational hurdles in NGS data management—storage, transfer, and analysis—and provides structured solutions for researchers and drug development professionals operating in data-intensive environments.
The core challenge stems from the fundamental nature of NGS technologies, which sequence millions to billions of DNA fragments in parallel [28]. A single human genome represents approximately 100 gigabytes of raw data, and global annual sequencing capacity has long since surpassed 13 quadrillion bases [79]. For research institutions and pharmaceutical companies engaged in large-scale genomic studies or population-scale initiatives like the Alliance for Genomic Discovery, which is analyzing hundreds of thousands of genomes, these data volumes present significant logistical challenges [30]. Effectively managing this data deluge requires sophisticated computational infrastructure, robust data management strategies, and efficient analytical workflows, which we will explore in detail throughout this application note.
Data Storage Architectures and Management Strategies
HPC Storage Hierarchy for NGS Data
High-Performance Computing (HPC) systems typically implement a tiered storage architecture to balance performance, capacity, and cost for NGS data management. Understanding this hierarchy is essential for optimizing storage strategies. Most HPC environments provide three primary storage locations, each with distinct purposes and characteristics [80]:
Home Directory: The home directory (typically /home/username/) serves as the initial landing point for users. This space is relatively small (often with 50-100GB quotas) and is designed for user profiles, scripts, configuration files, and important results rather than raw NGS data. Its key advantage is regular backup protection, making it suitable for storing critical analytical outputs and pipeline scripts [80].
Project/Work Directory: Project spaces (accessed via paths like /project/ or /work/) offer significantly larger quotas, often in the terabyte range. These shared spaces are ideal for processed data, analytical results, and collaborative projects. They may have some backup protection and are typically shared among project team members, facilitating collaboration while maintaining organized data governance [80].
Scratch Directory: Scratch space (often /scratch/) is designed specifically for temporary storage of massive datasets. This space provides very large capacity with high-speed I/O operations optimized for computational processing. However, it typically has no backup protection and may employ automatic file deletion policies. Scratch space is perfect for raw NGS data files and intermediate processing files during active analysis [80].
Table 1: HPC Storage Tiers for NGS Data Management
| Storage Tier | Typical Capacity | Primary Function | Backup Status | I/O Performance |
|---|---|---|---|---|
| Home Directory | 50-100 GB | User profiles, scripts, key results | Regularly backed up | Standard |
| Project/Work Directory | 1-10 TB+ | Processed data, results, collaboration | Some backup protection | Good |
| Scratch Directory | 100 TB+ | Raw NGS data, temporary files | No backup | High-speed |
Emerging Storage Solutions
As NGS data volumes continue to grow, researchers are increasingly turning to cloud-based storage solutions that offer scalability and flexibility. Cloud platforms enable dynamic allocation of storage resources based on project needs, implementing pay-as-you-go pricing models that can be more cost-effective than maintaining on-premises infrastructure for fluctuating workloads [79]. For large-scale collaborative initiatives, platforms like Globus provide robust data management capabilities, enabling secure and efficient data transfer across geographical and institutional boundaries while maintaining data integrity through comprehensive monitoring and verification mechanisms [79].
Data Transfer Protocols and Integrity Verification
Efficient Data Transfer Methods
Transferring massive NGS datasets between sequencing centers, computational resources, and collaborators requires specialized tools and protocols. Several transfer methods have been developed to address different scenarios:
Aspera Connect: IBM's Aspera platform uses the patented FASP (Fast and Secure Protocol) technology, a UDP-based transfer protocol that maximizes bandwidth utilization regardless of network conditions or distance. This technology is particularly valuable for transferring large NGS datasets from repositories like EBI's European Nucleotide Archive (ENA). A typical Aspera command for direct transfer to scratch space would be: ascp -QT -l 300m -P33001 era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR123/SRR12345678/SRR12345678_1.fastq.gz /scratch/your_username/ [80].
Globus Transfer: Globus provides research-focused data management services with high performance, secure, and reliable transfer capabilities. Its web interface and endpoint client simplify complex, large-scale data movements between institutions, with many research organizations maintaining dedicated Globus endpoints. The platform automates the task of moving files across administrative domains while providing superior data management capabilities for transferring big datasets from geographically distributed sequencing centers into cloud computing infrastructure [80] [79].
Direct Download Tools: For public data repositories, direct download tools remain essential. The SRA Toolkit is specifically designed for accessing data from NCBI's Sequence Read Archive, with commands like fasterq-dump SRR28119110 enabling direct downloads to HPC systems. Traditional tools like wget and curl also play important roles, with wget offering particular advantages for unstable connections due to its ability to automatically resume interrupted downloads [80].
Data Integrity Verification
Ensuring data integrity during transfer is critical for NGS workflows, as corrupted files can lead to invalid analytical results and erroneous scientific conclusions. MD5 checksums provide a cryptographic verification method that generates a unique 128-bit (16-byte) hash value, typically expressed as a 32-character hexadecimal number that serves as a file "fingerprint" [80].
The integrity verification process follows these essential steps:
Pre-Transfer Checksum Generation: Before transferring files, generate MD5 checksums for all NGS data files using the command: md5sum large_genome.fastq.gz > large_genome.fastq.gz.md5. For multiple files, use batch processing: md5sum *.fastq.gz > fastq_files.md5 [80].
Post-Transfer Verification: After transfer, verify file integrity by comparing with the original checksums: md5sum -c large_genome.fastq.gz.md5. The system will report "OK" for successful verification or "FAILED" for corrupted files, indicating the need for re-transfer [80].
Alternative Hashing Algorithms: While MD5 remains widely used in bioinformatics due to its speed and support, stronger algorithms like SHA-256 provide enhanced security against collisions: sha256sum large_genome.fastq.gz > large_genome.fastq.gz.sha256 [80].
Table 2: Data Integrity Verification Methods
| Method | Command Syntax | Strengths | Common Applications |
|---|---|---|---|
| MD5 | md5sum filename > filename.md5 |
Fast, widely supported | General NGS data verification |
| SHA-256 | sha256sum filename > filename.sha256 |
Stronger security guarantees | Sensitive or clinical data |
| Batch Verification | md5sum -c multiple_files.md5 |
Efficient for multiple files | Project-scale validation |
Analytical Workflows and Computational Platforms
Core NGS Analytical Steps
Clinical bioinformatics requires a standardized set of analyses to ensure accuracy, reproducibility, and comparability across samples and studies. Consensus recommendations from the Nordic Alliance for Clinical Genomics (NACG) outline essential analytical steps for production-scale NGS diagnostics [81] [82]:
Primary Data Processing: The initial phase converts raw sequencing output into analyzable sequence data through demultiplexing of pooled samples (BCL to FASTQ conversion) and alignment of sequencing reads to a reference genome (FASTQ to BAM conversion) [81] [82].
Variant Calling and Annotation: This critical phase identifies genomic variations through multiple specialized approaches: single nucleotide variants (SNVs) and small insertions/deletions (indels); copy number variants (CNVs) including deletions and duplications; structural variants (SVs) such as insertions, inversions, translocations, and complex rearrangements; short tandem repeats (STRs); loss of heterozygosity (LOH) regions indicating uniparental disomy; and mitochondrial SNVs/indels requiring specialized detection methods [81] [82].
Specialized Analytical Modules: Depending on research focus, additional analyses may include tumor mutational burden (TMB) quantification for immuno-oncology; microsatellite instability (MSI) analysis for DNA mismatch repair defects; homologous recombination deficiency (HRD) evaluation for PARP inhibitor response prediction; and polygenic risk scores (PRS) for complex disease predisposition assessment [81].
Workflow Management Systems
Bioinformatics workflow systems have become essential for managing the complexity of NGS data analysis. Galaxy provides a web-based platform that enables researchers to perform complex computational analyses without extensive programming expertise. Its accessible interface integrates numerous NGS tools while automating software installation and execution, making sophisticated analyses available to broader research teams [79].
The platform approach extends to cloud-based workflow platforms that combine Galaxy with provisioning tools like Globus Provision to create scalable, elastic environments for NGS analysis. These integrated solutions address key computational challenges through automated deployment of domain-specific tools, auto-scaling computational resources via schedulers like HTCondor, and high-performance data transfer capabilities [79].
For clinical applications, standardized practices are critical. Recommendations include using version-controlled code (git), containerized software environments (Docker, Singularity), and comprehensive testing protocols spanning unit, integration, system, and end-to-end validation. The hg38 genome build has emerged as the consensus reference for clinical alignment, providing improved representation of complex genomic regions compared to previous builds [81] [82].
Quality Assurance and Validation Frameworks
Quality Management Systems
Robust quality management systems (QMS) are essential for clinical and research NGS applications to ensure result accuracy and reproducibility. The Next-Generation Sequencing Quality Initiative (NGS QI) provides frameworks for laboratories implementing NGS, addressing personnel competency, equipment management, and process optimization [83]. These guidelines align with regulatory requirements such as CLIA (Clinical Laboratory Improvement Amendments) and ISO 15189 standards, creating a foundation for reliable genomic testing [83] [82].
Key components of an effective NGS QMS include standardized operating procedures (SOPs) for all analytical processes, regular competency assessments for bioinformatics personnel, systematic documentation practices, and change control procedures for pipeline updates. The NGS QI emphasizes the importance of validation tools and resources that help laboratories maintain compliance with evolving regulatory standards while accommodating rapid technological advances in sequencing platforms and analytical methods [83].
Validation Methods for Bioinformatics Pipelines
Rigorous validation is critical for ensuring the accuracy and reliability of NGS bioinformatics pipelines. The NACG recommendations specify multiple validation approaches that should be implemented concurrently [81] [82]:
Reference Materials Validation: Using standard truth sets such as Genome in a Bottle (GIAB) for germline variant calling and SEQC2 for somatic variant calling provides benchmark data with well-characterized variants. These resources enable objective assessment of pipeline performance across different variant types and genomic contexts [81] [82].
Clinical Sample Recall Testing: Supplementing reference materials with recall testing of real human samples previously characterized by validated methods provides critical assessment of clinical performance. This approach evaluates pipeline functionality with diverse sample types and quality levels encountered in routine practice [81] [82].
Sample Identity Verification: Implementing sample fingerprinting through genetically inferred identification markers (sex, ancestry, relatedness) and checks for sample contamination ensures sample integrity throughout the analytical process. Data integrity verification through file hashing (MD5, SHA-256) confirms that files have not been corrupted during processing or transfer [81] [82].
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Essential Research Reagents and Computational Solutions for NGS Bioinformatics
| Tool/Category | Specific Examples | Primary Function | Application Context |
|---|---|---|---|
| Sequencing Platforms | Illumina, PacBio, Oxford Nanopore | Generate raw sequencing data | Varies by read length, throughput, and error profile requirements [84] [28] |
| Data Transfer Tools | Aspera, Globus Transfer, SRA Toolkit | High-speed, reliable data movement | Transferring large NGS datasets between institutions and repositories [80] [79] |
| Workflow Management | Galaxy, Nextflow, Snakemake | Pipeline automation and reproducibility | Streamlining analysis and ensuring consistent results across projects [79] |
| Variant Callers | Multiple tools for SNV, CNV, SV detection | Identifying genomic variations | Comprehensive variant detection using complementary algorithmic approaches [81] [82] |
| Validation Resources | GIAB, SEQC2, in-house datasets | Pipeline performance assessment | Benchmarking analytical accuracy using reference materials [81] [82] |
| Containerization | Docker, Singularity, Conda | Software environment management | Ensuring reproducibility and simplifying dependency management [81] [82] |
| Quality Control Tools | FastQC, MultiQC, Samtools | Data quality assessment | Monitoring sequencing quality and analytical intermediate steps [81] |
Future Directions and Emerging Solutions
The NGS informatics landscape continues to evolve rapidly, with several emerging trends shaping future solutions. Multiomic integration represents a fundamental shift, combining genomic, epigenetic, transcriptomic, and proteomic data from the same sample to provide comprehensive biological insights [30]. This approach requires even more sophisticated computational infrastructure and analytical methods capable of correlating information across molecular domains.
Artificial intelligence and machine learning are playing increasingly important roles in NGS data analysis, enabling pattern recognition in high-dimensional datasets that exceeds traditional algorithmic approaches [30]. AI models require large-scale, accurate training data, driving demand for standardized, high-quality datasets and reproducible preprocessing workflows.
