Mitigating RNAi Off-Target Effects: A Strategic Guide for Robust Experimental Design and Therapeutic Development
This article provides a comprehensive guide for researchers and drug development professionals on addressing the pervasive challenge of off-target effects in RNA interference (RNAi) experiments.
Mitigating RNAi Off-Target Effects: A Strategic Guide for Robust Experimental Design and Therapeutic Development
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on addressing the pervasive challenge of off-target effects in RNA interference (RNAi) experiments. It explores the fundamental mechanisms behind these effects, including miRNA-like seed region binding and immune activation. The content details practical methodologies for designing specific RNAi triggers, from bioinformatic tools to chemical modifications. It further offers troubleshooting strategies for optimizing experimental conditions and validation frameworks to confirm on-target activity. By synthesizing foundational knowledge with advanced application and validation techniques, this guide aims to empower scientists to enhance the reliability of their functional genomics data and accelerate the development of safer, more effective RNAi-based therapeutics.
Deconstructing RNAi Off-Target Effects: Mechanisms, Sources, and Impact on Data Fidelity
FAQs: Understanding Off-Target Effects in RNAi
What are off-target effects in RNAi experiments? Off-target effects occur when your RNAi molecules, such as siRNA or miRNA, silence genes other than the intended target. This happens primarily through two mechanisms: sequence-dependent effects, where the guide strand has partial complementarity to non-target mRNAs (similar to how endogenous miRNAs function), and sequence-independent effects, where the RNAi molecules trigger innate immune responses, such as the interferon pathway [1] [2]. These effects can confound experimental results by producing misleading phenotypes and pose significant safety risks in therapeutic development.
Why should I be concerned about off-target effects? Off-target effects are a major concern because they can compromise the validity of your experimental data and the safety of potential therapeutics. In research, they can lead to incorrect conclusions about gene function [1]. In drug development, off-target activity can result in unexpected toxicities and adverse events, potentially halting the progression of promising RNAi drug candidates [2]. Managing these effects is therefore critical for both basic research success and clinical application.
How do RNAi off-target effects differ from those in CRISPR/Cas9? While both technologies face off-target challenges, their fundamental mechanisms differ. RNAi causes gene knockdown at the mRNA level, and its off-targets are often due to partial sequence complementarity, especially in the "seed region" (nucleotides 2-8 of the guide strand), leading to the degradation or translational repression of non-target mRNAs [3] [1]. In contrast, CRISPR/Cas9 creates permanent knockout mutations at the DNA level. Its off-target effects typically involve Cas9 cleaving genomic sites with sequence similarity to the guide RNA, potentially causing chromosomal rearrangements or unintended mutations [4] [5]. Recent comparative studies suggest that well-optimized CRISPR/Cas9 systems can have fewer off-target effects than RNAi [1].
What are the main strategies to minimize off-target effects? You can employ several key strategies to reduce off-target effects:
- Careful Oligo Design: Use bioinformatics tools to design siRNAs with high specificity, ensuring minimal sequence homology to other genes in the target organism [6] [7].
- Chemical Modifications: Utilize advanced chemistries like Stealth RNAi, which incorporate proprietary modifications to ensure only the antisense strand enters the RISC and to enhance stability, thereby reducing off-target potential [6].
- Optimal Concentration: Use the lowest effective concentration of siRNA during transfection, as high concentrations increase the likelihood of off-target binding [8].
- Pooling Strategies: Using pooled siRNAs can help dilute out individual sequences with high off-target potential, though this must be balanced with the ability to identify the specific effective sequence [3].
Troubleshooting Guide: Off-Target Effects
Problem: Suspected Phenotype from Off-Target Effects
Symptoms:
- Observed phenotypic effects that do not align with the known function of your target gene.
- Inconsistent results between different siRNA sequences designed for the same target gene.
- Failure to rescue the phenotype by expressing a target gene construct that is resistant to RNAi (e.g., with silent mutations).
Solutions:
- Validate with Multiple siRNAs: The most reliable approach is to test at least two or three distinct siRNA sequences targeting different regions of the same mRNA. If all independent siRNAs produce the same phenotypic result, it is more likely to be an on-target effect [8].
- Perform a Rescue Experiment: Co-transfect your siRNA with a plasmid expressing the target gene that has been engineered with silent mutations in the siRNA-binding site. This "recoded" gene will be resistant to RNAi. If the phenotype is reversed, it confirms on-target activity; if not, an off-target effect is likely.
- Conduct Transcriptomic Analysis: Use genome-wide expression profiling (e.g., RNA-seq) to compare cells treated with your siRNA to control cells. This will directly reveal which genes are being unexpectedly up or down-regulated, providing a map of potential off-targets [1].
- Measure Protein Levels: Always confirm knockdown at the protein level (e.g., by Western blot) in addition to mRNA levels. Persistent protein expression despite mRNA reduction could indicate a potent off-target effect driving the phenotype [6].
Problem: Poor Gene Knockdown Efficiency
Symptoms:
- Minimal reduction in target mRNA or protein levels after siRNA transfection.
- No observable phenotypic change.
Solutions:
- Check Transfection Efficiency: Ensure your siRNA is successfully entering the cells. Use a fluorescently labeled control siRNA (e.g., BLOCK-iT Fluorescent Oligo) and monitor uptake under a microscope. Optimize transfection parameters like reagent concentration, cell confluency, and complex formation time [8] [6].
- Verify Oligo Sequence and Quality: Sequence your siRNA or shRNA expression plasmid to confirm the insert is correct and has not acquired mutations during cloning. Use high-quality, HPLC- or PAGE-purified oligonucleotides [8].
- Re-design siRNA/shRNA: The initial target site or hairpin design may be suboptimal. Use a proprietary algorithm (e.g., RNAi Designer) to select a new target region with proven efficacy. For shRNAs, you can also try varying the stem length or loop sequence [8] [6].
- Switch Delivery Method: If using synthetic siRNA, consider switching to a viral delivery system (e.g., lentiviral shRNA) for more robust and sustained expression, especially in hard-to-transfect cells [6].
Key Reagents and Experimental Protocols
Research Reagent Solutions
| Reagent / Tool | Primary Function | Key Considerations for Reducing Off-Targets |
|---|---|---|
| Stealth RNAi [6] | Chemically modified siRNA duplex. | Proprietary modifications ensure only the antisense strand loads into RISC, reducing off-targeting from the sense strand. |
| Bioinformatics Design Tools (e.g., RNAi Designer) [6] | In silico design of siRNA sequences. | Uses algorithms to ensure sequence uniqueness and minimize homology to other genes in the selected organism. |
| Lentiviral shRNA Vectors [6] | Stable, long-term gene knockdown. | Allows generation of stable cell pools, minimizing clonal variation and enabling validation across multiple cell populations. |
| Inducible shRNA Systems (e.g., H1/TO promoter) [6] | Precise temporal control of shRNA expression. | Enabling short-term induction of RNAi can limit the duration of exposure, reducing the accumulation of off-target effects. |
| Control siRNAs (e.g., Scrambled, Non-targeting) [4] | Baseline for experimental comparisons. | Serves as a transfection control but is not a perfect off-target control, as different sequences have different off-target profiles. |
Protocol: Validating Knockdown Specificity
Purpose: To confirm that an observed phenotypic effect is due to on-target gene silencing and not off-target effects.
Materials:
- Validated siRNA sequences (at least 2-3) targeting your gene of interest.
- Non-targeting control siRNA.
- Plasmid for rescue experiment (target gene cDNA with silent mutations in the siRNA target site).
- Transfection reagent.
- Reagents for qRT-PCR and Western blotting.
Procedure:
- Transfert your target cells in separate wells with the different siRNAs and the non-targeting control.
- Assay for Phenotype: 48-72 hours post-transfection, assay for the phenotypic change of interest (e.g., cell viability, migration, differentiation).
- Confirm Knockdown: In parallel, harvest cells to confirm knockdown of the target mRNA (via qRT-PCR) and protein (via Western blot).
- Rescue Experiment: Co-transfect the most effective siRNA alongside the rescue plasmid expressing the mutated, RNAi-resistant target gene. Include controls with the rescue plasmid alone and the siRNA alone.
- Re-assay for Phenotype: 48-72 hours later, re-assay for the phenotypic change.
Interpretation: If the phenotype is consistently observed with all specific siRNAs and is reversed by the expression of the RNAi-resistant rescue construct, it is strong evidence for an on-target effect. Inconsistency between siRNAs or a failure to rescue suggests off-target activity.
Protocol: Detecting Transcriptome-Wide Off-Target Effects
Purpose: To identify all genes whose expression is inadvertently altered by your RNAi treatment.
Materials:
- Cells treated with siRNA and appropriate controls.
- RNA extraction kit.
- RNA-seq library prep kit and sequencing service.
Procedure:
- Treat and Harvest: Transfert cells with your target siRNA and a non-targeting control siRNA. Harvest total RNA 48 hours post-transfection. Perform this in biological triplicate.
- RNA Sequencing: Check RNA quality, prepare sequencing libraries, and perform RNA-seq on all samples.
- Bioinformatic Analysis:
- Map sequencing reads to the reference genome.
- Identify differentially expressed genes (DEGs) between the target siRNA and control siRNA samples.
- Filter the DEG list to focus on genes with partial complementarity to the siRNA's "seed region" (nucleotides 2-8 of the guide strand), as these are the most likely direct off-targets.
Interpretation: A large number of differentially expressed genes, particularly those with seed region matches to your siRNA, indicates significant off-target activity. This data can be used to re-design a more specific siRNA.
Visualizing the Pathways and Workflows
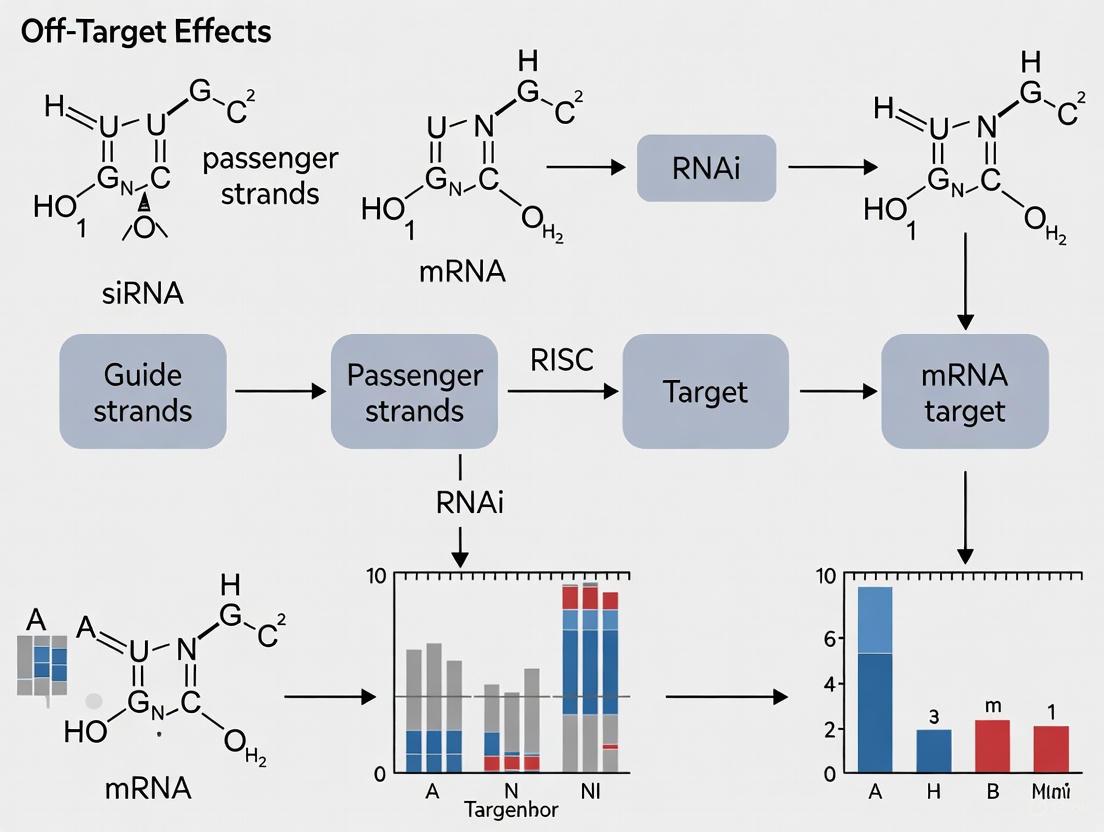
The diagram below illustrates the core RNAi mechanism and highlights where two major types of off-target effects can occur.
Experimental Workflow for Specific RNAi
This workflow outlines the key steps for conducting an RNAi experiment with built-in checks to identify and mitigate off-target effects.
MicroRNA (miRNA) mimicry has emerged as a powerful therapeutic strategy to restore the function of tumor-suppressing miRNAs that are frequently downregulated in diseases like cancer. The efficacy of these synthetic miRNA-like duplexes hinges on a fundamental principle: seed region complementarity. The seed region, a conserved sequence at the 5' end of the miRNA guide strand (typically nucleotides 2-8), is the primary determinant for recognizing and binding to target mRNAs [9] [10]. This binding, even with imperfect complementarity outside the seed region, can lead to translational repression or degradation of the target mRNA [10] [11]. While this mechanism allows a single miRNA to regulate a broad network of genes, it also introduces a significant risk of off-target effects, where the mimic unintentionally represses genes with complementary sequences in their 3' untranslated regions (3' UTRs) [12]. This technical support center provides a framework for understanding and troubleshooting these challenges to ensure the precision and success of your RNAi experiments.
FAQs on miRNA Mimicry and Seed Regions
What is the seed sequence of a miRNA and why is it so important?
The seed sequence is a conserved heptametrical sequence, mostly situated at positions 2-7 from the miRNA's 5' end [9]. This region is essential because it must be perfectly complementary for the initial binding of the miRNA to its target mRNA. It serves as the primary anchor for target recognition, directing the miRNA-induced silencing complex (miRISC) to its mRNA targets [10] [11].
How do off-target effects occur with miRNA mimics, and how do they differ from siRNA off-targets?
Off-target effects in miRNA mimicry are predominantly seed-mediated. When a synthetic mimic is introduced into a cell, its seed region can bind to the 3' UTRs of mRNAs that were not the intended primary targets, leading to their repression in a miRNA-like fashion [12]. While siRNAs are designed for perfect complementarity to a single target and can cause off-target effects through similar seed-mediated mechanisms, miRNA mimics are designed to leverage the natural, seed-dependent, multi-target regulatory network of endogenous miRNAs [11]. This makes the management of their off-target profile a central consideration.
What computational tools can help predict seed-mediated off-target effects?
SeedMatchR is an R package specifically developed to detect and visualize seed-mediated off-target effects of siRNA and miRNA using RNA-seq data [12]. It extends differential expression analysis tools by annotating results with predicted seed matches and provides statistical functions to test for cumulative changes in gene expression attributed to seed region activity.