Spatial biology and in situ sequencing are emerging frontiers, enabling direct sequencing of cells within their native tissue context [30]. These technologies generate complex datasets incorporating spatial coordinates alongside sequence information, creating new computational challenges for data integration, visualization, and analysis.
The decentralization of sequencing applications is moving NGS closer to point-of-care settings, necessitating simplified bioinformatics solutions that can be deployed by non-specialists while maintaining rigorous quality standards [30]. Cloud-based platforms and automated analytical systems will be essential for making sophisticated genomic analyses accessible across diverse healthcare and research settings.
As these technologies mature, the bioinformatics community must continue developing standardized practices, shared resources, and scalable computational infrastructure to ensure that the full potential of NGS can be realized across basic research, drug development, and clinical applications.
Next-generation sequencing (NGS) has revolutionized genomic research and clinical diagnostics by providing unprecedented capacity to analyze DNA and RNA at scale. The core principle of massive parallelism enables the simultaneous sequencing of millions of DNA fragments, dramatically reducing cost and time compared to traditional methods [85]. However, this high-throughput capability generates enormous datasets where distinguishing true biological variants from technical artifacts becomes a critical challenge. Data integrity in this context encompasses the complete NGS workflow, from nucleic acid extraction through final variant interpretation, ensuring that results accurately represent the sample's biological reality rather than technical noise.
For researchers and drug development professionals, maintaining data integrity is paramount for generating reproducible, clinically actionable results. Technical noise can arise from multiple sources, including library preparation artifacts, sequencing errors, alignment inaccuracies, and bioinformatic processing limitations. This application note provides detailed protocols and frameworks for identifying, quantifying, and mitigating these sources of error, with particular emphasis on variant interpretation in both germline and somatic contexts. By implementing rigorous quality control measures and validated bioinformatic pipelines, researchers can significantly enhance the reliability of their genomic findings and ensure compliance with evolving regulatory standards for precision medicine applications [86].
Origins of Technical Artifacts
Technical noise in NGS workflows originates from multiple procedural stages, each introducing distinct artifacts that can compromise variant interpretation. Understanding these sources is essential for developing effective filtering strategies.
During library preparation, artifacts may arise from PCR amplification biases, including duplicate reads and uneven coverage, especially in GC-rich or poor regions [85]. The quality of starting material significantly impacts downstream results; degraded RNA or cross-contaminated samples introduce substantial noise [87]. Adapter contamination occurs when DNA fragments are shorter than read length, causing sequencing of adapter sequences rather than biological material [87].
Sequencing instruments generate systematic errors dependent on the technology platform. Illumina systems exhibit increased error rates toward read ends and specific sequence contexts, while Oxford Nanopore technologies show higher indel rates in homopolymer regions [88]. These platform-specific error profiles must be accounted for in noise reduction strategies.
Bioinformatic processing introduces additional artifacts through alignment inaccuracies in complex genomic regions, improper base calling, and reference genome biases [89]. Batch effects between sequencing runs and sample indexing errors further contribute to technical variability that can mimic true biological signals.
Comprehensive Quality Control Metrics
Rigorous quality assessment throughout the NGS workflow is essential for identifying technical noise sources. The table below summarizes critical quality metrics and their interpretation:
Table 1: Essential Quality Control Metrics for NGS Data Integrity
| Analysis Stage | Metric | Target Value | Interpretation |
|---|---|---|---|
| Nucleic Acid Quality | A260/A280 Ratio (DNA) | ~1.8 | Lower values indicate protein contamination [87] |
| A260/A280 Ratio (RNA) | ~2.0 | Deviation suggests degradation or contamination [87] | |
| RNA Integrity Number (RIN) | ≥8 for RNA-seq | Values <7 indicate significant degradation [87] | |
| Sequencing Quality | Q Score | ≥30 | 99.9% base call accuracy; Q30 is standard for most applications [85] [87] |
| Cluster Passing Filter (%) | >80% | Lower values indicate issues with cluster density or purity [87] | |
| Phasing/Prephasing (%) | <0.5% | Measures signal loss per cycle in Illumina platforms [87] | |
| Alignment Quality | Mapping Rate | >90% | Low rates suggest contamination or poor library quality [81] |
| Duplication Rate | Variable by application | High rates indicate PCR bias or insufficient input [87] | |
| Insert Size Distribution | Matches library prep | Deviations suggest fragmentation issues [88] |
Systematic monitoring of these metrics enables early detection of technical issues before they compromise variant calling. Laboratories should establish quality thresholds based on their specific applications and document all quality assessments for regulatory compliance and reproducibility [86].
Diagram 1: NGS workflow with technical noise sources (red) and QC checkpoints (blue). This illustrates critical control points where quality assessment can identify and mitigate technical artifacts.
Bioinformatics Protocols for Noise Filtering
Pre-processing and Quality Control Protocol
Effective noise filtering begins with rigorous pre-processing of raw sequencing data. This protocol establishes a standardized approach for quality assessment and data cleaning prior to variant calling.
Protocol: Raw Read Quality Assessment and Trimming
Quality Metric Extraction
- Process FASTQ files through FastQC to generate comprehensive quality reports [87]
- Examine per-base sequence quality to identify quality decay at read ends
- Assess adapter contamination using dedicated adapter content modules
- Analyze sequence duplication levels to identify potential PCR biases
- Review per-sequence quality scores to identify outlier reads
Adapter Trimming and Quality Filtering
- Use CutAdapt or Trimmomatic with platform-specific adapter sequences
- Set quality threshold to Q20 (99% accuracy) for base trimming
- Remove reads with length <20 bases after trimming
- For paired-end data, ensure maintained pair synchronization
- Command example:
Post-trimming Quality Verification
- Re-run FastQC on trimmed FASTQ files
- Verify improvement in per-base quality scores
- Confirm reduction in adapter content and duplication rates
- Document all trimming parameters for reproducibility
This protocol typically processes 100 million reads in approximately 2-4 hours using standard computational resources. Post-trimming verification is critical to ensure that filtering steps have improved data quality without introducing additional biases [87].
Variant Calling and Filtering Protocol
Accurate variant calling requires sophisticated approaches to distinguish true biological variants from technical artifacts. This protocol leverages both traditional and AI-based methods for optimal performance.
Protocol: High-Confidence Variant Detection
Alignment-Based Artifact Mitigation
- Perform duplicate marking to remove PCR artifacts
- Apply base quality score recalibration (BQSR) to correct systematic errors
- Use local realignment around indels to minimize alignment artifacts
- For tumor-normal pairs, verify sample relatedness through fingerprinting
Variant Calling Implementation
- For germline variants: Utilize DeepVariant or GATK HaplotypeCaller
- For somatic variants: Implement paired tumor-normal analysis with Mutect2
- For complex regions: Supplement with multiple callers (Strelka2, VarDict)
- Command example for DeepVariant:
Post-calling Filtering Strategy
- Apply variant quality score recalibration (VQSR) using truth sets
- Filter by depth (minimum 10x for targeted, 30x for WGS)
- Filter by allele frequency in population databases (gnomAD)
- Remove variants with extreme strand bias or position effects
- Implement panel-of-normals filtering for recurrent technical artifacts
Table 2: Comparison of AI-Based Variant Calling Tools
| Tool | Methodology | Strengths | Limitations | Optimal Use Case |
|---|---|---|---|---|
| DeepVariant [89] | Deep learning via CNN on read pileup images | High accuracy, automatic filtering | High computational cost | Large-scale germline studies (e.g., UK Biobank) |
| DeepTrio [89] | Deep learning with family context | Improved de novo mutation detection | Requires trio data | Family-based studies, rare disease |
| DNAscope [89] | Machine learning enhancement of GATK | Computational efficiency, high SNP/InDel accuracy | ML-based (not deep learning) | Production environments with resource constraints |
| Clair3 [89] | Deep learning for long/short reads | Fast processing, good low-coverage performance | Less established community | Long-read technologies, rapid turnaround |
| GATK Mutect2 [90] | Statistical model for somatic calling | Excellent tumor-normal discrimination | Requires matched normal | Somatic variant detection in cancer |
The variant calling protocol typically requires 4-8 hours for whole exome data and 24-48 hours for whole genome sequencing, depending on computational resources. AI-based tools generally provide superior accuracy but require GPU acceleration for optimal performance [89].
Variant Interpretation and Clinical Translation
Annotation and Filtering Framework
After variant calling, comprehensive annotation and filtering are essential for biological interpretation. This framework enables prioritization of clinically relevant variants while excluding technical artifacts and population polymorphisms.
Variant Annotation Protocol
Functional Impact Prediction
- Annotate with Ensembl VEP or ANNOVAR for consequence prediction
- Include CADD scores for deleteriousness assessment
- Integrate SpliceAI for splice site effect prediction
- Add protein domain annotations from Pfam and InterPro
Population Frequency Filtering
- Cross-reference with gnomAD for allele frequencies
- Apply application-specific frequency thresholds (e.g., <0.1% for rare disease)
- Consider subpopulation-specific frequencies for diverse cohorts
- Filter common polymorphisms unless relevant to disease
Clinical Database Integration
- Annotate with ClinVar for clinical significance
- For cancer variants, include COSMIC and CIViC annotations
- Integrate drug-gene interactions from PharmGKB
- Add ACMG/AMP classification codes for clinical reporting
Technical Artifact Filtering Criteria
- Mapping quality: Remove variants with MQ < 40
- Strand bias: Filter variants with FS > 60 for SNPs or >200 for indels
- Read position: Exclude variants clustered at read ends
- Allelic depth: Require minimum AD for heterozygotes based on total depth
- Base quality: Filter variants with QUAL < 30 or low QD scores
This annotation framework typically adds 1-2 hours to the analysis pipeline but dramatically reduces manual curation time by automatically prioritizing potentially relevant variants [90].
Validation and Regulatory Compliance
For clinical and drug development applications, rigorous validation and regulatory compliance are essential components of the variant interpretation workflow.
Protocol: Assay Validation and Quality Assurance
Benchmarking Against Truth Sets
- Validate germline pipelines against GIAB reference materials
- For somatic variants, use SEQC2 reference datasets
- Supplement with in-house samples previously characterized by orthogonal methods
- Establish sensitivity and specificity targets (>99% for high-penetrance variants)
Implementation of Clinical Grade Bioinformatics
- Adopt hg38 genome build as standard [81]
- Implement containerized software environments for reproducibility
- Utilize version control for all pipelines and parameters
- Establish comprehensive documentation for audit trails
Regulatory Compliance Measures
Diagram 2: Variant interpretation workflow showing sequential annotation and filtering steps that transform raw variant calls into clinically actionable information.
Essential Research Reagents and Computational Tools
Successful implementation of noise filtering and variant interpretation requires carefully selected reagents and computational tools. The following table details essential resources for establishing robust NGS analysis workflows.
Table 3: Essential Research Reagents and Computational Tools for NGS Data Integrity
| Category | Specific Product/Tool | Function | Key Features |
|---|---|---|---|
| Library Prep Kits | Illumina DNA Prep | Library construction | Flexible input, robust performance |
| KAPA HyperPrep | PCR-free library prep | Low duplication rates, high complexity | |
| NEBNext Ultra II | RNA library preparation | Strand specificity, rRNA depletion | |
| Quality Control | Agilent TapeStation | Nucleic acid QC | RINe for RNA, sample integrity |
| Thermo Fisher NanoDrop | Concentration/purity | Rapid assessment, minimal sample use | |
| Qubit Fluorometer | Accurate quantification | DNA/RNA specificity, broad dynamic range | |
| Bioinformatics Tools | FastQC | Raw read QC | Comprehensive metrics, visual reports |
| Trimmomatic/CutAdapt | Read trimming | Adapter removal, quality filtering | |
| BWA-MEM2 | Sequence alignment | Speed, accuracy, GPU acceleration | |
| DeepVariant | Variant calling | AI-based, high accuracy [89] | |
| GATK Mutect2 | Somatic calling | Tumor-normal analysis, precision [90] | |
| Ensembl VEP | Variant annotation | Comprehensive, regularly updated | |
| Validation Resources | GIAB Reference Materials | Benchmarking | Characterized variants, gold standard |
| SeraCare Reference Standards | Somatic validation | Tumor-normal pairs, defined variants | |
| EQA Programs (EMQN) | Proficiency testing | Inter-laboratory comparison |
Laboratories should select reagents and tools based on their specific applications, throughput requirements, and regulatory needs. For drug development applications, emphasis should be placed on tools with established regulatory compliance profiles and comprehensive documentation [86].
Ensuring data integrity in NGS-based variant interpretation requires a systematic, multi-layered approach to identify and mitigate technical noise. This application note has detailed protocols for quality control, variant calling, and interpretation that collectively address the major sources of technical artifacts throughout the NGS workflow. By implementing these standardized methodologies, researchers and drug development professionals can significantly enhance the reliability of their genomic findings.
The integration of AI-based tools represents a substantial advancement in variant calling accuracy, though traditional methods remain relevant in well-characterized applications. As regulatory frameworks for NGS in clinical trials continue to evolve, establishing robust validation protocols and quality management systems will be increasingly important for generating compliant, reproducible data [86]. Future developments in single-molecule sequencing, long-read technologies, and integrated multi-omics approaches will present new challenges and opportunities for technical noise management, requiring ongoing refinement of these protocols.
By adopting the comprehensive framework presented here—encompassing wet-lab procedures, computational methods, and quality systems—research organizations can position themselves at the forefront of precision medicine while maintaining the highest standards of data integrity and analytical validity.
Ensuring Accuracy: NGS Validation, Benchmarking, and Clinical Integration
The integration of next-generation sequencing (NGS) into clinical oncology has fundamentally transformed cancer diagnosis and treatment, enabling molecularly driven cancer care [92]. Analytical validation ensures that NGS-based oncology tests perform reliably, accurately, and consistently in detecting clinically relevant genomic alterations. This process is fundamental to precision medicine, which utilizes information about an individual's genes, proteins, and environment to prevent, diagnose, and treat disease [93]. For researchers, scientists, and drug development professionals, adherence to established validation guidelines is critical for generating clinically actionable data, supporting regulatory submissions, and advancing therapeutic development.
The Association for Molecular Pathology (AMP) and the College of American Pathologists (CAP) have established consensus recommendations that provide a framework for validating NGS-based oncology panels. These guidelines aim to ensure that laboratories can confidently report variants with known performance characteristics regarding accuracy, precision, sensitivity, and specificity. This document outlines the core principles of these recommendations, provides detailed experimental protocols for validation studies, and demonstrates their application through case studies in accordance with the broader thesis that integrated DNA and RNA analysis provides a more comprehensive understanding of cancer biology.
Core Principles of AMP/CAP Validation Guidelines
The AMP/CAP guidelines provide a structured approach for establishing the analytical performance of NGS-based oncology panels. The core principles revolve around defining key performance metrics and ensuring the test reliably detects various variant types across the entire intended genomic scope.
Key Analytical Performance Metrics
Validation must establish and document a panel's performance across several fundamental metrics, as exemplified by recent large-scale validation studies [94] [95]. The target thresholds for these metrics can vary based on the test's intended use but must be rigorously demonstrated.
Table 1: Key Analytical Performance Metrics for NGS Oncology Panels
| Performance Metric | Definition | Typical Target Threshold | Validation Approach |
|---|---|---|---|
| Accuracy | Concordance with a reference method or ground truth | ≥99% for SNVs/Indels [94] | Comparison to orthogonal methods (e.g., Sanger sequencing) or reference materials |
| Precision | Reproducibility of results across replicates and runs | 100% for inter-run, intra-run, and inter-operator [96] | Repeated testing of the same samples under varying conditions |
| Analytical Sensitivity | Ability to detect true positives (low variant allele frequency) | >95% for SNVs at 5% VAF [94] [95] | Dilution series with cell lines or synthetic controls |
| Analytical Specificity | Ability to avoid false positives | >99% for SNVs/Indels [94] [95] | Testing known negative samples and calculating false positive rate |
| Reportable Range | Spectrum of variants the test can detect | All targeted genes and variant types | In silico analysis and wet-bench testing for coverage |
| Limit of Detection (LoD) | Lowest VAF reliably detected | VAF of 5% or lower, depending on application [94] | Probit analysis on serial dilutions to establish 95% detection threshold |
Scope of Validation: Covering Variant Types
A comprehensive validation must demonstrate performance across all variant classes the test is designed to report. The AMP/CAP guidelines emphasize that the following variant types require individual assessment:
- Single Nucleotide Variants (SNVs) and Insertions/Deletions (Indels): The validation must establish sensitivity and specificity across different genomic contexts and variant allele frequencies (VAFs). For example, one validated assay demonstrated >99% sensitivity and specificity for 3,042 validated SNVs [94] [95].
- Copy Number Variations (CNVs): Performance must be validated for detecting amplifications and deletions. This often requires testing a separate set of samples, as demonstrated by a study that validated 47,466 CNV calls [95].
- Gene Fusions/Translocations: For panels including RNA sequencing, the detection of fusion transcripts and rearrangements must be validated. Integrated DNA/RNA sequencing has been shown to improve fusion detection compared to DNA-only approaches [94] [97].
- Other Biomarkers: If the panel reports complex biomarkers like Tumor Mutational Burden (TMB), Microsatellite Instability (MSI), or Loss of Heterozygosity (LOH), each requires separate validation using samples with established status.
Experimental Design and Protocols for Validation
A robust validation study follows a multi-step process that utilizes well-characterized samples to challenge the NGS assay across its intended scope. The following protocols provide a detailed framework for conducting these experiments.
Sample Selection and Characterization
The foundation of a successful validation is the use of appropriate samples that represent the real-world specimens the laboratory will encounter.
- Sample Types: The validation should include the same sample types used in clinical testing, typically Formalin-Fixed, Paraffin-Embedded (FFPE) tissue, but also fresh frozen tissue, cytology specimens, and others. CAP guidelines note that IHC assays (and by extension, molecular assays) on cytology specimens fixed differently from initial validation tissues require separate validation with a minimum of 10 positive and 10 negative cases [98].
- Reference Materials: Use a combination of:
- Commercial Reference Standards: Cell lines or synthetic DNA mixes with known variants across a range of VAFs and variant types.
- Characterized Clinical Samples: Previously tested clinical samples with well-documented variants via orthogonal methods (e.g., Sanger sequencing, FISH, PCR).
- Variant Spectrum: The selected samples must encompass a wide range of variants (SNVs, Indels, CNVs, fusions) in the genes covered by the panel. One validation study used a custom reference sample containing 3,042 SNVs to ensure comprehensive coverage [94].
- Sample Purity: Include samples with varying tumor purity (e.g., down to 5-10%) to establish the assay's limit of detection in suboptimal specimens.
Wet-Lab Protocol: Library Preparation and Sequencing
This protocol covers the key steps in the NGS workflow, from nucleic acid extraction to sequencing, which must be standardized and controlled during validation.
Materials and Reagents
- Nucleic Acid Extraction Kits: e.g., AllPrep DNA/RNA FFPE Kit (Qiagen) for simultaneous DNA/RNA extraction from FFPE tissue [94].
- Library Preparation Kits: e.g., SureSelect XTHS2 DNA and RNA kits (Agilent) for exome capture, or targeted amplicon-based kits [94].
- Exome/Target Capture Probes: e.g., SureSelect Human All Exon V7 (Agilent) for whole-exome sequencing [94].
- Quality Control Instruments: Qubit fluorometer (Thermo Fisher) for quantitation, TapeStation (Agilent) for integrity analysis [94].
- Sequencing Platform: Illumina sequencers (e.g., NovaSeq 6000) are most common for clinical oncology panels [94].
Procedure
- Nucleic Acid Extraction and QC: Extract DNA and RNA from all validation samples. Quantify using a fluorometric method (Qubit) and assess quality. For FFPE-derived DNA/RNA, measure DNA integrity number (DIN) and RNA integrity number (RIN). Only proceed with samples passing pre-defined QC thresholds (e.g., RIN > 5 for RNA) [94].
- Library Preparation: For each sample, prepare sequencing libraries from 10-200 ng of input DNA or RNA according to the manufacturer's protocol. This includes steps of fragmentation, adapter ligation, and indexing. For integrated DNA/RNA assays, this process is performed in parallel [94] [97].
- Target Enrichment: For hybrid capture-based panels, perform hybridization with the target-specific probe set, followed by washing and amplification of captured libraries.
- Library QC and Pooling: Quantify the final libraries and assess average fragment size (e.g., via TapeStation). Pool libraries at equimolar concentrations for multiplexed sequencing.
- Sequencing: Load the pooled library onto the sequencer (e.g., Illumina NovaSeq 6000) to achieve a minimum mean coverage depth (e.g., 200x for tumor DNA, 100x for normal DNA, and higher for RNA-seq). Monitor sequencing run metrics, including % bases ≥Q30 and cluster density [94].
The following workflow diagram illustrates the key steps in the analytical validation process for an integrated DNA and RNA NGS assay:
Bioinformatics Protocol: Analysis and Variant Calling
The bioinformatics pipeline is a critical component of the NGS assay and must be validated with the same rigor as the wet-lab process.
Software and Tools
- Alignment: BWA MEM for DNA-seq; STAR for RNA-seq [94].
- Variant Calling: Strelka2 for somatic SNVs/Indels; Manta for structural variants; Pisces for RNA-seq variants [94].
- Quality Control: FastQC for raw read quality; Picard tools for duplicate marking; mosdepth for coverage metrics; RSeQC for RNA-seq QC [94].
Procedure
- Data Demultiplexing and QC: Convert base calls to FASTQ files and assess initial quality with FastQC.
- Alignment to Reference: Map sequencing reads to the human reference genome (hg38). For RNA-seq, also perform transcript-level quantification with tools like Kallisto [94].
- Post-Alignment Processing and QC: For DNA-seq, mark PCR duplicates and calculate coverage metrics (mean coverage, uniformity, on-target rate). For RNA-seq, assess metrics like ribosomal RNA content and strand specificity.
- Variant Calling: Call somatic SNVs/Indels from tumor-normal pairs. Call gene fusions from RNA-seq data. Call CNVs from DNA-seq data. For integrated assays, variants may also be called from RNA-seq to recover alterations missed by DNA-only analysis [94] [95].
- Variant Filtration and Annotation: Apply filters for depth, VAF, and quality scores (e.g., using a complex filter based on Strelka2 QSS and EVS scores) [94]. Annotate variants using relevant databases (e.g., COSMIC, ClinVar, gnomAD).
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation and validation of an NGS oncology panel require a suite of reliable reagents and computational tools. The following table details key solutions used in the featured validation studies.
Table 2: Key Research Reagent Solutions for NGS Assay Validation
| Category | Item | Specific Example(s) | Function in Validation |
|---|---|---|---|
| Nucleic Acid Extraction | DNA/RNA FFPE Kits | AllPrep DNA/RNA FFPE Kit (Qiagen) [94] | Simultaneous purification of high-quality DNA and RNA from challenging FFPE samples. |
| Library Preparation | Library Prep Kits | TruSeq stranded mRNA kit (Illumina); SureSelect XTHS2 (Agilent) [94] | Creates sequencing-ready libraries from input DNA or RNA with sample-specific barcodes. |
| Target Enrichment | Exome/Target Capture Probes | SureSelect Human All Exon V7 (Agilent) [94] | Enriches for the protein-coding exome or a targeted gene panel, enabling focused sequencing. |
| Sequencing | Sequencing Platform | NovaSeq 6000 (Illumina) [94] | High-throughput sequencing to generate the raw data (FASTQ files) for analysis. |
| Reference Materials | Characterized Cell Lines & Controls | Custom reference samples with known SNVs/CNVs [94] [95] | Provides ground truth for establishing accuracy, sensitivity, specificity, and LoD. |
Case Study: Validation of an Integrated DNA and RNA Assay
A landmark study validated an integrated RNA and DNA whole-exome assay across 2,230 clinical tumor samples, providing a practical model for implementing AMP/CAP principles [94] [95] [97]. The validation followed a three-step framework:
- Analytical Validation with Reference Standards: The assay was tested using custom reference samples containing 3,042 known SNVs and 47,466 CNVs, establishing exome-wide sensitivity and specificity. This step rigorously defined the assay's baseline performance metrics [95].