Can chemical modifications to the mimic reduce off-target effects?
Yes, strategic chemical modifications can mitigate off-target effects. Research has shown that modifications like a glycol nucleic acid (GNA) at position 7 (g7) of the seed region can significantly reduce cumulative off-target gene expression changes and associated toxicities, such as hepatotoxicity in rodent models, without completely abolishing on-target efficacy [12].
Troubleshooting Guides
Issue 1: Suspected Widespread Off-Target Effects in Transcriptomic Data
Problem: RNA-seq analysis after miRNA mimic transfection shows a significant, unintended downregulation of a large set of genes.
Diagnosis and Solutions:
| Step | Action | Rationale & Technical Details |
|---|---|---|
| 1. Confirm Effect | Run an ECDF (Empirical Cumulative Distribution Function) analysis on log2(fold change) data for genes with/without a seed match. | A significant leftward shift (Kolmogorov-Smirnov test) in the ECDF of genes with a seed match indicates a cumulative seed-mediated off-target effect [12]. |
| 2. Use Controls | Always include a scrambled sequence control mimic in experiments. | This helps distinguish sequence-specific effects from non-specific cellular responses to nucleic acid transfection [13]. |
| 3. Analyze Seed Context | Use tools like SeedMatchR to annotate your DE results with the number of seed matches in gene 3' UTRs. | This statistically tests if differential expression is associated with the presence of a seed match in the 3' UTR, confirming a seed-driven mechanism [12]. |
| 4. Optimize Dosage | Perform a dose-response curve and use the lowest effective concentration. | Suprapharmacological doses of RNAi triggers exacerbate seed-mediated off-target effects; using minimal effective dose mitigates this [12]. |
Issue 2: Lack of Intended On-Target Effect
Problem: The miRNA mimic fails to knock down the expression of its validated primary target genes.
Diagnosis and Solutions:
| Step | Action | Rationale & Technical Details |
|---|---|---|
| 1. Verify Delivery | Include a fluorescently-labeled control oligonucleotide to assess transfection efficiency. | If delivery failed, no knockdown will occur. This is a critical first step, especially in hard-to-transfect cells [13]. |
| 2. Check Expression | Confirm via qPCR that your target gene is expressed in the cell type used. | There is nothing to knock down if the target is not expressed in your model system [13]. |
| 3. Validate Mimic Design | Ensure the mimic's guide strand is the correct, biologically active sequence for the intended miRNA. | Using the passenger strand or an incorrect sequence will not produce the desired effect. Consult miRBase for canonical sequences. |
| 4. Try Consensus Mimics | Consider using a synthetic mimic based on a family consensus sequence. | For miRNA families (e.g., miR-15/107), a consensus-based mimic can exhibit enhanced growth inhibitory activity and target suppression compared to a native sequence mimic [14]. |
Research Reagent Solutions
The following table lists key reagents and tools essential for studying and implementing miRNA mimicry pathways.
| Reagent/Tool | Function & Application in miRNA Research |
|---|---|
| SeedMatchR [12] | An R package for identifying seed-mediated off-target effects from RNA-seq data. It annotates genes with seed matches and performs statistical tests on expression shifts. |
| Synthetic Consensus Mimics [14] | Engineered miRNA mimics based on the consensus sequence of a miRNA family (e.g., miR-15/107). They can offer enhanced therapeutic activity and broader target inhibition. |
| EGFR-Targeted Nanocells (EDV) [14] | A delivery system (e.g., EnGeneIC Dream Vector) that packages miRNA mimics and uses targeting moieties (e.g., against EGFR) for direct, efficient tumor cell delivery in vivo. |
| AUMantagomir sdASO [13] | A self-delivering antisense oligonucleotide (ASO) protocol for microRNA inhibition. Useful as a control to block endogenous miRNA function or to study mimic specificity. |
| Chemically Modified Mimics [12] | Mimics with specific chemical modifications (e.g., GNA at seed position g7) designed to reduce seed-mediated hepatotoxicity and other off-target effects while maintaining on-target activity. |
Experimental Protocols & Workflows
Detailed Protocol: Detecting Seed-Mediated Off-Target Effects with SeedMatchR
This protocol leverages the SeedMatchR package to analyze RNA-seq data from experiments involving miRNA mimics [12].
Inputs Required:
- A data frame of differential expression results (e.g., from DESeq2).
- A species-specific GTF file as a GRanges object.
- A species-specific DNAStringSet for genomic DNA.
- The siRNA/miMic guide sequence (≥ 8 nt).
Procedure:
- Visualize Seed Regions: Use the
plot_seeds()function to plot the miRNA mimic sequence and its default seed definitions. - Prepare Annotations: Generate the required GRanges and DNAStringSet objects from public databases (e.g., ENSEMBL) using built-in Bioconductor functions.
- Run SeedMatchR: Execute the primary
SeedMatchR()function. This function usesvcountpattern()to search the genomic annotations (focusing on 3' UTRs) for matches to the mimic's seed region and annotates your differential expression results with the count of matches per gene. - Statistical Analysis & Visualization:
- Use
de_fc_ecdf()to plot the Empirical Cumulative Distribution Functions (ECDFs) for the log2 fold changes of two gene sets: those with a seed match and those without. - Use
ecdf_stat_test()to perform a one-sided Kolmogorov-Smirnov (KS) test. A significant result indicates that the distribution of log2 fold changes for genes with a seed match is shifted towards downregulation compared to the background, confirming a cumulative seed-mediated off-target effect.
- Use
Workflow Diagram: miRNA Mimic Off-Target Analysis
Pathway Diagram: miRNA Mimic Mechanism & Seed-Mediated Off-Targets
RNA interference (RNAi) is a powerful tool for gene silencing, but its application is complicated by off-target effects that extend beyond the well-characterized seed-sequence-mediated miRNA-like effects. This technical support guide addresses two other major sources of experimental confounding: the activation of the innate immune response by double-stranded RNA (dsRNA) and the saturation of the endogenous RNAi machinery. Understanding these phenomena is crucial for designing robust RNAi experiments and accurately interpreting their results.
FAQs: Understanding the Core Challenges
Q1: How can dsRNA, the core trigger of RNAi, also confound my experiment through immune activation?
Double-stranded RNA is recognized by the mammalian innate immune system as a potential sign of viral infection. This triggers defense mechanisms that can mask or mimic your experimental phenotype.
Immune Sensing Pathways: Cytoplasmic dsRNA is primarily detected by two classes of sensors [3] [15]:
- RIG-I-like receptors (RLRs): RIG-I is activated by short dsRNAs with 5' triphosphates (ppp-dsRNA) or cap0 structures, leading to inflammatory signaling via the mitochondrial antiviral signaling (MAVS) protein and resulting in interferon and cytokine production [15].
- PKR and OAS/RNase L: These sensors recognize the presence of the RNA duplex itself, independent of 5' end modifications. PKR activation leads to a global shutdown of protein synthesis, while OAS/RNase L activation causes non-specific degradation of cellular RNA [15].
Consequences for Experiments: Activation of these pathways can lead to general cell growth inhibition, cytotoxicity, and widespread changes in gene expression that are independent of your specific target knockdown. This can create false positives or obscure genuine phenotypic effects [15] [16].
Q2: What does it mean for an RNAi experiment to "saturate the endogenous machinery," and why is it harmful?
The cellular machinery that processes your introduced short hairpin RNAs (shRNAs) or siRNAs is the same machinery required for the biogenesis and function of endogenous microRNAs (miRNAs). At high concentrations, your experimental RNAi triggers can overwhelm this shared pathway.
- Shared Limiting Factors: Key components like Exportin-5 (responsible for nuclear export of small RNA precursors) and Dicer (which processes dsRNA) can become rate-limiting [16].
- Phenotypic Impact: Saturation leads to a global disruption of endogenous miRNA-regulated gene networks. This can cause severe toxicity, as demonstrated in studies where high-dose shRNA expression in mice led to fatality, not from immune activation or specific off-targets, but from the collapse of essential miRNA functions [16].
- Distinguishing Feature: Unlike sequence-specific off-targets, saturation effects are largely sequence-independent. If multiple, distinct shRNAs against different targets cause similar toxic phenotypes at high doses, saturation of the machinery is a likely cause [16].
Q3: Are there specific experimental factors that make my system more prone to these effects?
Yes, several experimental parameters significantly influence your risk:
- Delivery Method and Dose: The use of viral vectors (especially for shRNA delivery) that lead to very high, persistent expression greatly increases the risk of both immune activation and saturation [16] [17]. Using the lowest possible effective dose of siRNA or shRNA is critical.
- dsRNA Structure and Modification: The immunogenicity of dsRNA is heavily influenced by its 5' end. Triphosphorylated (ppp) dsRNA and cap0 dsRNA are highly immunogenic, while cap1 dsRNA (with a 2'-O-methylation on the first transcribed nucleotide) is effectively masked from RIG-I and evades this inflammatory response [15].
- Cell Type: Different cell lines may have varying basal levels of immune pathway components and miRNA machinery, affecting their sensitivity to these off-target effects.
Troubleshooting Guides
Problem 1: Suspected Innate Immune Response Activation
Observed Symptoms: Unexplained cell death or growth inhibition; activation of interferon-stimulated genes in transcriptomic data; global translational shutdown; general inflammation in animal models.
| Diagnostic Check | Experimental Solution | Underlying Principle |
|---|---|---|
| Test for Interferon Response: Measure mRNA levels of classic interferon-stimulated genes (ISGs) like OAS1 or MX1 via qRT-PCR 24-48 hours after transfection/transduction [16]. | Use Chemically Modified or Cap1 dsRNA: When generating dsRNA in vitro, use methods that produce a cap1 5' structure to avoid RIG-I recognition [15]. | The cap1 structure (5' end with 2'-O-methylation) is a self-marker. Human dsRNA sensors like RIG-I do not recognize it as "non-self," preventing inflammatory pathway activation [15]. |
| Check for PKR Activation: Monitor for phosphorylation of eukaryotic initiation factor 2α (eIF2α) by Western blot, a downstream target of activated PKR. | Titrate RNAi Trigger Dose: Systematically reduce the concentration of siRNA/shRNA to the minimum required for effective knockdown [18] [19]. | Lower concentrations of dsRNA are less likely to reach the threshold required to activate the OAS/RNase L and PKR pathways, which are primarily sensitive to the presence of the duplex [15]. |
| Use Control RNAs: Include controls with known immunostimulatory (e.g., triphosphorylated dsRNA) and non-immunostimulatory (e.g., cap1 dsRNA) structures [15]. | Select Non-Immunostimulatory Sequences: Some sequence motifs can trigger immune responses. Use design tools to avoid these and employ pooled siRNAs to distribute the load [3] [18]. | Pooling multiple siRNAs against the same target allows you to use lower concentrations of each individual sequence, reducing the risk of any single siRNA triggering an immune sensor. |
Problem 2: Suspected Saturation of Endogenous RNAi Machinery
Observed Symptoms: Lethality or severe toxicity in vivo at high shRNA doses; disruption of normal cellular development and function; similar toxic phenotypes from multiple, unrelated shRNAs.
| Diagnostic Check | Experimental Solution | Underlying Principle |
|---|---|---|
| Monitor miRNA Function: Quantify the expression and activity of abundant endogenous miRNAs (e.g., let-7, miR-21) in treated vs. control cells [16]. A drop in mature miRNA levels or activity indicates saturation. | Use miRNA-Embedded shRNA Platforms: Express your shRNA within a native miRNA backbone (e.g., the Multi-miR platform) [20]. | Endogenous miRNA precursors are naturally optimized for efficient processing without saturating Exportin-5 or Dicer. Embedding your shRNA in this context exploits this natural efficiency [20]. |
| Assess Exportin-5 Dependence: Co-express Exportin-5. If this improves knockdown efficiency or reduces toxicity, it was likely a limiting factor [16]. | Switch to siRNA or Optimized shRNA Designs: For transient knockdown, use siRNAs, which bypass early steps of the miRNA pathway. For stable expression, use polymerase II-driven, miRNA-embedded shRNAs rather than simple Pol III-driven shRNAs [16] [20]. | siRNAs are directly loaded into RISC, bypassing the nuclear Exportin-5 and Dicer processing steps. This places less burden on the core miRNA biogenesis machinery [16]. |
| Perform Dose-Response Studies: Test a range of vector doses (MOI) or expression levels. Toxicity that is only apparent at high doses strongly suggests saturation. | Employ Inducible Expression Systems: Use tetracycline/doxycycline-inducible promoters to control the timing and level of shRNA expression, allowing transient rather than continuous high-level expression [8]. | Inducible systems prevent constant, high-level burden on the miRNA machinery and allow you to induce knockdown only during the experimental window, minimizing long-term saturation effects. |
Experimental Protocols for Validation
Protocol 1: Validating Immune Activation by dsRNA
Purpose: To determine if your RNAi trigger is activating the innate immune response. Key Materials: Cells, transfection reagent, synthetic siRNA/shRNA (test and control), TRIzol, qRT-PCR reagents.
- Treat Cells: Divide cells into three groups:
- Test Group: Transfected with your target siRNA/shRNA.
- Positive Control: Transfected with a known immunostimulatory RNA (e.g., in vitro transcribed ppp-dsRNA).
- Negative Control: Transfected with a non-immunostimulatory RNA (e.g., cap1-modified dsRNA or a commercial non-targeting control).
- Incubate: Incubate for 24-48 hours.
- Harvest RNA: Extract total RNA using TRIzol.
- Analyze by qRT-PCR: Synthesize cDNA and perform qRT-PCR for interferon-stimulated genes (ISGs) such as OAS1 and IFIT1. Use housekeeping genes (e.g., GAPDH, HPRT1) for normalization.
- Interpretation: A significant upregulation of ISGs in the test group compared to the negative control indicates immune activation. The positive control should show a strong response, while the negative control should not.
Protocol 2: Testing for Saturation of the miRNA Pathway
Purpose: To assess if your RNAi experiment is disrupting endogenous microRNA function. Key Materials: Cells, RNAi expression vector, control vector, reagents for RNA isolation and qRT-PCR, miRNA-specific assays.
- Establish Stable Cell Lines: Create cell lines stably expressing your shRNA (test) or a non-targeting control shRNA. Use inducible systems if possible.
- Extract RNA: Harvest total RNA, including the small RNA fraction, from both cell lines.
- Quantify Mature miRNAs: Using stem-loop RT-qPCR or similar specific assays, measure the levels of 2-3 highly expressed endogenous miRNAs (e.g., let-7a, miR-16).
- Quantify Primary miRNA Transcripts: Using standard qRT-PCR with primers flanking the Drosha processing site, measure the levels of the primary miRNA (pri-miRNA) transcripts for the same miRNAs.
- Interpretation: A significant decrease in the levels of mature miRNAs in the test group, without a corresponding decrease in their primary transcripts, is a classic signature of saturation. It indicates that the precursor miRNAs are being produced but their processing into mature forms is impaired due to competition with the experimental shRNA [16].
Signaling Pathways and Experimental Workflows
dsRNA Innate Immune Recognition Pathway
The diagram below illustrates how different features of dsRNA activate distinct branches of the innate immune response.
RNAi Saturation of Endogenous miRNA Machinery
This diagram shows how high levels of exogenous shRNA can compete with and disrupt the normal biogenesis of endogenous miRNAs.
The Scientist's Toolkit: Research Reagent Solutions
The following table lists key reagents and their specific roles in mitigating the off-target effects discussed in this guide.
| Research Reagent | Function & Application | Key Benefit |
|---|---|---|
| Cap1-modified dsRNA | An in vitro transcribed dsRNA with a 5' cap containing a 2'-O-methyl group on the first transcribed nucleotide [15]. | Effectively evades recognition by the RIG-I sensor, preventing the onset of dsRNA-induced cellular inflammation and interferon production [15]. |
| Chemically Modified siRNAs | siRNAs incorporating chemical modifications (e.g., 2'-O-methyl) in the sugar-phosphate backbone [3]. | Increases nuclease stability and can reduce immune activation. Some modifications also help to minimize seed-mediated off-target effects [3]. |
| miRNA-Embedded shRNA Vectors | Vector systems (e.g., Multi-miR) where the shRNA is expressed within the context of a natural microRNA backbone [20]. | Harnesses the cell's optimized miRNA biogenesis pathway for efficient processing, reducing competition and saturation of Exportin-5 and Dicer [20]. |
| Inducible shRNA Systems | shRNA expression systems (e.g., tetracycline/doxycycline-inducible) that allow precise temporal control over shRNA production [8]. | Enables transient knockdown, preventing long-term saturation of the RNAi machinery and allowing the study of essential genes whose permanent knockout would be lethal [8]. |
| Exportin-5 Expression Plasmid | A plasmid for co-expressing the Exportin-5 protein [16]. | Can be used as a diagnostic tool; if co-expression improves shRNA efficacy or reduces toxicity, it confirms that Exportin-5 was a saturated, limiting factor in the experiment [16]. |
FAQ: Understanding and Classifying RNAi Off-Target Effects
What are the main categories of RNAi off-target effects?
RNAi off-target effects are broadly classified into two categories based on their mechanism:
- Specific Off-Target Effects: These occur when the small interfering RNA (siRNA) or double-stranded RNA (dsRNA) exhibits partial complementarity to non-target mRNAs, leading to their degradation or translational repression. This is a sequence-dependent phenomenon [21] [22] [23].
- Non-Specific Off-Target Effects: These are sequence-independent effects triggered by the RNAi machinery or the dsRNA molecule itself. They include the activation of the innate immune response (e.g., interferon response), competition between siRNA and endogenous microRNAs (miRNAs) for the RNA-induced silencing complex (RISC), and saturation of the endogenous RNAi pathway [22] [23].
What sequence features determine specific off-target effects?
Specific off-target effects are primarily governed by the degree of sequence complementarity between the siRNA guide strand and non-target mRNAs. The following table summarizes key quantitative findings from research on dsRNA triggers, which are highly relevant for pest control and functional genomics in insects [22]:
| Feature | Description | Impact on Off-Target Risk |
|---|---|---|
| Overall Sequence Identity | The percentage of identical nucleotides between the dsRNA and a non-target gene over the entire length. | dsRNAs with >80% sequence identity to a non-target gene can trigger significant off-target knockdown [22]. |
| Contiguous Perfect Match | A stretch of perfectly matched bases between the siRNA (derived from dsRNA) and an off-target mRNA. | A segment of ≥16 bp of perfectly matched sequence is sufficient to trigger RNAi of the off-target gene [22]. |
| Almost Perfect Match with Mismatches | A long stretch of sequence with scarcely distributed mismatches. | A segment of >26 bp with one or two mismatches can trigger off-target effects. Single mismatches inserted between ≥5 bp matching segments, or mismatched couplets inserted between ≥8 bp matching segments, also pose a risk [22]. |
| Seed Region Complementarity | Nucleotides 2-8 at the 5' end of the siRNA guide strand. | Perfect complementarity in this region can lead to miRNA-like translational repression of off-target mRNAs, even without full sequence match [22] [23]. |
How can I minimize immune-related non-specific off-target effects?
Non-specific effects, such as immune activation, are often dose-dependent and more prevalent in certain organisms [22]. In vertebrates, dsRNA longer than 30 bp can trigger an interferon response [22]. To minimize this:
- Optimize Dosage: Use the lowest effective concentration of dsRNA/siRNA to reduce the risk of saturating the RNAi machinery or triggering immune sensors [22] [23].
- Consider Delivery Method: Direct application of dsRNA (e.g., SIGS) may present different risks compared to endogenous expression in GM plants (HIGS) [21] [24].
- Utilize Bioinformatic Tools: Tools like dsRIP (Designer for RNA Interference-based Pest Management) are being developed to help design optimal dsRNA sequences that maximize efficacy while minimizing risks to non-target organisms [25].
Why do some target genes show different susceptibility to RNAi in the same organism?
Genes are not equivalent targets for RNAi. The natural RNA metabolism of a target gene can influence its susceptibility. Research in C. elegans has shown that an intersecting network of RNAi regulators (e.g., MUT-16, RDE-10, NRDE-3) exists. Some genes require all regulators for efficient silencing, while others can be silenced even if one regulator is missing. This suggests that the cellular context and regulation of the target gene itself contribute to the RNAi outcome [26].
Troubleshooting Guide: Diagnosing Off-Target Effects
Problem: Observed Phenotype Does Not Match Expected Knockdown of Target Gene
This is a classic symptom of potential off-target effects. The following workflow outlines a systematic approach to diagnose the issue.
Experimental Protocols for Detection and Validation
Protocol 1: Comprehensive Bioinformatic Risk Assessment
This protocol is a prerequisite before conducting experiments to predict potential specific off-target effects [21] [22].
- Sequence Input: Obtain the full sequence of your dsRNA or siRNA trigger.
- Database Selection: Use a high-quality, annotated transcriptome or genome of the experimental organism. If using a model organism, a reference genome is ideal. For non-model organisms, a de novo assembled transcriptome may be used, acknowledging potential limitations [21].
- Alignment and Filtering:
- Use a local BLAST tool (e.g., BLASTn) to align the trigger sequence against the selected database.
- Apply filters to identify potential off-target candidates:
- Overall identity >80% over the entire length.
- Presence of contiguous perfect matches ≥16 bp.
- Presence of >26 bp segments with ≤2 mismatches.
- Manual Inspection: Manually inspect the top hits from the BLAST results to confirm the location and context of the matched sequences.
Protocol 2: Untargeted Transcriptomics for Off-Target Detection
This experimental protocol is used to empirically detect off-target effects by profiling global gene expression changes after RNAi treatment [21] [24].
- Experimental Design:
- Groups: Include a treatment group (dsRNA/siRNA) and a appropriate control group (scrambled RNA or buffer).
- Replicates: Use a minimum of three biological replicates per group to ensure statistical power.
- Sample Collection: Collect tissue or cells at a time point post-treatment that is relevant to the phenotype being studied.
- RNA Extraction & Sequencing:
- Extract total RNA using a method that preserves small RNAs (if interested in miRNA competition).
- Prepare RNA-seq libraries. For a comprehensive view, consider both standard mRNA-seq and small RNA-seq.
- Sequence the libraries on an appropriate platform (e.g., Illumina).
- Bioinformatic Analysis:
- Map sequencing reads to the reference genome/transcriptome.
- Perform differential gene expression analysis (e.g., using DESeq2 or edgeR).
- Identify Off-Targets: Look for significantly downregulated genes. Cross-reference these genes with the list of predicted off-target candidates from Protocol 1.
- Pathway Analysis: Use Gene Ontology (GO) or Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis to identify if downregulated genes are enriched in specific pathways, which may indicate a non-specific immune or stress response.
Visualization of the RNAi Pathway and Off-Target Mechanisms
The diagram below illustrates the core RNAi mechanism and points where specific and non-specific off-target effects can occur.
The Scientist's Toolkit: Research Reagent Solutions
The following table details key reagents and tools essential for studying and mitigating RNAi off-target effects.
| Research Reagent / Tool | Function in Off-Target Research | Key Considerations |
|---|---|---|
| Long dsRNA (>200 bp) | The primary trigger for RNAi in many invertebrate and plant systems; processed into a pool of siRNAs [25] [26]. | The length increases the potential for generating multiple siRNAs with different off-target potentials. Design should follow rules on sequence identity and contiguous matches [22]. |
| Synthetic siRNA (21-22 nt) | Allows for precise targeting with a defined sequence; commonly used in mammalian systems [23]. | Enables chemical modification (e.g., 2'-O-methyl) to reduce immune activation and improve stability. The seed sequence should be carefully evaluated [23]. |
| Bioinformatic Tools (e.g., dsRIP) | Web-based platforms for designing optimized dsRNA/siRNA sequences [25]. | Algorithms can predict highly efficient siRNA strands within a dsRNA based on features like thermodynamic asymmetry and GC content, and identify potential off-targets in non-target species [25]. |
| Dicer/DCR-1 Mutants | Genetic tools to dissect the steps in the RNAi pathway [26]. | Used to confirm that observed effects are dependent on the canonical RNAi pathway. |
| RISC Component Mutants (e.g., RDE-1, Ago2) | Used to validate on-target engagement and understand mechanisms of off-target silencing [26] [23]. | Loss of function can abolish RNAi. Different Argonautes have specialized roles (e.g., NRDE-3 in nuclear silencing) [26]. |
| Transcriptomics Kits (RNA-seq) | For genome-wide experimental detection of off-target mRNA degradation [21] [24]. | Essential for unbiased discovery of both specific and non-specific off-target effects. Requires a high-quality reference genome. |
Your Troubleshooting Guide to Mitigating RNAi Off-Target Effects
This guide helps you identify and troubleshoot the primary causes of off-target effects in RNAi experiments, which can lead to misleading data and false conclusions.
| Troubleshooting Guide | |||
|---|---|---|---|
| Problem | Possible Causes | Recommended Solutions | Key Controls to Implement |
| Sequence-Dependent Off-Targets | siRNA guide strand behaving like a microRNA, binding to and repressing transcripts with partial complementarity, especially in the "seed" region (nucleotides 2-8) [3] [2]. | - Bioinformatic Design: Use rigorous in silico tools to screen for complementarity to off-target transcripts, especially in the seed region [27] [25].- Chemical Modifications: Utilize siRNAs with chemical modifications (e.g., 2'-O-methyl) to reduce seed-mediated off-targeting [3].- Pooled siRNAs: Use a pool of several siRNAs targeting the same gene at lower concentrations; off-target effects are not synergistic while on-target effects are [3] [28]. | |
| Saturation of Endogenous RNAi Machinery | High concentrations of transfected siRNA can overwhelm the cellular RNAi pathway, disrupting natural microRNA regulation and causing phenotypic changes unrelated to your target [2]. | Titrate siRNA: Use the lowest effective siRNA concentration. For many lipid-mediated transfections, 10 nM is often sufficient, reducing off-targets while maintaining efficacy [28]. | |
| Activation of Innate Immune Response | Introduction of dsRNA can trigger interferon-activated pathways, leading to global changes in gene expression that are mistaken for specific RNAi effects [1]. | - Quality Control: Ensure siRNA is highly pure and free of long dsRNA contaminants [28].- Design: Avoid immunostimulatory sequences in your siRNA design.- Validation: Include controls to measure interferon response markers. | |
| Inaccurate Guide Strand Selection | The incorrect strand of the siRNA duplex (the passenger strand) is loaded into RISC, leading to the silencing of non-targeted genes [3] [25]. | Design for Thermodynamic Asymmetry: Design siRNAs so the antisense (guide) strand has a less stable 5' end than the passenger strand. This biases RISC loading towards the intended guide strand [3] [25]. |
Essential Protocols for Validation
Following these detailed methodologies is critical for confirming that your observed phenotypic effects are due to specific on-target silencing.
Protocol 1: Comprehensive Bioinformatic Analysis for Off-Target Prediction
This protocol is adapted from EFSA guidance for risk assessment of RNAi-based genetically modified plants and provides a conservative framework for predicting potential off-target transcripts in your experimental system [27].
- Step 1: Sequence Preparation. Compile the complete sequence of all 21-nucleotide small RNAs potentially processed from your dsRNA or siRNA trigger.
- Step 2: Transcriptome Alignment. Perform an in silico search against the most up-to-date transcriptome of your model organism. The search should use specific alignment parameters:
- No more than four mismatches in total (or three mismatches and one single-nucleotide gap).
- G:U wobble pairs count as half a mismatch.
- No mismatches or gaps at position 10 or 11 of the small RNA.
- No more than two mismatches in the first 12 nucleotides at the 5' end.
- A minimum free energy (MFE) ratio of the duplex to the perfect complement of greater than 0.75 [27].
- Step 3: Risk Assessment. Prioritize off-target transcripts with multiple potential binding sites for different small RNAs from your trigger. Analyze the established or predicted function of these off-target genes to assess potential impact on your experimental outcomes [27].
Protocol 2: Experimental Confirmation of On-Target Engagement
This protocol outlines a rescue experiment, which is considered the gold standard for validating the specificity of an RNAi-induced phenotype.
- Step 1: Design a Rescue Construct. Clone the cDNA of your target gene into an expression vector. Crucially, introduce silent mutations in the region targeted by the siRNA. These mutations should not change the amino acid sequence but should make the mRNA resistant to silencing by your specific siRNA [28].
- Step 2: Co-transfection. Co-transfect your cells with both the siRNA and the rescue plasmid expressing the modified, siRNA-resistant target mRNA [28].
- Step 3: Phenotypic Analysis. Monitor whether the expression of the modified mRNA rescues the phenotype caused by the siRNA alone.
- Step 4: Interpretation. A successful reversal of the phenotype strongly indicates that the observed effect was due to specific silencing of your intended target and not an off-target effect.
Visualizing the Pathways to Reliable and Unreliable Data
The diagrams below illustrate the intended RNAi mechanism versus the common routes to off-target effects that compromise data integrity.