- Orthogonal Confirmation with Patient Samples: Results from the novel integrated assay were compared to those from established, validated methods on real patient samples. This confirmed that the assay performed reliably in a clinical context.
- Assessment of Clinical Utility: The assay was applied to real-world cases, demonstrating that the combined RNA and DNA sequencing uncovered clinically actionable alterations in 98% of cases [97]. It improved the detection of gene fusions and allowed for the recovery of variants missed by DNA-only testing, directly impacting potential treatment strategies [94] [95].
This case highlights a key advancement in the field: moving beyond DNA-only analysis. The schematic below illustrates how integrating DNA and RNA data streams provides a more complete molecular portrait of a tumor, which is the cornerstone of modern precision oncology.
Adherence to the AMP/CAP analytical validation guidelines is not merely a regulatory hurdle but a fundamental scientific practice that ensures the reliability and clinical utility of NGS-based oncology panels. As demonstrated by the BostonGene study and others, a rigorous, multi-step validation process—encompassing analytical metrics, orthogonal confirmation, and clinical assessment—is achievable and essential for generating trustworthy data [94] [97].
The future of analytical validation will evolve with the technology. The field is moving toward multimodal assays that integrate DNA and RNA sequencing as a standard of care, providing a more comprehensive view of the tumor genome and transcriptome [92] [99]. Furthermore, the increasing use of liquid biopsies for non-invasive genomic profiling and the analysis of complex biomarkers like the tumor microenvironment using RNA-seq data will require updated validation frameworks [92]. Finally, the integration of artificial intelligence into bioinformatics pipelines promises to enhance variant interpretation and predictive biomarker discovery, but will also necessitate novel validation approaches to ensure these complex algorithms are transparent and robust [93] [97]. By adhering to the core principles outlined in this document, researchers and drug developers can confidently leverage NGS technologies to advance precision oncology and improve patient outcomes.
Next-generation sequencing (NGS) has revolutionized molecular diagnostics by enabling comprehensive genomic profiling, yet its performance relative to established gold-standard methods continues to be rigorously evaluated across various cancer types and genomic alterations. This assessment is particularly critical for therapeutically relevant mutations in the BRAF, EGFR, and KRAS genes, where detection accuracy directly impacts treatment decisions and patient outcomes [100] [101]. The transition from single-gene testing to multigene panels represents a paradigm shift in oncological testing, offering a more complete molecular portrait while conserving precious tissue samples [102]. Understanding the comparative analytical performance of these methodologies provides valuable insights for researchers and clinicians navigating the complex landscape of molecular diagnostics in the era of precision medicine.
Performance Comparison Across Key Mutations
BRAF V600E Detection in Thyroid Cancer
In papillary thyroid carcinoma (PTC), the BRAF V600E mutation serves as an important prognostic marker, necessitating highly accurate detection methods. A recent comparative study evaluating droplet digital PCR (ddPCR), immunohistochemistry (IHC), and direct Sanger sequencing revealed significant differences in detection sensitivity [100].
Table 1: Performance Comparison for BRAF V600E Detection in PTC
| Method | Detection Rate | Sensitivity Relative to SS | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Sanger Sequencing (SS) | 72.9% (35/48) | Reference | Well-established, broad mutation discovery | Lower sensitivity, requires high tumor purity |
| Immunohistochemistry (IHC) | 89.6% (43/48) | Significantly higher (P=0.001) | Cost-effective, rapid, visual localization | Subjective interpretation, antibody-dependent |
| Droplet Digital PCR (ddPCR) | 83.3% (40/48) | Significantly higher (P<0.001) | Absolute quantification, high sensitivity | Specialized equipment, limited multiplexing |
| Concordance All Methods | 83.3% (40/48) | N/A | High reliability when methods agree | Discordant cases require additional verification |
This study demonstrated that among discordant cases (all SS-negative), the majority exhibited low mutant allele frequencies (mean 14.5%) detectable by more sensitive methods, highlighting the critical importance of sensitivity thresholds in mutation detection [100].
EGFR Mutation Detection in NSCLC
In non-small cell lung cancer (NSCLC), epidermal growth factor receptor (EGFR) mutation status determines eligibility for tyrosine kinase inhibitor (TKI) therapies. While tissue biopsy remains the gold standard, liquid biopsy approaches have gained prominence for their non-invasive nature and ability to monitor resistance mutations such as T790M [103].
Table 2: EGFR Mutation Detection: Tissue vs. Liquid Biopsy
| Parameter | Tissue-Based NGS | Liquid Biopsy NGS | Combined Approach |
|---|---|---|---|
| T790M Detection Rate | 34.1% | 18.6% | 56.7% |
| Turnaround Time | 10-14 days | ~3 days | Varies |
| Advantages | Comprehensive profiling, histologic correlation | Minimally invasive, real-time monitoring | Maximized detection sensitivity |
| Limitations | Invasiveness, tumor accessibility | Lower sensitivity for some mutations | Increased cost, complex interpretation |
The incremental value of a plasma-first NGS approach in newly diagnosed advanced NSCLC has been reported at approximately 21%, supporting its role as a complementary tool to tissue-based testing [101]. Hybrid capture-based NGS assays demonstrated superior performance for detecting gene fusions compared to amplicon-based approaches [101].
KRAS Mutation Detection Across Methodologies
KRAS mutations, particularly the G12C variant prevalent in NSCLC and colorectal cancer, have emerged as actionable targets, underscoring the need for accurate detection [104]. The performance of different testing methodologies varies significantly based on the clinical context and sample type.
Table 3: KRAS Mutation Detection Performance Across Platforms
| Methodology | Application Context | Key Performance Metrics | Considerations |
|---|---|---|---|
| Large NGS Panels (e.g., TSO500) | Clonality assessment in NSCLC | 98-99% classification rate, 1% misclassification | Detects more mutations than IASLC recommendations |
| Small Oncogene Panels (12-gene) | Clonality assessment in NSCLC | 30% (LUAD) - 74% (LUSC) inconclusive rates | Limited by lack of detected mutations |
| Exosomal DNA Analysis (Intplex qPCR) | Early-stage CRC | 85% detection rate, 0.01% sensitivity | Superior to ctDNA in early-stage disease |
| NSCLC-Specific Panel (27-gene) | Clonality assessment | Reduces inconclusive to 5% | Addition of tumor suppressor genes improves performance |
Notably, exosomal DNA analysis for KRAS mutations in early-stage colorectal cancer demonstrated a median mutant allele frequency of 1.18% (range: 0.01-63.33%) with high concordance to tissue testing, suggesting its utility in early detection scenarios [105].
Experimental Protocols
NGS-Based Mutation Detection for Solid Tumors
Principle: Targeted NGS using multigene panels enables comprehensive genomic profiling of solid tumors by simultaneously assessing multiple genomic regions of interest through hybrid capture or amplicon-based enrichment [102].
Procedure:
- Nucleic Acid Extraction: Extract DNA from formalin-fixed paraffin-embedded (FFPE) tissue samples using validated kits (e.g., Maxwell RSC FFPE Plus DNA Kit). Quantify DNA using fluorometric methods and assess quality via fragment analysis.
- Library Preparation: For hybridization-capture based approaches (e.g., TTSH-oncopanel), fragment DNA to 200-300bp and ligate with sequencing adapters. Perform hybrid capture using biotinylated probes targeting 61 cancer-associated genes. Amplify captured libraries via PCR [102].
- Sequencing: Load libraries onto benchtop sequencers (e.g., MGI DNBSEQ-G50RS) following manufacturer's instructions. Target median coverage of 500-1000x with >98% of regions covered at ≥100x.
- Bioinformatic Analysis: Align sequencing reads to reference genome (GRCh37/38). Perform variant calling using specialized software (e.g., Sophia DDM). Filter variants based on quality metrics, read depth (>100x), and allele frequency (>5% for FFPE; >1% for liquid biopsy).
- Variant Interpretation: Annotate variants using clinical databases (ClinVar, COSMIC). Classify according to established guidelines (AMP/ASCO/CAP). Report clinically actionable mutations with therapeutic implications.
Quality Control:
- Minimum DNA input: ≥50ng
- Sensitivity threshold: 3% variant allele frequency
- Include positive and negative controls in each run
- Monitor sequencing metrics: >99% base call quality ≥Q20, >98% target coverage ≥100x
Liquid Biopy and Exosomal DNA Analysis for KRAS
Principle: Exosomal DNA carries tumor-specific mutations and offers enhanced stability compared to cell-free DNA, particularly in early-stage disease [105].
Procedure:
- Sample Collection and Processing: Collect blood in EDTA or Streck tubes. Process within 2 hours with centrifugation at 2,000 × g for 10 minutes at 4°C. Transfer plasma to new tubes and centrifuge at 5,000 × g for 10 minutes at 4°C. Aliquot and store at -80°C.
- Exosome Isolation: Purify exosomes from 500μL plasma using Size Exclusion Chromatography (Sephacryl S-400). Elute with phosphate-buffered saline and collect exosome-containing fractions.
- Exosomal DNA Extraction: Add SDS lysis buffer with Proteinase K to exosome fractions. Incubate at 55°C for 30 minutes. Extract DNA with phenol:chloroform:isoamyl alcohol (25:24:1). Precipitate DNA with sodium acetate and ice-cold absolute ethanol. Resuspend DNA in nuclease-free water.
- Mutation Detection: Perform initial PCR with outer primers to enhance mutant amplicon amplification. Use Intplex allele-specific quantitative PCR with mutation-specific primers and probes for KRAS G12, G13, and Q61 hotspots. Analyze using quantitative PCR systems with sensitivity to 0.01% mutant allele frequency.
Quality Control:
- Assess DNA quality by 1.2% agarose gel electrophoresis
- Verify exosome size distribution by atomic force microscopy
- Include no-template controls and positive controls for each run
- Establish minimum input requirements for reliable detection
Signaling Pathways and Molecular Context
EGFR Signaling in NSCLC
The epidermal growth factor receptor (EGFR) is a transmembrane receptor tyrosine kinase that regulates critical cellular processes including proliferation, differentiation, and survival. In NSCLC, activating mutations (primarily exon 19 deletions and L858R point mutations) lead to constitutive receptor activation, promoting oncogenic signaling through multiple downstream pathways [103].
KRAS Signaling in NSCLC
The Kirsten rat sarcoma viral oncogene homolog (KRAS) is one of the most frequently mutated oncogenes in NSCLC, particularly in adenocarcinomas. KRAS mutations (most commonly at codons G12, G13, and Q61) result in constitutive GTP binding and activation of multiple downstream effectors [104]. Different KRAS mutations activate distinct downstream signaling pathways; for example, G12A preferentially activates PI3K and MAPK pathways, while G12C and G12V preferentially activate the Ral-GDS signaling pathway [104]. This mutation-specific signaling bias has implications for therapeutic targeting and helps explain the varied clinical behaviors associated with different KRAS mutations.
Research Reagent Solutions
Table 4: Essential Research Reagents for Mutation Detection Studies
| Reagent/Category | Specific Examples | Research Application | Performance Considerations |
|---|---|---|---|
| NGS Panels | Oncomine Focus Assay (52 genes), TTSH-oncopanel (61 genes), Illumina TSO500 (523 genes) | Comprehensive genomic profiling, variant discovery | Sensitivity: 98.23%, Specificity: 99.99% for validated panels [102] |
| Digital PCR Systems | Bio-Rad QX200, droplet digital PCR | Absolute quantification, low-frequency variant detection | Sensitivity to 0.01% VAF, BRAF detection superior to Sanger sequencing [100] |
| Liquid Biopsy Kits | Oncomine Lung cfTNA, QIAamp Circulating Nucleic Acid Kit | Plasma-based mutation detection, therapy monitoring | Positive percent agreement: 56-79% vs. tissue; superior for SNVs [101] |
| IHC Antibodies | BRAF V600E (VE1) clone | Mutation-specific protein detection, spatial localization | 89.6% detection rate vs. 72.9% for Sanger in PTC [100] |
| Exosome Isolation | Size Exclusion Chromatography, Sephacryl S-400 columns | Exosomal DNA/RNA extraction, early detection | 85% KRAS detection in early-stage CRC, median VAF 1.18% [105] |
The comprehensive evaluation of NGS against gold-standard methods for BRAF, EGFR, and KRAS mutation analysis reveals a complex performance landscape where method selection must be guided by clinical context, required sensitivity, and available resources. For BRAF V600E detection, ddPCR and IHC demonstrate superior sensitivity compared to traditional Sanger sequencing (83.3% and 89.6% vs. 72.9%, respectively) [100]. In EGFR-driven NSCLC, liquid biopsy NGS offers rapid turnaround times (~3 days) and high concordance for single-nucleotide variants, though with lower detection rates for certain mutations compared to tissue testing (18.6% vs. 34.1% for T790M) [103]. For KRAS mutation analysis, large NGS panels (e.g., 523 genes) enable definitive clonality classification with minimal inconclusive results (0-1%), a significant improvement over smaller oncogene-only panels (30-74% inconclusive) [106] [107]. These findings underscore the importance of method-specific validation and contextual implementation to optimize mutation detection accuracy across diverse clinical and research scenarios.