Key Reagents for Robust RNAi Experiments
The following table details essential materials and controls required to conduct reliable RNAi experiments and effectively manage off-target risks.
| Research Reagent Solutions | |
|---|---|
| Reagent / Control | Function and Importance |
| Validated & Modified siRNAs | Use siRNAs with chemical modifications (e.g., 2'-O-methyl) to reduce seed-based off-target effects and increase stability [3]. |
| Bioinformatic Design Tools | Software (e.g., DEQOR, siDirect, dsRIP) is essential for selecting target sequences with high predicted efficacy and low potential for off-target binding [25]. |
| Positive Control siRNA | A siRNA known to achieve high knockdown of a standard gene (e.g., GAPDH). Validates your transfection and experimental conditions are working [28]. |
| Negative Control siRNA | A non-targeting (scrambled) siRNA with no significant homology to the transcriptome. Serves as the baseline for distinguishing specific silencing from non-specific effects [28]. |
| Fluorescent Transfection Control | A fluorescently-labeled oligonucleotide (e.g., BLOCK-iT Fluorescent Oligo) used to visually confirm and quantify transfection efficiency under the microscope [28]. |
| siRNA-Rescue Construct | A plasmid expressing the target mRNA with silent mutations in the siRNA-binding site. Gold-standard control to confirm phenotype specificity [28]. |
Frequently Asked Questions (FAQs)
Q: What is the fundamental difference between an off-target effect and a false positive in an RNAi screen? An off-target effect is the molecular event where an siRNA inadvertently silences genes other than its intended target. A false positive is the experimental consequence: you observe a phenotypic change and incorrectly attribute it to the silencing of your target gene, when it was actually caused by an off-target effect.
Q: I have used multiple siRNAs against the same target gene and they all produce the same phenotype. Does this rule out off-target effects? While this is a strong indicator of an on-target effect, it does not provide absolute proof. It is possible (though less likely) that each individual siRNA has its own unique set of off-target genes that, through different pathways, converge on the same phenotype. The most rigorous validation is a rescue experiment with an siRNA-resistant construct [28].
Q: My negative control siRNA is causing unexpected cytotoxicity. What could be wrong? This often points to issues with the siRNA preparation or transfection. The culprit could be:
- Chemical Contamination: The siRNA may contain salts or solvents from the synthesis process.
- Immunostimulation: The negative control sequence itself, or contaminants in the preparation, may be activating the innate immune response [28].
- Transfection Toxicity: The concentration of the transfection reagent may be too high. Re-optimize the reagent:siRNA ratio and always include an untransfected cell control.
Q: Are there advantages to using CRISPR-based knockdown (CRISPRi) over RNAi for gene silencing? Yes. CRISPRi functions at the DNA level by blocking transcription, and its guide RNA typically requires a perfect sequence match for binding, leading to significantly fewer sequence-based off-target effects compared to RNAi [1]. For loss-of-function studies where permanent knockout is not desired, CRISPRi can be a more specific alternative. However, the optimal choice depends on your experimental goals, as RNAi's ability to achieve partial, transient knockdown can be advantageous for studying essential genes [1].
Proactive Design Strategies for Enhancing RNAi Specificity and Efficacy
Troubleshooting Guides & FAQs
FAQ: Algorithm Selection and Data Interpretation
Q: My pre-designed siRNA is not producing the expected knockdown. What are the first parameters I should check?
A: Begin by troubleshooting the siRNA sequence itself and your experimental setup.
- Check siRNA Sequence Efficiency: siRNA efficacy is highly sequence-dependent. Review the GC content (optimal range is often 30-50%) and the stability of the 5'-terminal seed region (preference for A/U bases) [29].
- Verify mRNA Level Knockdown: Use real-time PCR to check mRNA levels at approximately 48 hours post-transfection. Ensure your qRT-PCR assay target site is positioned within a reasonable distance from the siRNA cut site to avoid issues with alternative splice transcripts [19].
- Confirm Transfection Efficiency: Always run a positive control siRNA in parallel to demonstrate that the transfection reagents are working and the siRNA is being delivered correctly to the cells [19].
Q: A key gene I need to target has a highly homologous gene family. How can I minimize off-target effects in this scenario?
A: This requires a multi-pronged bioinformatic and experimental approach.
- Leverage Advanced Prediction Tools: Use modern algorithms that incorporate deep learning, as they can better capture the complex patterns of off-target binding, including the critical importance of the seed region [30].
- Prioritize Specific Target Sites: During the design phase, use software that ranks potential target sequences based on the number of similar sites in the genome. Select the guide with the fewest and most dissimilar off-target candidates [31].
- Validate with Amplicon-Seq: After designing your siRNA, perform amplicon-based next-generation sequencing (NGS) on the top predicted off-target sites to empirically assess and confirm editing levels [32].
Q: What is the recommended strategy for comprehensively identifying off-target sites for a novel therapeutic siRNA candidate?
A: Relying on a single method is insufficient. A robust strategy involves a combination of in silico and experimental techniques [32].
- In Silico Prediction: Use one or more bioinformatic tools (e.g., Cas-OFFinder, CCTop) to generate an initial list of potential off-target sites based on sequence similarity [33] [30].
- Unbiased Experimental Discovery: Employ a genome-wide, unbiased assay such as GUIDE-seq or DISCOVER-seq to identify off-target sites that are actually cleaved in a cellular context, which accounts for factors like chromatin accessibility [33] [32].
- Targeted Quantification: Use the combined list from the above steps to perform deep amplicon sequencing, which serves as the gold standard for quantifying the frequency of off-target effects at these candidate loci [32].
Troubleshooting Guide: Common Experimental Pitfalls
| Problem Area | Potential Cause | Recommended Solution |
|---|---|---|
| Low Knockdown Efficiency | Suboptimal siRNA sequence; Low transfection efficiency; Incorrect assessment timing. | Test multiple siRNAs to the same target; Use a validated positive control; Perform a time-course experiment (check at 24h, 48h, 72h) [19]. |
| High Cell Toxicity | Toxicity from the transfection reagent itself; Excessive siRNA concentration. | Run a transfection reagent-only control; Titrate down the siRNA concentration and test different cell densities [19]. |
| Inconsistent Results | Protein turnover rate masking mRNA knockdown; Degraded RNA. | Run a longer time course to assess protein-level knockdown; Check RNA quality post-isolation [19]. |
| Unexpected Phenotypes | Off-target effects silencing genes with similar sequences. | Use a more stringent siRNA design algorithm; Employ a high-fidelity Cas nuclease variant if using CRISPR; Validate findings with multiple independent siRNAs/guides [34] [4]. |
Experimental Protocols for Off-Target Validation
Protocol: Candidate Site Validation via Amplicon Sequencing
This protocol is considered the gold standard for quantifying off-target editing frequency at sites identified through prediction or discovery methods [32].
Key Reagents:
- Primers: Design specific primers to amplify each candidate off-target locus (typically 200-300 bp amplicons).
- NGS Library Prep Kit: Use a high-fidelity polymerase and a compatible NGS library construction kit.
- Control DNA: Include a non-transfected/control sample to establish baseline sequencing noise.
Methodology:
- Design Primers: Design and validate PCR primers for all candidate off-target sites from your in silico prediction and genome-wide discovery assays.
- Extract Genomic DNA: Isolate high-quality genomic DNA from your treated and control cells.
- Amplify Targets: Perform PCR amplification of each candidate locus from both test and control samples.
- Prepare NGS Libraries: Pool the amplicons, prepare sequencing libraries, and sequence on an NGS platform with sufficient depth (e.g., >100,000x coverage) to detect low-frequency events.
- Analyze Data: Use bioinformatic tools (e.g., Inference of CRISPR Edits - ICE) to align sequences and quantify the percentage of indels at each site, comparing treated samples to the control baseline [34].
Protocol: Genome-Wide Off-Target Discovery with GUIDE-seq
GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by sequencing) is a cellular method that captures double-strand breaks (DSBs) in living cells, providing biologically relevant off-target data [33] [32].
Key Reagents:
- GUIDE-seq Oligo: A short, double-stranded, blunt-ended oligonucleotide that is incorporated into DSBs.
- Transfection Reagents: For co-delivery of the GUIDE-seq oligo with your CRISPR RNP or siRNA.
- NGS Library Prep & Sequencing Reagents.
Methodology:
- Co-transfect Cells: Co-deliver the programmable nuclease (e.g., Cas9/sgRNA) or siRNA and the GUIDE-seq oligo into your target cells.
- Genomic DNA Extraction: Harvest cells 48-72 hours post-transfection and extract genomic DNA.
- Library Preparation & Sequencing: Construct sequencing libraries that will capture the genomic regions flanking the integrated GUIDE-seq oligo. Sequence these libraries.
- Bioinformatic Analysis: Use the dedicated GUIDE-seq software pipeline to map the sequenced tags back to the reference genome, identifying all sites of DSB formation [33].
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function/Benefit |
|---|---|
| Silencer Select siRNA | A pre-designed siRNA format guaranteed to silence target mRNA by ≥70% when two siRNAs to the same target are used, providing a reliable starting point [19]. |
| High-Fidelity Cas9 Variants | Engineered Cas9 proteins (e.g., HypaCas9, eSpCas9) with reduced tolerance for mismatches, significantly lowering off-target cleavage while maintaining on-target activity [34] [4]. |
| Chemically Modified gRNAs | Synthetic guide RNAs with modifications (e.g., 2'-O-methyl analogs) that increase stability and can reduce off-target editing [34]. |
| Positive Control siRNA | A critical control (e.g., targeting GAPDH) used to confirm that transfection reagents and conditions are working optimally in your specific cell type [19]. |
| Negative Control Probe (dapB) | A bacterial gene probe used in assays like RNAscope to confirm the specificity of signal and the absence of background staining [35]. |
Visualizing Workflows and Relationships
Off-Target Analysis Decision Workflow
siRNA Design Parameters for Specificity
FAQs on RNAi Off-Target Effects
What are the primary sequence-based factors that cause RNAi off-target effects?
Off-target effects in RNAi experiments are primarily governed by two key sequence-based factors: the overall percentage of sequence identity between the dsRNA and non-target genes, and the presence of long stretches of perfectly or almost perfectly matched bases. Research shows that dsRNAs with >80% overall sequence identity to a non-target gene can efficiently trigger its knockdown [22]. Furthermore, the presence of even short perfectly matched segments within the dsRNA is a major driver of off-target effects; a contiguous match of ≥16 base pairs (bp) is sufficient to trigger RNAi, and a segment of >26 bp with just one or two mismatches can also cause significant off-target knockdown [22].
How do the rules for dsRNA off-target effects differ from those for siRNA?
While both dsRNA and siRNA can cause specific off-target effects through complementary base pairing, the assessment of risk can differ due to the processing of long dsRNA into multiple siRNAs. For siRNA, the seed region (nucleotides 2-8 of the guide strand) can mediate off-target effects, and as few as 11 contiguous complementary nucleotides can induce knockdown of off-target genes [22]. For dsRNA, which is more suitable for pest control applications, the risk assessment must account for the potential for any of the resulting siRNAs to have high complementarity to an off-target transcript. This makes the presence of long, highly complementary stretches within the parent dsRNA a critical factor [22].
Can a dsRNA with low overall identity still cause an off-target effect?
Yes, experimental evidence confirms that a dsRNA with low overall sequence identity can still cause significant off-target knockdown if it contains a long enough stretch of perfectly or nearly perfectly matched sequence. One study found that a dsRNA sharing only 53% overall identity with a target gene still caused 44.6% knockdown of the target mRNA because it possessed a 36 bp stretch of contiguous matching bases [22]. This underscores the critical importance of analyzing local sequence homology in addition to global percentage identity when designing dsRNAs.
Besides sequence factors, what other variables influence off-target risk?
Other factors include the expression level and renewal rate of the non-target gene's expression products [22]. Genes with lower expression levels may be less susceptible to observable knockdown in some contexts. Furthermore, the biological function of the gene and the dynamics of the cellular network it operates in can also play a role, as compensation effects from related genes can sometimes lead to the upregulation of non-target transcripts [22].
Troubleshooting Guides
Problem: Unexpected Phenotype or High Mortality in Control Groups
Potential Cause: Widespread off-target effects leading to the silencing of multiple essential genes beyond the intended target.
Diagnosis and Solution:
- Check Sequence Specificity: Re-analyze your dsRNA sequence against the most current genome assembly for the target organism. Use the thresholds in Table 1 to check for potential off-target genes.
- Validate with qPCR: Measure the expression levels of the primary target gene and the top potential off-target genes (those with high overall identity or long contiguous matches). An unexpected knockdown of multiple genes confirms an off-target problem.
- Redesign dsRNA: Design a new dsRNA targeting a different region of your intended gene, ensuring it minimizes both overall sequence identity (aim for <80%) and eliminates long contiguous matches (≥16 bp) with all other genes.
Problem: Inconsistent Knockdown Efficiency Between Biological Replicates
Potential Cause: Variable off-target effects interacting with genetic or physiological differences between replicates.
Diagnosis and Solution:
- Standardize Delivery: Ensure the concentration and delivery method (e.g., injection, feeding) of dsRNA are perfectly consistent across all replicates.
- Profile Gene Expression: Conduct a broader gene expression analysis (e.g., RNA-seq) on a subset of variable and consistent replicates. Look for correlated silencing of off-target genes in samples with atypical phenotypes.
- Use Multiple dsRNAs: If possible, use two or more distinct dsRNAs targeting the same gene. If they produce the same on-target phenotype without the inconsistencies, the original dsRNA was likely the source of the problem.
The following tables consolidate the key experimental findings on sequence parameters governing RNAi specificity.
Table 1: Thresholds for dsRNA-Mediated Gene Silencing
| Parameter | Threshold for Efficient Silencing | Experimental Context |
|---|---|---|
| Overall Sequence Identity | >80% | Random mutagenesis of dsRNA against target genes in T. castaneum [22] |
| Perfectly Matched Contiguous Sequence | ≥16 bp | Mutational analysis and off-target evaluation [22] |
| Nearly Perfect Contiguous Sequence | >26 bp with one or two mismatches scarcely distributed | Single mismatches inserted between ≥5 bp matches; mismatched couplets between ≥8 bp matches [22] |
Table 2: Experimental Evidence of Off-Target Effects
| Target Gene | Off-Target Gene | Overall Identity | Key Contiguous Match Feature | Observed Knockdown |
|---|---|---|---|---|
| CYP6BQ6 | CYP6BK13 | 71% | 26 bp of perfectly matched sequence | Significant [22] |
| CYP6BQ6 | CYP6BK7 | 68% | 24 bp stretch with only two single mismatches | Significant [22] |
| CYP6BK13 | CYP6BK13 (via dsCYP6BK13-53) | 53% | 36 bp stretch of contiguous matching bases | 44.6% [22] |
Experimental Protocols
Protocol: In Silico Assessment of dsRNA Off-Target Risk
This protocol describes a bioinformatics workflow to predict the risk of off-target effects for a candidate dsRNA sequence before synthesis.
I. Materials and Reagents
- Candidate dsRNA Sequence: The proposed sequence for your experiment.
- Bioinformatics Software: Access to a local or web-based BLAST suite.
- Reference Genome: The annotated genome sequence for your target organism and for any critical non-target organisms (e.g., beneficial insects in a pest control context).
- Sequence Analysis Tool: Software capable of performing pairwise sequence alignments (e.g., EMBOSS needle, custom Python/Bioperl scripts).
II. Methodology
- Sequence Preparation: Format your candidate dsRNA sequence in FASTA format.
- BLAST Analysis: Perform a BLASTN search of the dsRNA sequence against the reference genome database. Use a low word size (e.g., 7) and disable the filter for low complexity regions to maximize sensitivity.
- Identify Potential Off-Targets: Compile a list of all genes with a BLAST hit meeting an initial E-value cutoff (e.g., < 10). This is your initial risk list.
- Calculate Global and Local Identity:
- For each gene on the risk list, perform a global pairwise alignment with the dsRNA to determine the overall percentage sequence identity.
- Flag any gene with >80% overall identity as high risk.
- Scan for Contiguous Matches:
- For each gene on the risk list, perform a local alignment or use a sliding window algorithm to identify the longest stretch of perfectly matched bases.
- Similarly, identify the longest stretch with one or two mismatches.
- Flag any gene containing a contiguous perfect match of ≥16 bp, or an almost perfect match of >26 bp with the mismatch pattern described in Table 1, as high risk.
- Final Risk Assessment: A candidate dsRNA should be considered specific only if it has no high-risk off-target genes based on the criteria in steps 4 and 5.
Protocol: Experimental Validation of Off-Target Knockdown
This protocol outlines how to experimentally confirm suspected off-target effects in a laboratory setting.
I. Materials and Reagents
- dsRNA: The candidate dsRNA and a negative control dsRNA (e.g., targeting GFP or a gene from a different organism).
- Experimental Organisms: The insects or cells used in your assay.
- RNA Extraction Kit: A standard kit for high-quality total RNA isolation.
- cDNA Synthesis Kit: A reverse transcription kit.
- qPCR Equipment and Reagents: SYBR Green or TaqMan master mix, and a real-time PCR system.
- Gene-Specific Primers: Validated qPCR primers for the intended target gene and for the potential off-target genes identified in the in silico assessment.
II. Methodology
- Treatment and Sampling: Treat experimental groups with the candidate dsRNA and the negative control dsRNA using your standard method (e.g., microinjection or feeding). Perform the treatment in at least three biological replicates.
- RNA Extraction and cDNA Synthesis: At an appropriate timepoint post-treatment (e.g., 24-72 hours), harvest tissue or cells and extract total RNA. Synthesize cDNA from equal amounts of RNA for each sample.
- Quantitative PCR (qPCR):
- Perform qPCR reactions using primers for your target gene and the potential off-target genes.
- Include at least one stable reference gene for normalization.
- Use a standard relative quantification method (like the 2^(-ΔΔCq) method) to calculate the fold-change in gene expression in the treated group compared to the control group.
- Data Interpretation: A statistically significant reduction (e.g., >50%) in the mRNA level of a gene other than the primary target confirms an off-target effect. The results should correlate with your in silico predictions.
Signaling Pathways and Workflows
Decision Workflow for dsRNA Specificity
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Resources for RNAi Specificity Research
| Item | Function in Research | Application Note |
|---|---|---|
| Long dsRNA (200-500 bp) | The primary trigger for the RNAi pathway in insects. More effective than siRNA for many insect systems [22]. | Should be designed using the specificity rules outlined above. Can be synthesized in vitro. |
| Bioinformatics Software (BLAST) | To identify potential off-target genes by searching for sequences with high similarity to the dsRNA [22]. | A critical first step for any dsRNA-based experiment. Use sensitive parameters. |
| qPCR Primers & Reagents | To quantitatively measure the knockdown efficiency of both the target gene and potential off-target genes [22]. | Essential for experimental validation of specificity. Requires stable reference genes. |
| Reference Genome Assembly | A high-quality, annotated genome for the target organism is necessary for a comprehensive in silico off-target prediction [22]. | The quality of the genome directly impacts the reliability of off-target predictions. |
A significant challenge in RNA interference (RNAi) research and therapy is the phenomenon of off-target effects, where a small interfering RNA (siRNA) unintentionally silences genes other than its intended target. This occurs primarily through a mechanism that mimics microRNA (miRNA) activity: the siRNA guide strand can bind to partially complementary sequences, particularly in the 3' untranslated regions (UTRs) of off-target mRNAs [36]. The seed region (nucleotides 2-8 from the 5' end of the guide strand) is critically important for this unintended binding [37] [36]. Off-target effects can lead to misleading results in functional genomics studies and pose a substantial risk for therapeutic applications. Consequently, developing robust strategies to suppress off-target silencing is a central focus in advancing RNAi technology.
FAQs: Understanding 2'-O-Methyl Modifications
Q1: How does a 2'-O-methyl (2'-O-Me) ribosyl substitution specifically reduce off-target effects?
A 2'-O-methyl (2'-O-Me) modification involves adding a methyl group to the 2' hydroxyl group of the ribose sugar in an RNA nucleotide [38]. When placed at specific positions on the siRNA guide strand—most critically at position 2—this chemical alteration disrupts the stable binding between the siRNA seed region and off-target mRNAs that have only partial complementarity [37]. It functions by weakening the RNA-RNA interaction in the seed region, thereby preventing the silencing of these unintended transcripts. Importantly, because the binding to perfectly matched, on-target mRNAs is more stable, this modification does not significantly compromise the intended gene silencing effect [37].
Q2: What is the optimal position for incorporating a 2'-O-methyl modification to minimize off-targeting?
Research has demonstrated a sharp position dependence for the 2'-O-methyl modification. Introducing the modification at position 2 of the guide strand has the most potent effect in reducing off-target silencing [37]. While modifications at positions 1 and 2 together are also effective, modification of position 1 alone shows little benefit [37]. The high specificity for position 2 suggests a unique role for this nucleotide in the guide strand that is distinct from its simple base-pairing function.
Q3: Do 2'-O-methyl modifications affect the on-target potency of an siRNA?
When applied correctly, 2'-O-methyl modifications do not negatively impact the silencing of the intended, perfectly matched target. Studies have confirmed that siRNAs with a 2'-O-Me modification at position 2 of the guide strand maintain full on-target silencing efficacy across a wide range of concentrations [37].
Q4: How do 2'-O-methyl modifications compare to seed region mismatches for improving specificity?
While introducing mismatches in the seed region can also reduce off-target effects, this approach has a broader position dependence and a major drawback: it can create a new, non-native seed sequence. This new sequence may then silence a new set of off-target transcripts complementary to the mutated sequence [37]. In contrast, the 2'-O-methyl modification at position 2 suppresses silencing of the original off-target transcripts without inducing a new off-target signature, making it a superior strategy for enhancing siRNA specificity [37].
Troubleshooting Guides
Problem: Persistent Off-Target Effects After Modification
Potential Causes and Solutions:
Cause 1: Incorrect Modification Placement The efficacy of the 2'-O-methyl modification is highly position-specific.
- Solution: Verify that the modification is placed on the guide (antisense) strand at position 2. Consider also modifying position 1 of the guide strand and positions 1 and 2 of the passenger (sense) strand to further reduce passenger strand activity [37].
Cause 2: High siRNA Concentration Even with modifications, excessively high siRNA concentrations can saturate the RNAi machinery and exacerbate residual off-target binding.
- Solution: Perform a dose-response curve. Use the lowest possible siRNA concentration that still achieves the desired level of on-target knockdown. Testing concentrations between 5 nM and 100 nM is generally recommended [19].
Cause 3: Inherently Promiscuous Seed Sequence Some seed sequences may have high complementarity to many transcripts.
- Solution: Redesign the siRNA. Use multiple siRNAs targeting the same gene to confirm that observed phenotypes are due to on-target effects. Employ computational tools (e.g., BLAST) to screen for homology with other genes during the design phase [36].
Problem: Poor On-Target Knockdown After Modification
Potential Causes and Solutions:
Cause 1: Over-modification Excessively modifying the guide strand, especially outside the seed region, can interfere with its loading into RISC or its ability to cleave the target mRNA.
Cause 2: Assay-Related Issues The problem may not lie with the modification itself but with the experimental setup for measuring knockdown.
- Solution: Check mRNA levels using a sensitive method like quantitative RT-PCR. Ensure the qRT-PCR assay target site is not too far from the siRNA cut site (>3,000 bases away) to avoid issues with alternative splicing. Always use a validated positive control siRNA to confirm transfection efficiency and assay functionality [19].
Experimental Protocols
Protocol 1: Validating the Reduction of Off-Target Effects Using Microarrays
This protocol is based on the seminal study that established the role of 2'-O-methyl modifications [37].
1. Design and Synthesis:
- Design the siRNA against your target of interest.
- Synthesize two versions: an unmodified siRNA and a modified siRNA with a 2'-O-methyl ribosyl substitution at position 2 of the guide strand. Ensure the 5'-end of the guide strand is phosphorylated.
- Recommended Control: Include a 2'-O-methyl modification at positions 1 and 2 of the sense strand to minimize its contribution to off-target silencing.
2. Transfection:
- Culture appropriate cells (e.g., HeLa cells).
- Transfect cells with both the modified and unmodified siRNA duplexes. A range of concentrations (e.g., 10-50 nM) is recommended to assess dose-dependency.
3. Gene Expression Analysis:
- Time Point: Harvest cells 48 hours post-transfection.
- Method: Isolate total RNA and analyze global gene expression using microarray technology.
- Comparison: Compare the expression profiles of cells treated with unmodified siRNA versus modified siRNA.
4. Data Interpretation:
- On-Target Efficacy: Confirm that silencing of the intended target mRNA is comparable between modified and unmodified siRNAs.
- Off-Target Reduction: Identify transcripts significantly down-regulated by the unmodified siRNA but not regulated (or regulated to a lesser extent) by the modified siRNA. The study by Jackson et al. showed that this modification can reduce silencing of approximately 80% of off-target transcripts, with the magnitude of their regulation reduced by an average of 66% [37].
Protocol 2: Confirming Reduction of Off-Target Effects at the Protein Level
1. siRNA Treatment:
- Perform a dose titration of both modified and unmodified siRNA, as described in Protocol 1.
2. Protein Measurement:
- Time Point: Harvest cells for protein analysis at 48-72 hours post-transfection (or later, depending on the protein's half-life).
- Method: Use Western blotting with commercial antibodies to measure levels of both the on-target protein and a selected off-target protein.
- Selection of Off-Target Protein: Choose a protein whose transcript was identified as an off-target in microarray data and shares seed region complementarity with the siRNA [37].
3. Data Analysis:
- Compare the potency and maximal extent of silencing for both the on-target and off-target proteins between the modified and unmodified siRNAs. The modification should specifically reduce the silencing of the off-target protein without affecting the on-target protein [37].
Data Presentation
Table 1: Quantitative Impact of 2'-O-Methyl Modification on siRNA Specificity
| Parameter | Unmodified siRNA | 2'-O-Me Modified siRNA | Experimental Context |
|---|---|---|---|
| On-Target Silencing | Unaffected / Full efficacy | Unaffected / Full efficacy | MAPK14 siRNA; mRNA level [37] |
| Off-Target Transcripts Regulated | Baseline (100%) | Reduced by ~80% | 10 different siRNAs; microarray [37] |
| Magnitude of Off-Target Regulation | Baseline (100%) | Reduced by ~66% (average) | 10 different siRNAs; microarray [37] |
| Off-Target Protein Silencing | Potent silencing | Potent reduction in silencing | PIK3CB siRNA; YY1 protein level [37] |
| False-Positive Phenotypes | Observed (e.g., growth inhibition) | Significantly reduced | Functional cell-based assays [37] |
Table 2: The Scientist's Toolkit: Key Reagents for Specific siRNA Design
| Tool / Reagent | Function / Description | Role in Suppressing Off-Targets |
|---|---|---|
| 2'-O-Methyl (2'-O-Me) Modification | A common chemical modification that alters the ribose sugar of specific nucleotides [38]. | Weaken RISC binding to partially complementary mRNAs when placed in the seed region (esp. position 2) of the guide strand [37]. |
| Phosphorothioate (PS) Linkage | Replaces a non-bridging oxygen with sulfur in the phosphate backbone [38]. | Increases nuclease resistance and improves pharmacokinetics, allowing for lower effective doses which can reduce off-target exposure [38] [36]. |
| Silencer Select/Validated siRNAs | Commercially pre-designed and validated siRNAs (Thermo Fisher) [19]. | Providers often guarantee a minimum level of on-target knockdown (e.g., ≥70%), and their design algorithms incorporate strategies to enhance specificity. |
| Computational Design Tools (BLAST, ML models) | Algorithms to predict siRNA efficacy and off-target potential [36]. | Identifies and eliminates siRNA candidates with high sequence homology to off-target transcripts during the design phase. |
| Positive Control siRNA | A validated siRNA known to robustly knock down a common gene (e.g., GAPDH) [19]. | Critical for troubleshooting; confirms that experimental conditions (transfection, RNA isolation, qPCR) are working, ensuring any lack of effect is not due to technical failure. |
| Negative Control siRNA | A scrambled sequence siRNA with no specific target in the transcriptome [19]. | Serves as a baseline to normalize mRNA knockdown data and identify non-sequence-specific effects. |
Diagram and Visualizations
Diagram 1: Mechanism of 2'-O-Methyl Modification in Blocking Off-Target Effects
Diagram 2: Experimental Workflow for Off-Target Validation
RNA interference (RNAi) is a powerful tool for gene silencing, but its utility in both research and therapeutics can be compromised by off-target effects. These unintended silencing events occur when the small interfering RNA (siRNA) directs the RNA-induced silencing complex (RISC) to cleave or repress non-target mRNAs. The primary mechanisms include:
- Passenger Strand Off-Targeting: The sense (passenger) strand of the siRNA can be loaded into RISC instead of the guide strand, leading to silencing of genes with complementarity to the sense sequence [3] [39].
- Seed Region-Based Off-Targeting: The guide strand can behave like a microRNA (miRNA). A 6-8 nucleotide "seed" sequence (bases 2-7/8 of the guide strand) can bind to partially complementary sites, typically in the 3' untranslated region (3' UTR) of off-target mRNAs, leading to their translational repression or degradation [3] [40] [36].
The following diagram illustrates the core RNAi pathway and the primary sources of these off-target effects.
Frequently Asked Questions (FAQs) on Advanced siRNA Design
Q1: What are the main advantages of using asymmetric siRNAs (aiRNAs) over traditional siRNAs?
Asymmetric siRNAs are designed with a full-length guide strand (typically 19-21 nt) and a truncated passenger strand (commonly 15-16 nt). This design offers two key benefits [39] [41] [42]:
- Improved Strand Specificity: The shortened passenger strand is too small to be efficiently loaded into RISC, virtually eliminating off-target effects caused by the passenger strand.
- Maintained Potency: Well-designed aiRNAs can silence their targets as efficiently as, and in some cases better than, traditional 19+2 siRNAs.