Next-generation sequencing (NGS) has revolutionized genomic research by enabling the high-throughput, parallel sequencing of millions of DNA fragments, dramatically reducing the cost and time required for comprehensive genetic analysis [10] [7]. The selection of an appropriate NGS platform is a critical strategic decision that directly influences the feasibility and success of research and clinical projects [27]. This application note provides a detailed comparison of contemporary NGS platforms, focusing on their accuracy, sensitivity, and limitations for detecting various genetic variants, along with standardized protocols to guide researchers and drug development professionals in optimizing their sequencing approaches for DNA and RNA analysis.
Platform Categories and Key Characteristics
NGS technologies are broadly categorized into second-generation (short-read) and third-generation (long-read) platforms, each with distinct operational principles and performance characteristics [108] [27]. Second-generation platforms, dominated by Illumina's sequencing-by-synthesis technology, produce massive volumes of short reads (typically 75-300 bp) with exceptionally high accuracy [27] [7]. Third-generation technologies, exemplified by Pacific Biosciences (PacBio) Single-Molecule Real-Time (SMRT) sequencing and Oxford Nanopore Technologies (ONT), sequence single DNA molecules in real-time, generating much longer reads (averaging 10,000-30,000 bp) that facilitate the resolution of complex genomic regions [27] [7].
Table 1: Key Specifications of Major NGS Platforms
| Platform | Technology | Read Length | Accuracy Rate | Primary Error Type | Throughput | Best Application |
|---|---|---|---|---|---|---|
| Illumina | Sequencing-by-Synthesis | 75-300 bp | >99.9% (Q30) [108] | Substitution [109] | High to ultra-high | Variant calling, transcriptomics, targeted sequencing |
| PacBio (HiFi) | SMRT Sequencing | 10,000-25,000 bp | >99.9% (Q30) [108] | Random indel | Medium | De novo assembly, structural variants, haplotype phasing |
| Oxford Nanopore | Nanopore Sensing | 10,000-30,000+ bp | ~99.2% (Q28) to Q30 [108] | Context-dependent indel | Variable (MinION to PromethION) | Real-time sequencing, structural variants, direct RNA sequencing |
| Element AVITI | Sequencing-by-Binding | 100-200 bp | >99.99% (Q40) [108] | Low | High | Rare variant detection, cancer genomics |
| PacBio Onso | Sequencing-by-Binding | 100-200 bp | >99.99% (Q40) [108] | Low | High | Rare variant detection, low-pass WGS |
Recent advancements have significantly improved performance metrics across platforms. While a quality score of Q30 (99.9% accuracy) has been the standard for short-read platforms, newer systems like Element Biosciences' AVITI and PacBio's Onso now achieve Q40 (99.99% accuracy), reducing error rates by an order of magnitude [108]. This enhanced accuracy is particularly valuable for applications like rare variant detection in cancer and shallow whole-genome sequencing. Long-read technologies have also seen remarkable improvements, with PacBio and Oxford Nanopore now claiming Q30 and Q28 standards respectively, making them increasingly suitable for clinical applications [108].
Variant Detection Performance
The performance of NGS platforms varies significantly across different variant types due to their underlying biochemistry and detection mechanisms.
Table 2: Platform Performance for Different Variant Types
| Variant Type | Illumina | PacBio HiFi | Oxford Nanopore | Key Considerations |
|---|---|---|---|---|
| SNPs/Point Mutations | Excellent sensitivity/specificity [110] | High accuracy with HiFi reads | Good accuracy with latest models | Base quality scores crucial; high accuracy short-reads ideal for low-frequency variants |
| Small Indels | Excellent with optimized bioinformatics | High accuracy with HiFi reads | Good with homopolymer challenges | Homopolymer regions problematic for some technologies; ONT shows context-dependent indel errors [7] [109] |
| Structural Variants | Limited detection | Excellent | Excellent | Long reads essential for detecting large rearrangements, duplications, deletions |
| Gene Fusions/Rearrangements | Limited to targeted approaches | Excellent for novel discovery | Excellent for novel discovery | Short-read requires prior knowledge; long-read ideal for de novo detection |
| Copy Number Variations | Good with sufficient coverage | Good | Good | Uniform coverage critical for short-read; long-read less affected by mapping biases |
In clinical oncology applications, NGS demonstrates high diagnostic accuracy for identifying actionable mutations. In tissue samples, NGS shows 93% sensitivity and 97% specificity for EGFR mutations, and 99% sensitivity and 98% specificity for ALK rearrangements in non-small cell lung cancer [110]. However, in liquid biopsy applications, while NGS performs well for point mutations (80% sensitivity, 99% specificity for EGFR, BRAF V600E, KRAS G12C, and HER2), it has limited sensitivity for detecting ALK, ROS1, RET, and NTRK rearrangements [110].
Experimental Protocols
Comprehensive DNA Variant Detection Workflow
Objective: To comprehensively detect multiple variant types (SNPs, indels, CNVs, structural variants) from human genomic DNA using complementary short-read and long-read sequencing approaches.
Materials:
- High-quality genomic DNA (≥50 kb fragments)
- Illumina DNA Prep kit or equivalent
- PacBio SMRTbell library preparation kit
- Qubit fluorometer and Agilent TapeStation
- Illumina sequencing platform (NovaSeq X, MiSeq, or equivalent)
- PacBio Revio or Sequel IIe system
Procedure:
Quality Control and DNA Shearing
- Assess DNA quality using Qubit and TapeStation. DNA integrity number (DIN) should be ≥7.
- For Illumina: Fragment 100-500 ng DNA to 350 bp using Covaris sonicator.
- For PacBio: Use large (>30 kb) DNA fragments without shearing.
Library Preparation
- Illumina Library:
- Perform end repair, A-tailing, and adapter ligation using Illumina DNA Prep kit.
- Clean up using SPRI beads.
- Amplify library with 4-6 PCR cycles.
- Validate library size distribution (TapeStation).
- PacBio Library:
- Repair DNA and ligate SMRTbell adapters.
- Remove failed ligation products with exonuclease treatment.
- Size-select using BluePippin or SageELF (≥10 kb cutoff).
- Illumina Library:
Sequencing
- Illumina:
- Normalize and pool libraries.
- Sequence on NovaSeq X using 2×150 bp cycles.
- Target 30-50× coverage for whole genome.
- PacBio:
- Bind polymerase to SMRTbell templates.
- Sequence on Revio system with 30-hour movies.
- Target 15-25× coverage for whole genome.
- Illumina:
Data Analysis
- Illumina Data:
- Demultiplex with bcl2fastq.
- Align to reference genome (GRCh38) using BWA-MEM.
- Call variants with GATK (SNPs/indels) and CNVkit (copy number variants).
- PacBio Data:
- Process subreads to generate HiFi reads using CCS algorithm.
- Align to reference genome using pbmm2.
- Call structural variants using pbsv.
- Integrated Analysis:
- Combine variant calls from both technologies.
- Resolve discordances using manual inspection in IGV.
- Illumina Data:
Troubleshooting:
- Low library yield: Check initial DNA quality and quantification.
- Insufficient coverage: Optimize library concentration for sequencing.
- High duplication rate: Increase input DNA or reduce PCR cycles.
Targeted RNA-Seq for Expressed Mutation Detection
Objective: To detect and validate expressed mutations using targeted RNA sequencing, complementing DNA-based mutation screening [111].
Materials:
- High-quality total RNA (RIN ≥7)
- Agilent Clear-seq or Roche Comprehensive Cancer panels
- RNA library preparation kit (Illumina TruSeq RNA Access)
- Hybridization and capture reagents
- Bioanalyzer or TapeStation
Procedure:
RNA Quality Control and Library Preparation
- Assess RNA quality using Bioanalyzer. RIN should be ≥7.
- Convert 100 ng total RNA to cDNA using reverse transcriptase.
- Synthesize second strand to create double-stranded cDNA.
- Perform end repair, A-tailing, and adapter ligation.
Target Enrichment
- Hybridize library to panel-specific biotinylated probes (e.g., Agilent Clear-seq or Roche Comprehensive Cancer panels).
- Capture target regions using streptavidin-coated magnetic beads.
- Wash to remove non-specific binding.
- Amplify captured library with 10-12 PCR cycles.
Sequencing and Analysis
- Sequence on Illumina platform with 2×100 bp reads.
- Target 5-10 million reads per sample for focused panels.
- Align reads to reference genome using STAR aligner.
- Call variants using optimized pipeline (VarDict, Mutect2, LoFreq) [111].
- Apply filters: VAF ≥2%, total DP ≥20, alternative ADP ≥2.
Validation and Integration
- Compare RNA variants with DNA sequencing results.
- Prioritize variants with functional evidence (expression).
- Exclude false positives from RNA editing sites and alignment artifacts.
Quality Control Metrics:
- >80% of reads on target
- Uniform coverage across targets
- Sufficient coverage depth (>200× for low-expressed genes)
Visualization of Experimental Workflows
Comprehensive NGS Variant Detection Workflow
Platform Selection Decision Pathway
Research Reagent Solutions
Table 3: Essential Research Reagents for NGS Applications
| Reagent/Category | Function | Example Products | Key Considerations |
|---|---|---|---|
| Library Prep Kits | Convert nucleic acids to sequencing-ready libraries | Illumina DNA Prep, PacBio SMRTbell Prep | Select based on input material, application, and platform compatibility |
| Target Enrichment Panels | Capture specific genomic regions of interest | Agilent Clear-seq, Roche Comprehensive Cancer | Probe length affects coverage; 120 bp vs 70-100 bp designs [111] |
| Quality Control Tools | Assess nucleic acid and library quality | Agilent TapeStation, Qubit fluorometer | Critical for successful sequencing; DIN ≥7 for DNA, RIN ≥7 for RNA |
| Hybridization & Capture Reagents | Isolate targeted sequences | IDT xGen Lockdown Probes, Twist Target Enrichment | Optimization needed for GC-rich regions and uniform coverage |
| Enzymatic Mixes | Amplify and modify nucleic acids | NEBNext Ultra II, KAPA HiFi HotStart | High-fidelity polymerases essential for variant detection accuracy |
| Normalization Beads | Size selection and cleanup | SPRIselect, AMPure XP | Ratios affect size selection; optimize for each application |
| Sequencing Reagents | Enable the sequencing reaction | Illumina SBS chemistry, PacBio SMRTbell binding | Platform-specific; major cost component in sequencing workflows |
The selection of NGS platforms must be guided by the specific research objectives, variant types of interest, and available resources. Short-read technologies like Illumina remain the gold standard for detecting single nucleotide variants and small indels with high accuracy and throughput, while long-read platforms excel in resolving structural variants and complex genomic regions [27] [109]. The emerging Q40 platforms (Element AVITI, PacBio Onso) offer exceptional accuracy for applications requiring ultra-high precision, such as rare variant detection in cancer [108].
For comprehensive genomic characterization, a hybrid approach combining both short-read and long-read technologies provides the most complete picture, leveraging the strengths of each platform [109]. Additionally, integrating DNA and RNA sequencing, particularly through targeted RNA-seq approaches, helps bridge the gap between genetic alterations and functional expression, providing critical insights for precision oncology [111].