Q2: How does pooling multiple siRNAs reduce off-target effects, and what is the critical design rule?
siRNA pools reduce off-target effects through dilution of individual seed sequences. When a pool of siRNAs, each with a unique seed sequence, is used at a given total concentration, the effective concentration for any single seed sequence is dramatically lowered. This minimizes the miRNA-like off-target silencing for any individual seed in the pool [40]. The critical design rule is to ensure the pool is composed of siRNAs with distinct seed sequences targeting the same mRNA.
Q3: My siRNA shows strong mRNA knockdown but no phenotype. Could this be an off-target effect?
Yes. It is possible that the observed phenotype in your initial experiment was primarily driven by seed-based off-target effects silencing critical genes in the pathway, rather than the on-target knockdown itself [40]. Always confirm phenotypes using multiple, distinct siRNAs or an alternative gene silencing method.
Q4: At what siRNA concentration should I perform my experiments to minimize off-target effects?
There is a narrow window where on-target silencing is effective but off-target effects are minimized. Off-target effects are largely absent at very low concentrations (e.g., 100 pM) but so is on-target silencing [40]. The goal is to use the lowest possible concentration that still achieves robust on-target knockdown. Perform a dose-response curve to find this optimum, which is often in the low nanomolar range (1-5 nM) [19] [40].
Troubleshooting Guides
Problem: High Off-Target Effects Suspected in Experiments
| Symptom | Possible Cause | Solution |
|---|---|---|
| Phenotype observed with a single siRNA but not with other siRNAs targeting the same gene [43]. | Seed-based off-target effects from the first siRNA. | Use at least two, and preferably three, siRNAs with different seed sequences for the same target [43] [44]. |
| Gene expression profiling shows deregulation of genes without perfect complementarity to the siRNA [3]. | miRNA-like off-target effects via the guide strand's seed sequence. | Switch to asymmetric siRNAs (aiRNA) to eliminate passenger strand effects and use lower concentrations to minimize guide strand seed effects [39] [41]. |
| Poor correlation between mRNA knockdown and phenotypic readout [43]. | Off-target effects are driving the phenotype, or the protein has a slow turnover rate. | Validate with multiple siRNAs. For protein analysis, allow more time post-transfection for depletion [19]. |
Problem: Low Gene Silencing Efficacy
| Symptom | Possible Cause | Solution |
|---|---|---|
| No knockdown (<10%) with any siRNA against your target. | The assay or transfection is not working. | Use a validated positive control siRNA (e.g., targeting GAPDH) to confirm transfection efficiency and assay functionality [19]. |
| Low knockdown with one siRNA, but others work. | The siRNA design is suboptimal or the target region is inaccessible. | Always test multiple (3-4) siRNAs against different regions of the target mRNA [43]. |
| mRNA is knocked down, but protein level is unchanged. | The protein has a long half-life (slow turnover rate). | Perform a time-course experiment and analyze protein levels at later time points (e.g., 72 or 96 hours) post-transfection [19]. |
Experimental Protocols for Validating and Implementing Advanced Triggers
Protocol 1: Evaluating Asymmetric siRNA (aiRNA) Efficacy and Specificity
This protocol outlines steps to compare the performance of novel aiRNAs against traditional siRNAs.
1. Design and Synthesis:
- Design a traditional 19+2 siRNA (19 bp duplex with 2-nt 3' overhangs) against your target [39].
- Design a corresponding aiRNA with a 19-nt guide strand and a 15-16 nt passenger strand [41] [42].
- Control: Include a non-targeting siRNA with a scrambled sequence.
2. Transfection and mRNA Analysis:
- Transfert cells (e.g., HEK293T or GHOST cell lines) with a concentration series (e.g., 0.1 nM, 1 nM, 5 nM, 10 nM) of both the traditional siRNA and the aiRNA [39].
- 48 hours post-transfection, harvest total RNA using a reagent like TRIzol [39].
- Perform reverse transcription followed by quantitative PCR (qPCR) to measure target mRNA levels. Use the ΔΔCt method for relative quantification, normalizing to a housekeeping gene (e.g., GAPDH) [39] [19].
3. Assessing Strand Specificity:
- Use a dual-luciferase reporter system. Clone sequences that are perfectly complementary to the guide strand and the passenger strand into separate reporter vectors.
- Co-transfect each reporter with the siRNA or aiRNA.
- Measure luciferase activity. Effective aiRNAs should only silence the reporter containing the guide strand complement, not the passenger strand complement [39].
Protocol 2: Implementing siRNA Pools to Minimize Seed-Based Off-Targets
1. Designing a Smart Pool:
- Select 3-5 pre-validated siRNAs that target different regions of your gene of interest.
- Critical Step: Use BLAST or a similar tool to verify that each siRNA has a unique seed sequence (nucleotides 2-8 of its guide strand) [40] [36].
2. Transfection and Phenotypic Analysis:
- Prepare two sets of transfections:
- Set A: Individual siRNAs (e.g., at 5 nM each).
- Set B: The siRNA pool, where the total concentration is 5 nM, meaning each individual siRNA is present at a lower concentration (e.g., 1.67 nM for a pool of 3) [43].
- Perform your phenotypic assay (e.g., cell proliferation, apoptosis, imaging) 48-96 hours post-transfection.
- Key Analysis: Compare phenotypes between the individual siRNAs and the pool. A true on-target phenotype should be consistent across most individual siRNAs and the pool. If one individual siRNA shows a strong, unique phenotype, it is likely driven by its specific seed-based off-targets [43].
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| Asymmetric siRNA (aiRNA) | Gene silencing trigger with truncated passenger strand to prevent its loading into RISC. | Reducing passenger strand-mediated off-target effects in therapeutic siRNA development [41] [42]. |
| Complex siRNA Pools (siPOOLs) | A pool of many (e.g., 30) siRNAs targeting the same gene, each with a unique seed sequence. | Diluting individual seed effects for highly specific phenotypic screening in functional genomics [40]. |
| Chemically Modified siRNAs | Incorporation of modified nucleotides (e.g., 2'-O-methyl) to improve stability and reduce immunogenicity. | Enhancing serum stability and potency of siRNAs for in vivo applications [36]. |
| Positive Control siRNA | A validated siRNA known to robustly knock down a common gene (e.g., GAPDH). | Verifying transfection efficiency and assay functionality in every experiment [19]. |
| Negative Control siRNA | A non-targeting siRNA with a scrambled sequence that does not target any known gene. | Establishing a baseline for gene expression and phenotypic changes in an experiment [19] [43]. |
Data Presentation: Comparative Performance of siRNA Formats
The following table summarizes quantitative data from key studies comparing traditional siRNAs, asymmetric siRNAs, and siRNA pools.
Table 1: Comparison of siRNA Trigger Designs for Specificity and Efficacy
| siRNA Format | Reported On-Target Efficacy | Impact on Off-Target Effects | Key Supporting Evidence |
|---|---|---|---|
| Traditional siRNA (19+2) | Variable; ~82% of algorithm-designed siRNAs achieve >70% knockdown [43]. | High; passenger strand and seed-based effects are common [3] [40]. | Microarray analysis shows siRNA transfection deregulates many off-target genes [39]. |
| Asymmetric siRNA (aiRNA) | Can match or exceed traditional siRNA potency; effective at doses as low as 100 pM [41]. | Greatly reduced; short passenger strand prevents its entry into RISC, cutting sense-strand off-targets [39] [41]. | Reporter assays show aiRNA only silences antisense targets, not sense strand targets [39]. |
| siRNA Pools (Simple) | Can be compromised if less active siRNAs interfere with potent ones [43]. | Still high; if seeds are not unique, the pool concentrates multiple off-target profiles. | Phenotypic screens show high false positive and false negative rates (>40%) [43]. |
| Complex siRNA Pools (siPOOL) | High and robust; combines many effective siRNAs for consistent on-target knockdown [40]. | Very low; unique seeds dilute individual seed concentration below effective threshold [40]. | Phenotypes correlate highly between siRNAs targeting the same gene, not between siRNAs sharing a seed [40]. |
Troubleshooting Guide: Addressing Common LNP and Conjugation Issues
This guide addresses frequent challenges researchers encounter when developing and working with lipid nanoparticles (LNPs) and targeted conjugates for RNAi experiments.
Problem 1: Low Gene Silencing Efficiency in Target Cells
- Potential Cause: Inefficient cellular uptake or poor endosomal escape of the LNP.
- Solution: Optimize the LNP formulation. Ensure the ionizable lipid has an appropriate pKa (typically between 6.2-6.5) for efficient endosomal disruption. Supplementing formulations with helper lipids like DOPE can enhance membrane fusion and escape [45]. For targeted delivery, confirm the activity and orientation of the targeting ligand on the LNP surface [46].
Problem 2: High Off-Target Effects (Liver Accumulation)
- Potential Cause: Non-specific uptake by the mononuclear phagocyte system (MPS) and adsorption of serum proteins like ApoE, which naturally directs particles to the liver [47] [48].
- Solution: Actively target LNPs to extrahepatic tissues. Employ exogenous targeting ligands, such as antibodies or GalNAc, to redirect LNPs away from hepatic pathways [47] [49]. Adjust the PEG-lipid content and chain length in the LNP formulation to reduce non-specific interactions and "shield" the particle until it reaches its target [45].
Problem 3: Inconsistent Targeting with Antibody-Conjugated LNPs
- Potential Cause: Loss of antibody binding affinity due to random orientation or harsh conjugation chemistry on the LNP surface [46].
- Solution: Use an oriented conjugation strategy. Implement a capture system, such as an anti-Fc nanobody (e.g., TP1107) incorporated into the LNP, which binds the antibody's Fc region. This preserves the antigen-binding domains and improves targeting efficiency by more than eightfold compared to random conjugation methods [46].
Problem 4: siRNA Instability and Rapid Clearance
- Potential Cause: Degradation by serum nucleases or activation of the innate immune response [45].
- Solution: Incorporate chemically modified nucleotides into the siRNA design. Modifications such as 2'-O-methyl (2'-OMe) or 2'-fluoro (2'-F) can significantly enhance stability and reduce immunogenicity without compromising silencing activity [50].
Frequently Asked Questions (FAQs)
Q1: Why do most standard LNP formulations primarily target the liver? The liver has a fenestrated endothelium, allowing nanoparticles to easily access hepatocytes. Furthermore, LNPs, particularly those with ionizable lipids, adsorb apolipoprotein E (ApoE) from the blood. ApoE acts as an endogenous targeting ligand, binding to receptors on hepatocytes (like the LDL receptor) and mediating efficient cellular uptake. This process is a primary reason for strong hepatic accumulation [47].
Q2: What are the main strategies to achieve organ-selective delivery beyond the liver? The two primary strategies are:
- Endogenous Targeting: Relying on the natural tropism of the LNP, often by manipulating the LNP's lipid composition, size, and surface charge to influence which serum proteins (the "protein corona") adsorb to it, thereby directing it to specific tissues or cells [47] [48].
- Exogenous Targeting: Actively conjugating a targeting ligand to the LNP surface. This includes:
- Antibodies or antibody fragments that recognize cell-specific surface markers [46] [49].
- Small molecule ligands like GalNAc, which has a high affinity for the asialoglycoprotein receptor (ASGPR) on hepatocytes and is a well-established approach for liver targeting [47].
- Peptides or other molecules that bind to receptors on the target cell type [48].
Q3: How can I confirm that observed phenotypic effects are due to on-target gene silencing and not off-target effects? The most robust validation is an RNAi rescue experiment. This involves co-expressing an siRNA-resistant version of the target gene (e.g., a synthetic gene with synonymous codon changes) alongside the siRNA. If the phenotypic effect is reversed, it confirms the effect is due to specific silencing of the intended gene [51]. Other critical methods include using multiple, distinct siRNAs against the same target and profiling global gene expression to identify siRNA-specific, rather than gene-specific, changes [51] [50].
Q4: What is the impact of the "seed region" in siRNA off-target effects? The seed region (nucleotides 2-8 from the 5' end of the guide strand) is critical for initial target recognition. Partial complementarity between this seed region and non-target mRNAs can lead to miRNA-like off-target effects, where translation is inhibited or the mRNA is degraded. This is a major contributor to off-targeting in RNAi experiments [3] [50].
Q5: Our antibody-conjugated LNPs show good in vitro binding but poor in vivo performance. What could be wrong? This is often related to the in vivo stability and orientation of the antibody on the LNP. Traditional conjugation chemistries (e.g., NHS ester reactions with lysines) can randomize antibody orientation, blocking antigen-binding sites and reducing affinity. Furthermore, the antibody may detach from the LNP surface in circulation. Switching to a stable, oriented capture system (like nanobody-based Fc capture) can significantly improve in vivo performance by ensuring optimal antibody presentation and stability [46].
The following tables summarize key quantitative findings from recent literature on LNP performance and off-target effect mitigation.
Table 1: Efficacy of Targeted LNP Delivery Systems
| Targeting System | Target Cell/Receptor | Reported Outcome | Key Metric |
|---|---|---|---|
| Anti-Fc Nanobody Capture [46] | T cells | Enhanced protein expression vs. conventional methods | >8x higher |
| Anti-Fc Nanobody Capture [46] | T cells | Enhanced protein expression vs. non-targeted LNPs | >1,000x higher |
| ApoE-mediated targeting [47] | Hepatocytes (in HeLa cells) | Enhanced cellular uptake with ApoE supplementation | ~20-fold increase |
| GalNAc-based exogenous targeting [47] | Hepatocytes (via ASGPR) | Potent, ligand-dependent gene silencing | Demonstrated in vivo |
| TfR1-targeted Antibody-LNP [49] | Muscle | Increased drug exposure in muscle tissue | >15-fold higher |
Table 2: Strategies to Minimize RNAi Off-Target Effects
| Mitigation Strategy | Method | Key Outcome / Rationale |
|---|---|---|
| Rational siRNA Design [51] [50] | Bioinformatics tools to select highly specific sequences; avoid regions with high homology to other genes. | Minimizes base-pairing with non-target mRNAs. |
| Chemical Modifications [3] [50] | Incorporate 2'-O-methyl (2'-OMe) groups in the seed region of the guide strand. | Reduces miRNA-like off-target effects without compromising on-target activity. |
| Optimal siRNA Concentration [51] | Titrate siRNA to use the lowest effective concentration (e.g., < 30 nM). | Mitigates nonspecific silencing effects often seen at high concentrations (≥ 100 nM). |
| Multiple siRNA Validation [51] | Use two or more distinct siRNAs against the same target gene. | Phenotypes consistent across siRNAs are likely on-target; siRNA-specific effects are off-target. |
Experimental Protocols
Protocol 1: Validating siRNA Specificity Using a Rescue Experiment This is a definitive method to confirm that an observed phenotype is due to specific silencing of your target gene [51].
- Design a Rescued Construct: Synthesize a cDNA of your target gene that is resistant to the siRNA. This is achieved by introducing silent mutations into the siRNA target site without altering the amino acid sequence.