As NGS technologies continue to evolve, with improvements in accuracy, read length, and cost-effectiveness, researchers must stay informed about the latest developments to maximize the return on their sequencing investments. Standardization of workflows, rigorous quality control, and appropriate bioinformatic analysis remain crucial for generating reliable, reproducible results across all platforms.
Next-generation sequencing (NGS) has revolutionized molecular diagnostics and precision oncology by enabling the simultaneous detection of diverse genomic alterations from a single assay. The clinical utility of NGS hinges on the rigorous analytical validation of its capability to accurately detect multiple variant types, each presenting unique technical challenges. This document provides detailed application notes and protocols for the validation of NGS panels for the detection of single nucleotide variants (SNVs), small insertions and deletions (indels), copy number variations (CNVs), and gene fusions, framed within the broader context of advancing genomic research and therapeutic development.
Each variant class requires distinct bioinformatic approaches and wet-lab methodologies. SNVs and small indels are now routinely detected with high accuracy, whereas CNVs and fusions present greater technical hurdles due to their size and structural complexity [112]. The growing importance of comprehensive genomic profiling in both clinical trials and routine practice necessitates standardized validation frameworks that ensure reliable detection of all clinically relevant alteration types, ultimately facilitating matched therapeutic interventions and improving patient outcomes [113].
Performance Standards and Validation Metrics
Establishing performance metrics for each variant type is fundamental to NGS assay validation. These metrics define assay reliability and determine its suitability for clinical application.
Table 1: Recommended Analytical Validation Metrics for NGS Oncology Panels
| Variant Type | Recommended Sensitivity | Recommended Specificity | Key Parameters | Recommended Read Depth |
|---|---|---|---|---|
| SNVs | >99% | >99% | Variant Allele Frequency (VAF) | ≥500x [112] |
| Indels | >95% | >99% | Variant Allele Frequency (VAF), homopolymer regions | ≥500x [112] |
| CNVs | >90% (single exon); >97% (multi-exon) [114] | >99% [114] | Tumor purity, ploidy, coverage uniformity | Dependent on method [115] |
| Gene Fusions | >98% PPA [113] | ~99.99% NPA [113] | Breakpoint location, supporting reads | RNA-seq often used with DNA [113] |
Validation requires testing a sufficient number of samples to statistically demonstrate assay performance. The Association for Molecular Pathology (AMP) and College of American Pathologists (CAP) recommend using well-characterized reference materials and cell lines to establish accuracy for each variant type [112]. The limit of detection (LOD), particularly the minimum detectable variant allele frequency (VAF), must be established through dilution series. For example, one validated NGS panel demonstrated the ability to detect SNVs and indels at VAFs as low as 2.9% [102], while advanced liquid biopsy assays have reported detection of SNVs/indels at 0.5% allele frequency with 96.92% sensitivity [116].
Experimental Protocols for Validation
Sample Preparation and QC
Requirements: Fresh frozen or FFPE tissue, cytology specimens, or liquid biopsy samples. For solid tumors, pathologist review for tumor cell percentage is mandatory [112]. Procedure:
- Sample Review: A certified pathologist must review hematoxylin and eosin (H&E)-stained slides to confirm tumor presence, mark regions for macrodissection if needed, and estimate tumor cell percentage [112].
- Nucleic Acid Extraction: Extract DNA and RNA using validated kits. For FFPE samples, assess DNA integrity (e.g., DNA Integrity Number - DIN) and RNA quality (e.g., DV200%) [117].
- Quality Control: Quantify DNA/RNA using fluorometric methods. For DNA, a minimum input of 50 ng is often required for targeted NGS [102]. For RNA, ensure RIN/eRIN values or DV200% meet pre-established thresholds. Notes: Cytology specimens preserved in nucleic acid stabilizers can yield high-quality results, with success rates up to 98.4% in prospective studies [117]. Tumor purity estimation is critical for accurate CNV and fusion interpretation [112].
Library Preparation and Sequencing
Two primary library preparation methods are used for targeted NGS: hybrid capture-based and amplicon-based approaches [112]. Hybrid Capture Protocol (e.g., for comprehensive profiling):
- Library Construction: Fragment DNA and ligate with sequencing adapters.
- Target Enrichment: Hybridize with biotinylated oligonucleotide probes complementary to genomic regions of interest. Capture using streptavidin-coated magnetic beads.
- Amplification: Perform PCR amplification of captured libraries.
- Sequencing: Pool libraries and sequence on platforms such as Illumina NovaSeq or MGI DNBSEQ-G50RS [102]. Amplification-Based Protocol (e.g., for focused panels):
- Target Amplification: Use multiplex PCR with primers flanking regions of interest.
- Library Construction: Ligate amplified products with sequencing adapters and barcodes.
- Sequencing: Pool libraries and sequence on platforms such as Illumina MiSeq [117]. Method Selection: Hybrid capture more effectively covers wide genomic regions and avoids allele dropout, while amplicon approaches are often more efficient for focused panels [112]. For fusion detection, RNA sequencing using amplification-based methods is highly sensitive [113].
Bioinformatic Analysis
SNVs and Indels:
- Alignment: Map sequencing reads to a reference genome (e.g., GRCh37/hg19, GRCh38/hg38) using tools like BWA or STAR.
- Variant Calling: Use callers such as MuTect2 or VarScan for SNVs, and Pindel or VarScan for indels.
- Filtering: Filter variants based on depth, allele frequency, and quality scores. Annotate using databases like ClinVar and COSMIC. CNVs:
- Coverage Normalization: Normalize sample coverage against a control set of normal samples [115].
- CNV Calling: Utilize read-depth-based tools like CNVkit, ExomeDepth, or FACETS [115] [118]. For targeted panels, a sliding window approach (e.g., 75 bp windows) increases resolution for small CNVs [115].
- Ploidy Assessment: Determine genome ploidy, as inaccurate ploidy estimation is a major source of CNV calling error, particularly in hyper-diploid genomes [118]. Gene Fusions:
- DNA-based Detection: Use structural variant callers (e.g., LUMPY) on DNA sequencing data, requiring intronic probe coverage [113].
- RNA-based Detection: Employ fusion-specific callers (e.g., STAR-Fusion) on RNA-seq data, which identifies expressed fusion transcripts independent of breakpoint location [113].
- Validation: Integrate DNA and RNA findings. Combined DNA and RNA sequencing increases fusion detection by 21% compared to DNA alone [113].
Diagram 1: NGS Validation Workflow. This diagram outlines the comprehensive workflow for validating an NGS panel, from sample preparation through to integrated variant reporting.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Successful NGS validation requires carefully selected reagents, controls, and analytical tools.
Table 2: Essential Research Reagents and Platforms for NGS Validation
| Category | Specific Examples | Function in Validation |
|---|---|---|
| Reference Materials | HD701 Reference Standard, Cell Line DNA (e.g., HCC1395) | Provides known variants for establishing accuracy and precision for SNVs, indels, CNVs [102] [118] |
| Nucleic Acid Stabilizers | GM Tube (Ammonium sulfate-based stabilizer) | Preserves nucleic acid integrity in cytology specimens during transport/storage [117] |
| Library Prep Kits | Sophia Genetics Library Kit, Illumina Nextera Flex | Prepares sequencing libraries with optimized adapter ligation and amplification [102] |
| Target Enrichment | Hybrid Capture Probes (Biotinylated), Amplicon Panels | Enriches genomic regions of interest prior to sequencing [112] |
| QC Instruments | Qubit Fluorometer, TapeStation, Bioanalyzer | Quantifies and qualifies nucleic acids and libraries pre-sequencing [117] |
| CNV Calling Tools | CNVkit, ExomeDepth, FACETS, DRAGEN | Detects copy number changes from NGS data using read-depth approaches [115] [118] |
| Fusion Calling Tools | STAR-Fusion, LUMPY, AGFusion | Identifies gene fusions from DNA and/or RNA sequencing data [113] |
Advanced Applications and Integrated Analysis
Complementary Technologies for Complex Variants
While NGS is powerful, orthogonal technologies can enhance SV detection. Optical Genome Mapping (OGM) provides high-resolution mapping of large SVs, including those in non-coding regions, and can reveal complex rearrangements missed by short-read NGS [119]. For challenging cases, combining OGM with RNA-seq allows both detection and functional interpretation of SVs through transcriptome analysis [119]. Long-read sequencing technologies (e.g., nanopore) can resolve complex regions and precisely determine breakpoints.
Diagram 2: Multi-Technology Integration. This diagram illustrates how combining OGM and RNA-seq addresses the challenge of unsolved neurodevelopmental disorders (NDDs) after exome sequencing (ES), enabling both detection and functional interpretation of structural variants.
Tumor-Agnostic Applications and Drug Development
The expanding landscape of tumor-agnostic therapies necessitates comprehensive genomic profiling that reliably detects predictive biomarkers across cancer types. Notably, RET and NTRK1/2/3 fusions have FDA-approved therapies regardless of tumor origin [113]. For drug development professionals, understanding the prevalence of targetable fusions in non-approved cancer types (29% of fusions in one pan-cancer study [113]) identifies potential cohorts for clinical trial expansion. Robust NGS validation ensures reliable patient selection for these targeted therapies.
Rigorous validation of NGS panels for all alteration types is a cornerstone of modern precision oncology and genomic research. The protocols and metrics outlined provide a framework for establishing assays that meet the demands of both clinical diagnostics and therapeutic development. As the landscape of targetable alterations grows—particularly for complex structural variants and fusions—the integration of multiple technologies, including RNA-seq and long-read sequencing, will become increasingly important. Ensuring the accuracy, sensitivity, and reproducibility of NGS detection for SNVs, indels, CNVs, and fusions ultimately empowers researchers and clinicians to fully leverage genomic information for drug discovery and personalized patient care.
Implementing Quality Control and Ongoing Monitoring in a Clinical Setting
Next-generation sequencing (NGS) has become a cornerstone of modern clinical diagnostics and biomedical research, enabling unparalleled insight into the biology, evolution, and transmission of both infectious and non-infectious diseases [120]. However, the complexity of NGS workflows, from sample preparation to data analysis, introduces significant challenges for ensuring consistent, reliable, and clinically actionable results. A powerful diagnostic tool like NGS demands a robust Quality Management System (QMS) to guarantee data quality, which is essential for informed clinical and public health decisions that impact patient and community health [121] [120]. The implementation of a rigorous quality control (QC) and ongoing monitoring regimen is not merely a regulatory formality but a critical component for clinical accuracy, influencing diagnosis, prognosis, and ultimately, patient outcomes [120] [122]. This document outlines practical protocols for establishing and maintaining such a system within the context of a clinical NGS laboratory.
Foundational Elements of a Quality Management System
A robust QMS for NGS is built upon coordinated activities to direct and control an organization regarding quality [120]. The CDC and APHL's Next-Generation Sequencing Quality Initiative (NGS QI) provides a foundational framework based on the Clinical & Laboratory Standards Institute's (CLSI) 12 Quality System Essentials (QSEs) [120]. This framework helps laboratories navigate complex regulatory environments like the Clinical Laboratory Improvement Amendments (CLIA) and meet accreditation standards [121] [83].
For laboratories implementing NGS, key challenges addressed by a QMS include personnel management, equipment management, and process management [121] [83]. A particular challenge is the retention of proficient personnel, as testing personnel may hold their positions for less than four years on average, creating a need for continuous training and competency assessment [121]. The NGS QI offers over 100 freely available tools, including the widely adopted QMS Assessment Tool and Identifying and Monitoring NGS Key Performance Indicators SOP, to help labs build their systems from the ground up or enhance existing ones [121] [120]. All processes within the QMS must be locked down once validated, and any changes, such as transitioning to new sequencing platforms or updated bioinformatic pipelines, require thorough revalidation [121] [83].
Key Performance Indicators and Quality Metrics
Ongoing monitoring requires the definition and tracking of Key Performance Indicators (KPIs). These metrics should be monitored throughout the entire NGS workflow—pre-analytic, analytic, and post-analytic—to ensure the process remains in control.