- Co-transfection: In your cell model, perform the following transfections:
- Group A: Non-targeting control siRNA.
- Group B: siRNA against your target gene.
- Group C: siRNA against your target gene + the siRNA-resistant expression construct.
- Analysis:
- mRNA Level: Use qRT-PCR with two primer sets—one that detects only the endogenous (wild-type) mRNA and another that detects only the rescued (mutated) mRNA.
- Protein Level: Assess protein levels via Western blot or immunofluorescence.
- Phenotype: Measure the functional phenotype (e.g., cell viability, migration, etc.).
- Interpretation: Successful rescue is demonstrated in Group C by the restoration of protein levels and a reversal of the phenotypic effect seen in Group B, confirming the specificity of the siRNA.
Protocol 2: Incorporating a Targeting Nanobody onto LNPs for Oriented Antibody Display This protocol describes a method for creating actively targeted LNPs with optimal antibody orientation [46].
- Nanobody Engineering: Express a nanobody (e.g., TP1107) with a site-specific incorporation of an azide-bearing synthetic amino acid (e.g., p-azido-phenylalanine) at a position determined by structural analysis to be optimal for conjugation without disrupting Fc binding.
- Conjugate to Lipid: React the modified nanobody with a DBCO-functionalized lipid (e.g., DSPE-PEG2000-DBCO) via a click chemistry reaction. Purify the nanobody-lipid conjugate.
- LNP Formulation: Prepare LNPs using standard methods (e.g., microfluidics) containing an ionizable lipid, helper lipids (DSPC, cholesterol), and a PEG-lipid.
- Surface Functionalization: Incubate the pre-formed LNPs with the nanobody-lipid conjugate, allowing it to insert into the LNP membrane via its lipid anchor.
- Antibody Capture: Finally, add the desired antibody to the nanobody-decorated LNPs. The nanobody will capture the antibody via its Fc region, ensuring optimal orientation for target binding. No further purification is needed.
Signaling Pathways and Workflows
siRNA Off-target Validation Pathway
LNP Targeted Delivery Mechanisms
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for LNP and RNAi Specificity Research
| Item | Function / Application |
|---|---|
| Ionizable Lipids (e.g., DLin-MC3-DMA, SM102) | Core component of LNPs; enables siRNA encapsulation and endosomal escape. Their pKa is critical for in vivo performance and tissue tropism [45] [47]. |
| PEG-Lipids (e.g., DMG-PEG2000, DSPE-PEG2000) | Provides a hydrophilic stealth layer, modulating LNP stability, circulation time, and biodistribution. Shorter anchors (C14) allow for quicker dissociation and targeting ligand exposure [46] [45]. |
| Anti-Fc Nanobodies (e.g., TP1107) | Used for optimal, oriented antibody capture on LNP surfaces without chemical modification, significantly improving targeting efficiency [46]. |
| DBCO-PEG-DSPE Lipid | A conjugation-ready lipid used with azide-modified targeting ligands (like nanobodies) for click chemistry-based surface functionalization of pre-formed LNPs [46]. |
| GalNAc (N-acetylgalactosamine) | A high-affinity small molecule ligand for the asialoglycoprotein receptor (ASGPR); used for targeted delivery of siRNA conjugates or LNPs to hepatocytes [47]. |
| Chemically Modified siRNAs (2'-OMe, 2'-F) | siRNA with modified ribose sugars to enhance nuclease stability, reduce immunogenicity, and critically, to minimize seed-region mediated off-target effects [3] [50]. |
| siRNA-Rescue Gene Constructs | Synthetic genes with synonymous codon changes used in rescue experiments to definitively validate the specificity of an siRNA's phenotypic effect [51]. |
Troubleshooting and Refining RNAi Experiments: A Practical Optimization Guide
Core Concept: How siRNA Dose Influences Off-Target Effects
FAQ: Why does reducing siRNA concentration minimize off-target effects?
High concentrations of siRNA increase the likelihood that the RNA-induced silencing complex (RISC) will load the siRNA and target mRNAs with only partial complementarity, particularly to short 6-8 nucleotide "seed" sequences [3] [52]. This miRNA-like effect can unintentionally silence hundreds of non-target genes [52]. Using the lowest effective concentration reduces this promiscuous binding while maintaining sufficient levels for effective on-target gene knockdown [53].
FAQ: What is the typical effective concentration range for siRNA?
While the optimal concentration must be determined empirically for each experiment, a general starting range is 5 nM to 100 nM [53]. The goal of titration is to find the lowest concentration within this range that achieves the desired on-target knockdown.
Experimental Protocol: siRNA Titration for Off-Target Minimization
The following workflow provides a systematic method for determining the minimum effective dose for your siRNA experiments.
Detailed Methodology
Basic Protocol: Titration and Evaluation of siRNA Dose-Response [54] [55] [53]
- siRNA Preparation: Begin with a high-concentration stock solution of your validated siRNA. Prepare a serial dilution in nuclease-free buffer to create a range of working concentrations. A typical series includes 100 nM, 50 nM, 25 nM, 10 nM, and 5 nM [53].
- Cell Transfection:
- Plate cells at an optimal density (typically 60-90% confluency) to ensure healthy growth and high transfection efficiency [55] [53].
- Using a transfection reagent optimized for siRNA delivery (e.g., Lipofectamine RNAiMAX), complex the siRNA at each concentration in the dilution series [55].
- Transfer the complexes to the plated cells. Include essential controls: a non-targeting siRNA (negative control), a known effective siRNA (positive control), and a mock-transfected well (transfection reagent only) [53].
- Incubation and Time Course: Incubate the cells for 24-96 hours. The earliest silencing effects are typically observable at 24 hours, but the maximum effect may take 48-72 hours. The duration can be cell-type and target-dependent [53].
- Efficacy and Specificity Analysis:
- On-Target Efficacy: Measure the reduction in target mRNA levels using qRT-PCR and/or target protein levels via Western blot or immunofluorescence [53].
- Off-Target Assessment: To directly test for seed-mediated off-target effects, measure the expression levels of genes (e.g., via qRT-PCR) whose 3'UTRs contain matches to the siRNA's seed region (nucleotides 2-7 or 2-8 of the guide strand) [52].
Data Interpretation and Decision Matrix
The table below summarizes how to interpret the results from the titration experiment to identify the optimal siRNA concentration.
| siRNA Concentration | On-Target Knockdown | Off-Target Signature | Interpretation & Action |
|---|---|---|---|
| High (e.g., 50-100 nM) | Strong (>90%) | Significant | Unacceptable. High off-target risk. Proceed to lower concentrations. |
| Medium (e.g., 10-25 nM) | Strong (>80%) | Low/Undetectable | Ideal. This is the Minimum Effective Dose. Use for future experiments. |
| Low (e.g., 5 nM) | Suboptimal (<70%) | Low/Undetectable | Ineffective. On-target efficacy is insufficient. Increase concentration. |
Advanced Troubleshooting FAQs
FAQ: After titration, my on-target knockdown is still insufficient. What can I do?
- Verify siRNA Design: Ensure your siRNA has a GC content between 30-50%, is 21-23 nt in length, and lacks homology to other coding sequences [53].
- Check Transfection Efficiency: Use a fluorescently labeled negative control siRNA to visualize and quantify the percentage of cells that are taking up the siRNA [55] [53].
- Re-optimize Transfection Parameters: Critically evaluate cell density, the amount of transfection reagent, and the complex formation time, as these are the most common points of failure [55] [8].
FAQ: How can I be sure that observed phenotypic changes are due to on-target effects?
- Use Multiple siRNAs: The gold standard for confirming on-target effects is to demonstrate that two or more distinct siRNAs targeting the same gene produce the same phenotypic outcome [52].
- Correlate Knockdown with Phenotype: Ensure that the siRNA causing the strongest mRNA/protein knockdown also produces the strongest phenotype. If a poorly functional siRNA gives better knockdown than a functional one, the effect is likely off-target [52].
- Rescue Experiment: Express a functional, siRNA-resistant version of the target gene. If the phenotype is reversed, the effect is on-target.
FAQ: Beyond titration, what other strategies can reduce off-target effects?
- Chemical Modifications: Incorporate acyclic artificial nucleic acids (e.g., Serinol Nucleic Acid, SNA) or 2'-O-methyl modifications into the siRNA sense and antisense strands. These modifications can enhance strand selectivity for RISC loading and reduce miRNA-like off-targeting without compromising on-target activity [54] [3].
- Pooling siRNAs: Using a pool of several siRNAs against the same target, each at a low concentration, can achieve effective on-target knockdown while diluting out the individual seed-mediated off-target effects of any single siRNA [3].
| Item | Function & Rationale |
|---|---|
| Lipofectamine RNAiMAX | A popular transfection reagent specifically formulated for high-efficiency delivery of small RNAs with low cytotoxicity [55]. |
| Fluorescently Labeled siRNA | A negative control siRNA used to visually optimize transfection protocols and quantify transfection efficiency via microscopy or flow cytometry [55] [53]. |
| Validated Positive Control siRNA | An siRNA targeting a common housekeeping gene (e.g., GAPDH) used to confirm that the entire transfection and assay system is working properly [55]. |
| Non-Targeting Scrambled siRNA | The critical negative control that accounts for non-sequence-specific effects of introducing siRNA and transfection reagents into cells [53]. |
| Serinol Nucleic Acid (SNA) | An acyclic artificial nucleic acid used to chemically modify siRNAs, improving strand selectivity and suppressing off-target effects [54]. |
| RNase Decontamination Solution | Essential for maintaining an RNase-free work environment to prevent degradation of siRNA stocks, which can reduce efficacy and introduce experimental variability [53]. |
FAQ: Identifying and Troubleshooting RNAi Off-Target Effects
What are the primary causes of off-target effects in RNAi experiments?
Off-target effects in RNAi experiments occur when small interfering RNAs (siRNAs) silence genes other than the intended target. Two main mechanisms are responsible for this:
miRNA-like off-target effects: This is the most common cause, occurring when the "guide strand" of the siRNA exhibits partial complementarity to non-target mRNAs. Similar to endogenous microRNAs (miRNAs), siRNAs can recognize these mRNAs through homology in the "seed sequence" (nucleotides 2-8 of the guide strand), leading to translational inhibition or mRNA degradation [3] [23] [2]. Even a small region of complementarity (as few as 14-15 consecutive nucleotides) can be sufficient to cause an off-target effect [51].
Sequence-independent immune activation: In some cell types, siRNAs can trigger innate immune responses, such as the interferon pathway, leading to widespread changes in gene expression that are not sequence-specific [1].
What are the first steps I should take if I suspect off-target effects?
If your RNAi experiment yields unexpected phenotypic changes, follow these initial diagnostic steps:
- Confirm the On-Target Effect: First, verify that your siRNA effectively knocks down your intended target gene. Use qRT-PCR to measure mRNA levels and/or immunoblotting to assess protein levels [51].
- Review siRNA Design: Check the sequence of your siRNA for potential homology to other genes. Modern siRNA design algorithms incorporate stringent specificity checks to minimize this risk [51].
- Titrate siRNA Concentration: High siRNA concentrations (often above 30 nM) can exacerbate both sequence-dependent and immune-related off-target effects. Repeat the experiment using the lowest effective concentration of siRNA [51].
How can I definitively confirm that an observed effect is off-target?
To conclusively determine that a phenotype is due to an off-target effect, a combination of bioinformatic and experimental validation is required.
- Gene Expression Profiling: The most comprehensive method is to use DNA microarrays or RNA sequencing (transcriptomics) to analyze global changes in gene expression. True off-target effects will appear as "siRNA-specific" changes—i.e., different siRNAs against the same target gene will cause different expression profiles, rather than converging on a unified, gene-specific signature [51].
- Rescue Experiments: A powerful validation involves expressing an siRNA-resistant version of your target gene. This is typically a modified gene where the coding sequence has been altered (e.g., through codon optimization) to avoid siRNA recognition while still producing a wild-type protein. If the phenotypic effect is rescued (reversed) by this modified gene, it confirms that the effect was specific to the intended target knockdown. If the effect persists, it is likely caused by an off-target event [51].
- Use Multiple siRNAs: Always test at least two or more distinct siRNAs targeting different regions of the same mRNA. If they all produce the same phenotypic change, confidence in the result being on-target is high. If phenotypes differ, it strongly indicates off-target effects [51] [56].
The following workflow outlines a systematic approach to diagnosing and rescuing an RNAi experiment with suspected off-target effects:
What strategic controls should be included in every RNAi experiment?
Incorporating the correct controls from the beginning is crucial for robust and interpretable results. The table below summarizes the essential controls for a well-designed RNAi experiment.
Table 1: Essential Control Experiments for RNAi
| Control Type | Description | Purpose |
|---|---|---|
| Positive Control siRNA [56] | A known siRNA that provides high knockdown of a well-characterized gene. | Verifies that the experimental system (transfection, reagents, etc.) is working optimally. |
| Negative Control siRNA [51] [56] | A non-silencing siRNA with no significant homology to any gene in the target organism (e.g., scrambled sequence). | Distinguishes specific silencing effects from non-specific effects caused by the transfection process or the siRNA molecule itself. |
| Transfection Control [56] | A fluorescently labeled siRNA or an siRNA targeting an essential gene. | Measures transfection efficiency and cell viability post-transfection. |
| Mock Transfection [56] | Cells are treated with the transfection reagent but no siRNA. | Identifies any non-specific effects caused by the transfection reagent alone. |
| Untreated Cells [56] | Cells that undergo no treatment whatsoever. | Provides the baseline, normal level of gene expression for comparison. |
What tools and reagents are key for troubleshooting off-target effects?
A successful troubleshooting process relies on specific reagents and tools. The following table lists essential solutions for validating your RNAi data.
Table 2: Research Reagent Solutions for RNAi Validation
| Reagent / Tool | Function | Application in Troubleshooting |
|---|---|---|
| Validated & Pre-designed siRNAs [51] | siRNAs designed with advanced algorithms that include specificity checks. | Reduces the risk of off-target effects from the start of the experiment. |
| siRNA-Resistant Gene Construct [51] | A codon-optimized version of the target gene that is not recognized by the siRNA. | The definitive experiment to confirm on-target effects via phenotypic rescue. |
| Multiple siRNAs per Target [51] [56] | At least two unique siRNAs targeting different sites on the same mRNA. | Confirms that a phenotype is consistent and not specific to a single siRNA sequence. |
| Fluorescently Labeled siRNA [56] | An siRNA conjugated to a fluorophore (e.g., FITC). | Allows direct visualization and quantification of transfection efficiency. |
| Microarrays / RNA-seq [51] | Tools for genome-wide expression profiling. | Identifies sequence-specific off-target effects by revealing all genes whose expression is altered. |
| Interferon Response Assays [51] | Assays to monitor markers like 2'-5' oligoadenylate synthetase or STAT1. | Detects non-specific, immune-related off-target effects activated by the siRNA. |
Off-target effects pose a significant challenge in functional genomics screens, often arising from unintended interactions between short "seed" sequences within guide RNAs and partially complementary genomic regions. These seed sequences—typically the 9-12 nucleotide PAM-proximal region of a guide RNA—can mediate widespread unintended effects that compromise experimental integrity. Recent research demonstrates that CRISPRi off-target effects are more common than previously recognized, with seed sequence matches as short as 8-9 base pairs sufficient to produce detectable phenotypic effects [57]. Similarly, siRNA screens have demonstrated high false-positive rates attributable to seed-based off-target effects, requiring extensive experimental triage to distinguish true hits [58].