Table 1: Essential Quality Metrics for Clinical NGS Workflows
| Workflow Stage | Metric | Definition / Calculation Method | Typical Threshold (Example) | Primary Data Source |
|---|---|---|---|---|
| Pre-Analytic | DNA/RNA Integrity Number (DIN/RIN) | Measures nucleic acid degradation. Calculated by instrument software (e.g., Agilent TapeStation). | DIN ≥ 7.0 (for WGS) [122] | Bioanalyzer/TapeStation |
| Nucleic Acid Concentration | Quantification via fluorometry (e.g., Qubit). | Sufficient for library prep protocol | Fluorometry | |
| Sequencing | Q-score | $Q = -10 \times \log_{10}(P)$ where P is the probability of an incorrect base call. | ≥ 30 (99.9% accuracy) [88] | FASTQ files |
| Cluster Density | Number of clusters per mm² on the flow cell. | Within platform-specific optimal range (e.g., 1700-220K/mm²) | Sequencing Platform SW | |
| % Bases ≥ Q30 | Percentage of bases with a Phred quality score of 30 or higher. | ≥ 80% [122] | FASTQ files / FastQC | |
| Alignment | Mapping Rate | $\frac{\text{Uniquely Mapped Reads}}{\text{Total Reads}} \times 100$ | Varies by application (e.g., RNA-seq > 80%) [122] | BAM files |
| Duplication Rate | $\frac{\text{PCR Duplicate Reads}}{\text{Total Reads}} \times 100$ | Dependent on sample type and depth | BAM files / Picard | |
| Application-Specific | Fraction of Reads in Peaks (FRiP) | $\frac{\text{Reads in Called Peaks}}{\text{Total Mapped Reads}}$ For ChIP-seq. | > 1% (e.g., for transcription factors) [122] | BED/Peaks files |
| 3'-Bias | Measures skew in RNA-seq fragment distribution across transcripts. | < 2-3 (dependent on protocol) | RSeQC |
It is crucial to note that threshold values are not universal; they can vary significantly depending on the assay (e.g., RNA-seq vs. ChIP-seq), sample type, and specific laboratory conditions [122]. Laboratories should derive their own condition-specific thresholds based on internal validation data and statistical guidelines derived from large public datasets [122].
Experimental Protocols for Quality Control
Protocol: Initial Method Validation for an NGS Assay
Prior to implementing any new or substantially modified NGS assay in the clinical workflow, a comprehensive validation must be performed. The NGS QI's NGS Method Validation Plan and NGS Method Validation SOP provide standard templates for this process [121].
1. Objective: To establish and document the analytical performance characteristics of a new NGS assay, ensuring it meets regulatory requirements (e.g., CLIA) and is fit for its intended clinical purpose [121] [83].
2. Materials:
- Reference Materials: Certified reference standards (e.g., from Coriell Institute, Genome in a Bottle Consortium) with known variants.
- Sample Types: A panel of clinical samples representing the expected sample matrix (e.g., blood, tissue, FFPE).
- Instrumentation: NGS sequencer, qPCR instrument for quantification.
- Bioinformatics: Validated analysis pipeline, reference genome.
3. Procedure:
- Accuracy and Precision: Sequence reference materials and a set of clinical samples in replicate (n≥3) across multiple runs and days. Compare called variants to known truth sets. Calculate SNP concordance and indel concordance.
- Analytical Sensitivity: Determine the limit of detection (LOD) by sequencing samples with variants at known, low allele frequencies (e.g., 5%, 2%, 1%).
- Analytical Specificity: Assess the false-positive rate by analyzing samples known to be negative for the variants of interest.
- Robustness: Introduce minor, deliberate variations in pre-analytical conditions (e.g., input DNA quantity, incubation times) to test the assay's resilience.
- Reportable Range: Verify the performance across the entire range of genetic targets the assay is designed to detect.
4. Data Analysis: Compile all data into a validation summary report. Performance metrics must meet or exceed pre-defined acceptance criteria before the assay can be used for patient testing.
Protocol: Ongoing Quality Control with a Sequencing Run
This protocol describes the routine QC performed with every clinical sequencing run to monitor process stability.
1. Objective: To ensure that each individual sequencing run produces data of sufficient quality for clinical analysis.
2. Materials:
- Control Materials: A positive control (e.g., a characterized DNA sample) and a negative control (e.g., nuclease-free water) included in every library preparation.
- Software Tools: FastQC, Picard Tools, in-house monitoring dashboards.
3. Procedure:
- In-Run Monitoring: Monitor real-time metrics from the sequencer, such as cluster density, intensity, and phasing/pre-phasing rates.
- Post-Run FASTQ QC: Execute FastQC on the generated FASTQ files. Critically review the Per Base Sequence Quality plot and Adapter Content plot.
- Alignment QC: After mapping reads to a reference genome (resulting in a BAM file), use tools like Picard to calculate metrics like mapping rate, duplication rate, and insert size.
- Control Assessment: Verify that the positive control shows the expected variants and coverage, and that the negative control is free of contamination.
4. Data Analysis and Acceptance Criteria: Compare all QC metrics against the laboratory's established KPIs (see Table 1). The run may only proceed to clinical interpretation if all controls and key metrics meet the predefined criteria. Any deviation must be documented and investigated.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Reagents for Clinical NGS QC
| Item | Function / Explanation | Example Vendors / Products |
|---|---|---|
| Certified Reference Standards | Provides a ground truth for assay validation and ongoing accuracy monitoring. Essential for establishing analytical sensitivity and specificity. | Genome in a Bottle (GIAB), Coriell Institute, Seraseq |
| Library Prep QC Kits | Fluorometric assays for accurate quantification of DNA/RNA and final libraries. Critical for determining optimal loading concentrations for sequencing. | Qubit dsDNA HS/BR Assay (Thermo Fisher) |
| Fragment Analyzers | Assesses nucleic acid size distribution and integrity (DIN/RIN). Poor integrity is a major source of assay failure. | Agilent TapeStation, Fragment Analyzer |
| Universal Human Reference RNA | Standard for RNA-seq assay validation, allowing for cross-lab comparison of performance metrics like 3'-bias and gene detection. | Agilent, Thermo Fisher |
| Multiplexed QC Spike-in Controls | Sequencer-independent synthetic oligonucleotides spiked into samples to detect technical biases in library prep and sequencing. | ERCC RNA Spike-In Mix (Thermo Fisher) |
| Bioinformatics QC Suites | Software packages that generate standardized quality reports from raw data (FASTQ) and aligned data (BAM). The first line of defense in data QC. | FastQC, Picard, Qualimap |
Workflow and Data Relationships
The following diagram illustrates the logical flow of a clinical NGS sample through the major stages of the workflow, highlighting the key QC checkpoints and decision points.
NGS QC Checkpoint Workflow
Statistical Guidelines and Classification for QC
Merely collecting QC metrics is insufficient; laboratories must implement data-driven guidelines to accurately classify data quality. Studies analyzing thousands of public NGS files from repositories like ENCODE have shown that universal, fixed thresholds (e.g., "minimum of 30 million reads") are often inadequate for differentiating between high- and low-quality files across different experimental conditions [122].
A more robust approach involves using machine learning-based decision trees that consider multiple quality features in combination, tailored to specific assay types and conditions (e.g., RNA-seq in liver cells versus blood cells) [122]. The most relevant features for accurate quality classification often come from genome mapping statistics and, for functional genomics assays like ChIP-seq, features like the Fraction of Reads in Peaks (FRiP) [122]. Laboratories should leverage these publicly available, condition-specific statistical guidelines and refine them with their own internal validation data to build accurate and reliable QC classifiers [122].
Implementing a comprehensive quality control and ongoing monitoring system is a non-negotiable requirement for the clinical application of NGS. This involves building a robust QMS framework, defining and tracking condition-specific KPIs, rigorously validating methods, and performing stringent run-to-run QC. By leveraging publicly available resources like those from the NGS Quality Initiative and adopting data-driven statistical guidelines, clinical laboratories can navigate the complexities of NGS technology. This ensures the generation of high-quality, reproducible, and reliable genomic data that is essential for accurate diagnosis, effective patient management, and advancements in drug development.
Conclusion
Next-generation sequencing has fundamentally reshaped genomic research and clinical diagnostics, offering unprecedented insights into DNA and RNA. Its applications in oncology, rare diseases, and drug development are paving the way for personalized medicine. Future progress will hinge on overcoming current challenges in data analysis, integrating AI for enhanced variant calling, and establishing robust, standardized validation frameworks to ensure reliable clinical application. As sequencing costs continue to fall and long-read technologies mature, NGS is poised to become an even more integral tool in biomedical research, ultimately improving patient outcomes through more precise diagnostics and targeted therapies.
References
- 1. DNA Sequencing Technologies–History and Overview [https://www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/next-generation-sequencing/dna-sequencing-history.html]
- 2. The evolution of next-generation sequencing technologies [https://pmc.ncbi.nlm.nih.gov/articles/PMC10246072/]
- 3. Applications of NGS in Drug Research [https://www.psomagen.com/blog/next-generation-sequencing-revolutionizes-drug-discovery-and-development]
- 4. Next-Generation Sequencing (NGS) | Explore the technology [https://www.illumina.com/science/technology/next-generation-sequencing.html]
- 5. A History of Sequencing [https://frontlinegenomics.com/a-history-of-sequencing/]
- 6. DNA Sequencing: How to Choose the Right Technology [https://frontlinegenomics.com/dna-sequencing-how-to-choose-the-right-technology/]
- 7. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 8. A Primer on NGS Technologies and Their Specs (2025) [https://blog.latch.bio/p/a-primer-on-ngs-technologies-and]
- 9. Comprehensive comparison of the third-generation ... [https://www.nature.com/articles/s41467-025-59187-2]
- 10. Next-generation sequencing: 2025 Revolution [https://lifebit.ai/blog/next-generation-sequencing/]
- 11. Next generation DNA Sequencing data analysis and its ... [https://www.sciencedirect.com/science/article/abs/pii/S034403382500473X]
- 12. Comparative analysis of Illumina, PacBio, and nanopore ... [https://www.frontiersin.org/journals/microbiomes/articles/10.3389/frmbi.2025.1587712/full]
- 13. Sequencing systems [https://www.pacb.com/sequencing-systems/]
- 14. London Calling 2025 Technology Update [https://nanoporetech.com/news/london-calling-2025-technology-update]
- 15. Applications for Nanopore sequencing [https://nanoporetech.com/applications]
- 16. Powered by PacBio: Selected publications from May 2025 [https://www.pacb.com/blog/powered-by-pacbio-selected-publications-from-may-2025/]
- 17. Comparative analysis of illumina and oxford nanopore ... [https://www.nature.com/articles/s41598-025-18768-3]
- 18. Applications of NGS [https://www.illumina.com/science/technology/next-generation-sequencing/beginners/applications.html]
- 19. Comparative evaluation of sequencing platforms [https://pmc.ncbi.nlm.nih.gov/articles/PMC12365774/]
- 20. Whole-genome sequencing of 490640 UK Biobank ... [https://www.nature.com/articles/s41586-025-09272-9]
- 21. Advancements in Whole-Genome Sequencing Protocols [https://pmc.ncbi.nlm.nih.gov/articles/PMC12598290/]
- 22. Whole Exome Sequencing and Whole Genome Sequencing ... [https://guidelines.carelonmedicalbenefitsmanagement.com/whole-exome-sequencing-and-whole-genome-sequencing-2025-07-26-updated-10-01/]
- 23. Navigating single-cell RNA-sequencing: protocols, tools, ... [https://pubmed.ncbi.nlm.nih.gov/40382658/]
- 24. 2025 and Beyond: The Future of Genomic Data Analysis ... [https://blog.crownbio.com/2025-and-beyond-the-future-of-genomic-data-analysis-and-innovations-in-genomics-services]
- 25. New Generation of Clinical Epigenetics Analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12192280/]
- 26. Whole genome sequencing [https://www.pacb.com/products-and-services/applications/whole-genome-sequencing/]
- 27. Next-Generation Sequencing: A Review of Its Transformative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12523276/]
- 28. Next Generation Sequencing Platform: Ultimate Guide 2025 [https://lifebit.ai/blog/next-generation-sequencing-platform/]
- 29. Roche unveils a new class of next-generation sequencing ... [https://www.roche.com/media/releases/med-cor-2025-02-20]
- 30. 2025 Trends: NGS [https://www.genengnews.com/topics/genome-editing/2025-trends-ngs/]
- 31. Precision Medicine Trends 2025: 5 Game-Changers [https://lifebit.ai/blog/the-future-is-personalprecision-medicine-trends-you-cant-ignore-in-2025/]
- 32. United States Next Generation Sequencing (NGS) Market ... [https://crisprmedicinenews.com/press-release-service/card/united-states-next-generation-sequencing-ngs-market-research-2025-2033-personalized-medicine-res/]
- 33. Sample Preparation for NGS – A Comprehensive Guide [https://frontlinegenomics.com/sample-preparation-for-ngs-a-comprehensive-guide/]
- 34. Next generation DNA sequencing data analysis and its ... [https://pubmed.ncbi.nlm.nih.gov/41177096/]
- 35. Volta Labs Unveils 2025 Application Roadmap for NGS ... [https://www.prnewswire.com/news-releases/volta-labs-unveils-2025-application-roadmap-for-ngs-sample-prep-at-agbt-302382988.html]
- 36. Clinical Next-Generation Sequencing (NGS) Data Analysis [https://www.openpr.com/news/4274679/comprehensive-clinical-next-generation-sequencing-ngs-data]
- 37. NGS library preparation [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/ngs/dna-sequencing/library-preparation]
- 38. Hybridization capture vs amplicon sequencing | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/targeted-sequencing-hybridization-capture-or-amplicon-sequencing/]
- 39. Amplicon Sequencing vs. Hybridization Capture [https://www.cd-genomics.com/diseasepanel/amplicon-sequencing-vs-hybridization-capture-the-comparison-of-target-enrichment-methods-for-disease-panel.html]
- 40. Explaining DNA vs. RNA sequencing [https://dispendix.com/blog/explaining-dna-vs-rna-sequencing]
- 41. Method Matters: Comparing Common Targeted NGS ... [https://seqwell.com/method-matters-comparing-common-targeted-ngs-techniques/]
- 42. A simplified hybrid capture approach retains high ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12403294/]
- 43. Master NGS library preparation for high-quality sequencing [https://www.idtdna.com/pages/technology/next-generation-sequencing/library-preparation]
- 44. Next-Generation Sequencing in Oncology—A Guiding Compass for Targeted Therapy and Emerging Applications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11988731/]
- 45. Is next generation sequencing for the diagnosis of rare ... [https://pubmed.ncbi.nlm.nih.gov/41222854/]
- 46. The implementation of genome sequencing in rare genetic ... [https://www.sciencedirect.com/science/article/pii/S2666606525000100]
- 47. Next-Generation Sequencing and Bioinformatics ... [https://wwwnc.cdc.gov/eid/article/30/14/24-0306_article]
- 48. Genomic surveillance methods to track infectious disease [https://www.illumina.com/areas-of-interest/microbiology/public-health-surveillance/genomic-surveillance.html]
- 49. Next-Generation Sequencing in Drug Discovery Market ... [https://www.cervicornconsulting.com/next-generation-sequencing-in-drug-discovery-market]
- 50. Advancing Drug Resistance Detection: Comparative ... [https://www.mdpi.com/2813-9038/2/3/14]
- 51. NGS is evolving: collaboration and tech lead the way [https://www.drugtargetreview.com/article/168383/ngs-is-evolving-collaboration-and-tech-lead-the-way/]
- 52. Advances in biomarkers of resistance to KRAS mutation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12508421/]
- 53. Multi-omics advances in understanding cancer drug ... [https://www.sciencedirect.com/science/article/pii/S2949723X25000510]
- 54. The Role of Biomarkers in AML Drug Discovery [https://blog.crownbio.com/the-role-of-biomarkers-in-aml-drug-discovery]
- 55. Application of next-generation sequencing in the detection ... [https://www.sciencedirect.com/science/article/abs/pii/S0009898125003997]
- 56. Next-generation sequencing applications in food science [https://pmc.ncbi.nlm.nih.gov/articles/PMC12405390/]
- 57. Harnessing genomics for sustainable food systems with ... [https://link.springer.com/article/10.1007/s44279-025-00354-w]
- 58. Biologicals [https://www.sciencedirect.com/science/article/pii/S1045105625000508]
- 59. Circulating DNA / RNA Patent Landscape Report [https://www.expertmarketresearch.com/patent-analysis/circulating-dna-rna-patent-landscape]
- 60. Mission Bio Announces Single-Cell Targeted DNA + RNA ... | Mission Bio [https://missionbio.com/press/targeted-dna-rna/]
- 61. Pre-Analytical Considerations for Successful Next-Generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5037844/]
- 62. Input DNA/RNA QC [https://nanoporetech.com/document/input-dna-rna-qc]
- 63. RNA-seq Sample Guidelines | Genomics Core Facility [https://www.huck.psu.edu/core-facilities/genomics-core-facility/sample-recommendations/rna-seq-sample-guidelines]
- 64. Getting Started with RNA-Sequencing (RNA-Seq) [https://www.neb.com/en-us/tools-and-resources/usage-guidelines/getting-started-with-rna-seq?srsltid=AfmBOopyibWIzHUiYIQt6FreTshvMC_BQW2X2e_1moX6DFWAQFqUsG_O]
- 65. A Beginner's Guide to Analysis of RNA Sequencing Data [https://pmc.ncbi.nlm.nih.gov/articles/PMC6096346/]
- 66. Improved Tumor Purity Metrics in Next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6887630/]
- 67. Deriving tumor purity from cancer next generation ... [https://www.sciencedirect.com/science/article/pii/S0893395222002617]
- 68. PUREE: accurate pan-cancer tumor purity estimation from ... [https://www.nature.com/articles/s42003-023-04764-8]
- 69. Real-world data analysis for factors influencing the quality ... [https://www.nature.com/articles/s41598-025-85846-x]
- 70. dupRadar: a Bioconductor package for the assessment of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5073875/]
- 71. Characterization and mitigation of artifacts derived from NGS ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10157-w]
- 72. Multiplexed targeted next generation sequencing coverage [https://www.idtdna.com/page/support-and-education/decoded-plus/minimizing-duplicates-and-obtaining-uniform-coverage-in-multiplexed-target-enrichment-sequencing/]
- 73. Read Duplicates [https://www.biostars.org/p/159597/]
- 74. NGS Library Preparation - 3 Key Technologies [https://www.illumina.com/techniques/sequencing/ngs-library-prep.html]
- 75. 5 Best Practices for NGS Library Prep Success [https://dispendix.com/blog/5-best-practices-for-ngs-library-prep-success]
- 76. Get ready for short tandem repeats analysis using long ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1610026/full]
- 77. NGS considerations: coverage, read length, multiplexing [https://irepertoire.com/ngs-considerations-coverage-read-length-multiplexing/]
- 78. Integrating Artificial Intelligence in Next-Generation Sequencing [https://pmc.ncbi.nlm.nih.gov/articles/PMC12191491/]
- 79. Cloud-based bioinformatics workflow platform for large- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4203338/]
- 80. HPC Data Management for NGS Analysis: Storage, Transfer ... [https://ngs101.com/hpc-data-management-for-ngs-analysis-storage-transfer-and-sharing-best-practices/]
- 81. Recommendations for bioinformatics in clinical practice [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-025-01543-4]
- 82. Recommendations for bioinformatics in clinical practice - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12535132/]
- 83. The Next-Generation Sequencing Quality Initiative and ... [https://wwwnc.cdc.gov/eid/article/31/13/24-1175_article]
- 84. A comparison of tools for the simulation of genomic next- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5224698/]
- 85. Next Generation Sequence Testing: Essential 2025 Guide [https://lifebit.ai/blog/next-generation-sequence-testing/]
- 86. Clinical Quality Considerations when Using Next-Generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8332578/]
- 87. How-to: NGS Quality Control [https://frontlinegenomics.com/how-to-ngs-quality-control/]
- 88. The Complete Guide to NGS Data Types and Formats [https://ngs101.com/the-complete-guide-to-ngs-data-types-and-formats-from-raw-reads-to-analysis-ready-files/]
- 89. Artificial intelligence in variant calling: a review [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1574359/full]
- 90. A Simplified Approach to Somatic Analysis in NGS [https://www.euformatics.com/blog-post/a-simplified-approach-to-somatic-analysis-in-ngs]
- 91. Technote: Implementing Data Integrity and 21 CFR Part 11 ... [https://www.avancebio.com/technote-implementing-data-integrity-and-21-cfr-part-11-compliance-in-ngs-workflows/]
- 92. Next-generation sequencing in cancer diagnosis and ... [https://pubmed.ncbi.nlm.nih.gov/40253661/]
- 93. Top Oncology Innovations That Shaped the First Half of 2025 [https://binaytara.org/cancernews/article/top-oncology-innovations-that-shaped-the-first-half-of-2025]
- 94. Clinical and analytical validation of a combined RNA ... [https://www.nature.com/articles/s43856-025-00934-3]
- 95. Clinical and analytical validation of a combined RNA ... [https://pubmed.ncbi.nlm.nih.gov/40523952/]
- 96. Clinical Validation of Duoseq, a novel assay for ... [https://www.sciencedirect.com/science/article/abs/pii/S1525157825002557]
- 97. BostonGene Publishes Landmark Study Providing Clinical ... [https://bostongene.com/news-and-publications/news/bostongene-publishes-landmark-study-providing-clinical-and-analytical-validation-for-its-multimodal-rna-and-dna-assay]
- 98. Principles of Analytic Validation of Immunohistochemical ... [https://www.cap.org/protocols-and-guidelines/cap-guidelines/current-cap-guidelines/principles-of-analytic-validation-of-immunohistochemical-assays]
- 99. Celebrating milestones and the future of oncology in China [https://www.illumina.com/company/news-center/feature-articles/CSCO-2025.html]
- 100. Accuracy comparison of direct Sanger sequencing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12621767/]
- 101. Detecting actionable mutations from matched plasma-based ... [https://jeccr.biomedcentral.com/articles/10.1186/s13046-025-03480-x]
- 102. A targeted next-generation sequencing panel for ... [https://www.nature.com/articles/s41598-025-08039-6]
- 103. From missed mutations to liquid clues: a new era of EGFR ... [https://medcraveonline.com/JSRT/from-missed-mutations-to-liquid-clues-a-new-era-of-egfr-diagnostics-in-nsclc.html]
- 104. Literature review of advances and challenges in KRAS G12C ... [https://tlcr.amegroups.org/article/view/102803/html]
- 105. Performance of KRAS mutation detection in plasma exosomes ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06710-0]
- 106. Performance and considerations in the use of diagnostic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12141053/]
- 107. Performance and considerations in the use of diagnostic ... [https://www.sciencedirect.com/science/article/pii/S205970292500941X]
- 108. The Latest Developments in Sequencing Technologies [https://frontlinegenomics.com/the-latest-developments-in-sequencing-technologies/]
- 109. A practical comparison of the next-generation sequencing ... [https://www.life-science-alliance.org/content/6/4/e202201744]
- 110. Diagnostic accuracy of next-generation sequencing (NGS) ... [https://link.springer.com/article/10.1007/s12094-025-04040-7]
- 111. Augmenting precision medicine via targeted RNA-Seq ... [https://www.nature.com/articles/s41698-025-00993-8]
- 112. Guidelines for Validation of Next-Generation Sequencing–Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6941185/]
- 113. Molecular Characterization of Oncogenic Gene Fusions in a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12589939/]
- 114. Validation of copy number variation analysis for next- ... [https://www.nature.com/articles/ejhg201742]
- 115. Detecting copy number variation in next generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8406611/]
- 116. Analytical Validation of a Pan-Cancer NGS Assay for In- ... [https://www.sciencedirect.com/science/article/pii/S152515782500220X]
- 117. Prospective multicenter validation of a next-generation ... [https://bmccancer.biomedcentral.com/articles/10.1186/s12885-025-14770-0]
- 118. Evaluation of somatic copy number variation detection by NGS ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03294-8]
- 119. Combining optical genome mapping and RNA-seq for structural variants detection and interpretation in unsolved neurodevelopmental disorders | Genome Medicine | Full Text [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-024-01382-9]
- 120. The Next Generation Sequencing Quality Initiative [https://www.cdc.gov/lab-quality/php/ngs-quality-initiative/index.html]
- 121. The Next-Generation Sequencing Quality Initiative and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12078536/]
- 122. Statistical guidelines for quality control of next-generation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8408346/]