Understanding and identifying these seed-based effects is therefore essential for proper interpretation of high-throughput screening data, particularly in drug development contexts where erroneous target identification carries significant financial and clinical consequences.
Technical Support & Troubleshooting Guides
Frequently Asked Questions
What are the primary indicators of seed-based off-target effects in my screening data? The most telling indicator is observing multiple different sgRNAs or siRNAs with identical seed sequences clustering as top hits in your screen. Additionally, unexpected phenotypic effects that cannot be explained by the intended target genes suggest potential off-target activity. Statistical analysis of seed sequence enrichment across your entire dataset provides the most reliable identification method [58].
How short can a seed sequence match be while still producing off-target effects? Research has demonstrated that perfect complementarity in PAM-proximal sequences as short as 8-9 base pairs can produce detectable off-target effects in CRISPRi screens. However, it's important to note that not all guides with these minimal seed matches will produce effects, as other factors influence off-target susceptibility [57].
Are seed-based off-target effects consistent across different CRISPR systems? No, significant variation exists between different CRISPR systems. CRISPRi and CRISPRa systems demonstrate substantially more pronounced seed-based off-target effects compared to nuclease-active CRISPR-Cas9 knockout systems. In comparative studies, CRISPRn screens did not show the same seed sequence-mediated off-targeting observed in CRISPRi screens [57].
What computational approaches can help identify potential seed-based effects before conducting experiments? Standard practice includes performing genome-wide searches for exact matches to the 9-bp PAM-proximal seed sequence of each guide RNA, particularly focusing on regions within 2000 bp of transcription start sites. This pre-screening helps flag guides with high potential for off-target effects prior to library construction [57].
How can I optimize my sgRNA design to minimize seed-based off-target effects? Focus on selecting guides with minimal off-target seed matches in the genome, particularly within promoter regions of genes likely to produce confounding phenotypes in your specific assay. Additionally, using multiple sgRNAs per gene and comparing results helps distinguish true on-target effects from seed-based artifacts [57].
What percentage of sgRNAs in a typical CRISPRi library exhibit seed-based off-target effects? The proportion varies by library and screen, but studies have identified approximately 35-68% of predicted off-target sgRNAs mapping to promoters of known pathway mediators across different screens. In one pyroptosis resistance screen, 35% of predicted off-target guides mapped to promoters of just two key pathway genes (GSDMD and CASP4) [57].
Common Experimental Issues and Solutions
| Problem | Possible Cause | Solution |
|---|---|---|
| Unexpected gene hits in screening | Seed sequence matches to promoters of unrelated genes | Perform Common Seed Analysis (CSA) to identify enriched seed sequences among hits [58] |
| High false-positive rates | Off-target binding causing unintended transcriptional changes | Implement more stringent seed match filters in guide design; use orthogonal validation approaches [57] |
| Inconsistent results between guides targeting same gene | Variable seed-mediated off-target effects between different guides | Include multiple independent guides per gene; exclude guides with promiscuous seed sequences [57] |
| Poor correlation between screening results and validation experiments | Seed-based effects manifest only in specific cellular contexts | Conduct secondary assays using chemically distinct modulators (e.g., small molecules) for hit confirmation [58] |
| Unexpected pathway enrichment | Seed-mediated regulation of multiple genes in same pathway | Analyze seed sequence matches to all pathway component promoters; redesign affected guides [57] |
Key Experimental Protocols
Common Seed Analysis (CSA) for siRNA Screens
Common Seed Analysis provides a systematic approach for identifying seed-based off-target effects in siRNA screening data through the following methodology:
Procedure:
- Sequence Grouping: Collect all siRNA sequences from your screening results and group them by their seed sequences (nucleotides 2-8 from the 5' end of the guide strand)
- Statistical Analysis: For each unique seed sequence, perform statistical testing to determine if siRNAs sharing that seed demonstrate consistently enriched phenotypes compared to the library-wide distribution
- Hit Identification: Apply false discovery rate correction to identify seed sequences with statistically significant effects on your assay readout
- Filtering: Flag these promiscuous seed sequences and exclude them from subsequent hit confirmation studies [58]
Expected Outcomes: Application of CSA to primary screening data of the Wnt pathway enabled identification of 158 distinct seed sequences with statistically significant effects on the assay, allowing researchers to discount these promiscuous sequences in follow-up experiments [58].
CRISPRi Off-Target Identification Protocol
This protocol enables systematic identification of seed-mediated off-target effects in CRISPRi screens:
Procedure:
- Guide-level Analysis: Perform initial screening analysis at the individual guide level rather than gene level to identify sgRNAs with unusual enrichment patterns
- Seed Sequence Extraction: For each significantly enriched sgRNA, extract the 9-bp PAM-proximal seed sequence
- Genome Alignment: Perform exact match searches for each seed sequence across the entire genome, retaining only matches with canonical NGG PAM sequences
- Promoter Mapping: Filter matches to include only those located within 2000 bp of transcription start sites
- Pathway Mapping: Cross-reference identified promoter matches with genes known to be involved in your screened pathway or phenotype [57]
Validation: In necroptosis resistance screens, this approach revealed that 68% (35/51) of predicted off-target guides mapped to promoters of four key pathway genes (RIPK3, RIPK1, MLKL, or TNFRSF1A), confirming the biological relevance of the identified off-target effects [57].
Off-Target Effect Prevalence Across Screen Types
| Screen Type | Off-Target Guides Mapped to Pathway Genes | Minimum Seed Match Length | Key Statistical Findings |
|---|---|---|---|
| CRISPRi Pyroptosis | 35% (27/76 guides) mapped to GSDMD/CASP4 promoters | 8-9 bp | 21 guides with perfect 9-bp seed matches to GSDMD promoter showed fold-change equivalent to on-target guides [57] |
| CRISPRi Necroptosis | 68% (35/51 guides) mapped to RIPK3/RIPK1/MLKL/TNFRSF1A | 8-9 bp | Only 4 of 532 sgRNAs with seed matches to TNFRSF1A promoter showed effects equivalent to on-target guides [57] |
| siRNA Wnt Pathway | 158 distinct seed sequences with statistically significant effects | 7 bp (nt 2-8) | Promiscuous seed sequences identified through Common Seed Analysis [58] |
| CRISPRn Pyroptosis | No evidence of seed sequence-mediated off-targeting | N/A | Contrasts with CRISPRi, suggesting system-specific differences [57] |
Seed Sequence Match Efficiency
Seed Match Efficiency in CRISPRi Screens
Visualization of Key Concepts
CRISPRi Off-Target Identification Workflow
CRISPRi Off-Target Identification Workflow
Seed Sequence Binding Mechanism
Seed Sequence Binding Mechanism
Research Reagent Solutions
| Essential Material | Function in Seed Effect Analysis | Implementation Considerations |
|---|---|---|
| Whole-genome CRISPRi sgRNA library | Systematic gene silencing with coverage of promoter regions | Ensure multiple guides per gene to distinguish true hits from seed effects [57] |
| dCas9-KRAB expressing cell lines | Transcriptional repression without DNA cleavage | Stable expression required for CRISPRi screens; verify repression efficiency [57] |
| Control siRNA/sgRNA libraries | Distinguish sequence-specific from seed-mediated effects | Include non-targeting controls and seeds with known effects [58] |
| Sequence-verified oligonucleotides | Ensure accuracy of guide RNA sequences | Up to 20% of clones may contain mutated inserts without proper verification [8] |
| High-efficiency transfection reagents | Ensure consistent delivery of RNAi constructs | Optimize DNA:reagent ratios and avoid antibiotics during transfection [8] |
| Tet-repressor expressing cell lines | For inducible RNAi systems | Ensure fetal bovine serum is tetracycline-free to prevent unintended induction [8] |
| One Shot Stbl3 Competent E. coli | Stable propagation of lentiviral RNAi vectors | Reduces recombination in plasmids containing direct repeats [8] |
Seed sequence-mediated off-target effects present a substantial challenge in functional genomics screens, particularly in CRISPRi and siRNA applications. The methodologies outlined here provide systematic approaches for identifying, quantifying, and mitigating these effects. Key recommendations include: (1) implementing Common Seed Analysis as a standard component of screening data analysis; (2) utilizing multiple independent guides per gene with careful attention to seed sequence properties during library design; (3) employing orthogonal validation approaches for screening hits; and (4) recognizing that seed sequence match alone is necessary but not sufficient for off-target activity, with additional contextual factors influencing phenotypic outcomes. By integrating these practices, researchers can significantly improve the reliability of target identification in both basic research and drug development applications.
What is the primary challenge in RNAi screening for TRAIL pathway genes?
The major challenge is the prevalence of off-target effects (OTEs), where siRNAs reduce the expression of unintended genes, leading to false positives and complicating the identification of genuine hits. In TRAIL apoptosis screens, these OTEs often arise because siRNAs can function like microRNAs (miRNAs), causing non-specific gene silencing through partial sequence complementarity [59].
Troubleshooting Guide: Identifying Off-Target Effects
How can I determine if my screen results include false positives from off-target effects?
Systematic analysis reveals that off-target effects in TRAIL screens are frequently linked to specific 6-7 nucleotide "seed" sequences in the siRNA. The workflow below outlines a method to identify these effects [59] [60]:
Key Indicators of Potential Off-Target Effects:
- Multiple top-scoring siRNAs share the same seed sequence despite targeting different genes [59].
- Inconsistent phenotypes from multiple siRNAs targeting the same gene, where the siRNA with the strongest phenotypic effect does not yield the best target mRNA knockdown [59].
- Enrichment of specific hexamer/heptamer sequences in high-scoring siRNAs beyond what would occur by chance [59].
What experimental validation is required to confirm genuine hits?
A robust confirmation protocol is essential. The following table summarizes the key validation steps used in the systematic TRAIL screen analysis [59]:
Table 1: Experimental Protocol for Hit Confirmation
| Step | Methodology | Purpose | Acceptance Criteria |
|---|---|---|---|
| Secondary Assay | Measure Caspase-3/7 activity after TRAIL treatment [59]. | Confirm phenotype using a different apoptosis readout. | ≥2 siRNAs per gene show significant effect. |
| mRNA Knockdown Validation | qPCR to measure target mRNA levels [59]. | Verify on-target efficiency. | siRNAs with phenotypic effect show stronger knockdown than non-effective siRNAs. |
| Seed Sequence Analysis | Bioinformatic analysis of siRNA seed regions [59]. | Identify miRNA-like off-target potential. | Confirmed hits lack over-represented seed sequences. |
| Functional miRNA Testing | Transfect miRNA mimics [59]. | Test if seed-driven effects recapitulate phenotype. | miRNA mimic should mimic the siRNA's protective effect. |
FAQ: Addressing Common Researcher Questions
What are the molecular mechanisms behind these off-target effects?
Off-target effects occur primarily through the "miRNA-like" mechanism. The siRNA guide strand is loaded into the RNA-Induced Silencing Complex (RISC). Instead of perfect complementarity with the intended target, the "seed sequence" (nucleotides 2-8 at the 5' end of the guide strand) binds imperfectly to complementary sites in the 3' Untranslated Regions (UTRs) of off-target mRNAs, leading to their degradation or translational repression [3] [2]. The diagram below illustrates this mechanism.
Which specific miRNAs were implicated in off-target effects in the TRAIL screen?
The systematic analysis identified that three specific miRNA seed sequences were enriched in high-scoring siRNAs from the TRAIL screen. Transfection of mimics for these miRNAs protected cells from TRAIL-induced death, confirming their functional role in the pathway [59] [60].
Table 2: miRNAs Implicated in TRAIL-Induced Apoptosis via Off-Target Analysis
| miRNA | Role in TRAIL-Induced Apoptosis | Experimental Validation |
|---|---|---|
| miR-26a | Protective effect | Transfection of miR-26a mimic protected several cell types from TRAIL-induced cell death [59]. |
| miR-145 | Protective effect | Transfection of miR-145 mimic protected several cell types from TRAIL-induced cell death [59]. |
| miR-384 | Protective effect | Transfection of miR-384 mimic protected several cell types from TRAIL-induced cell death [59]. |
What strategies can I use to minimize off-target effects in my RNAi screen?
Proactive design and analytical strategies are key to mitigating OTEs [3] [2].
Table 3: Strategies to Minimize Off-Target Effects
| Strategy | Description | Implementation in TRAIL Screen |
|---|---|---|
| Bioinformatic Design | Design siRNAs with minimal seed sequence complementarity to off-target 3' UTRs. | Use pre-designed algorithms to filter siRNAs with common or problematic seed sequences. |
| Pooling siRNAs | Using pools of multiple siRNAs targeting the same gene. | A lower concentration of each individual siRNA reduces seed-driven effects while maintaining on-target efficacy. |
| Chemical Modifications | Incorporating chemical modifications (e.g., 2'-O-methyl) into the siRNA seed region. | This can reduce the affinity of the seed sequence for off-target mRNAs. |
| Use Multiple siRNAs per Gene | Requiring consistent phenotypes from ≥2 independent siRNAs for hit confirmation. | This was a core part of the validation protocol in the TRAIL screen case study [59]. |
| Systematic Seed Analysis | Post-hoc bioinformatic analysis of enriched seed sequences in hit lists. | As performed in the TRAIL screen, this identifies siRNAs likely acting via off-target effects [59]. |
The Scientist's Toolkit: Key Research Reagents
Table 4: Essential Research Reagents for RNAi Screens on TRAIL Apoptosis
| Reagent / Tool | Function / Purpose | Example from Case Study |
|---|---|---|
| siRNA Library | Genome-wide or pathway-focused collection of siRNAs for screening. | "Druggable genome" library of 12,190 siRNAs [59]. |
| Recombinant TRAIL | The apoptosis-inducing ligand used to stimulate the death pathway. | Used to treat cells to induce apoptosis in the primary screen [59]. |
| Caspase-Glo 3/7 Assay | Luminescent assay to measure activation of executioner caspases. | Used as a secondary assay to confirm hits from the primary screen [59]. |
| miRNA Mimics | Synthetic small RNAs that mimic mature endogenous miRNAs. | Used to validate the role of miR-26a, -145, and -384 in TRAIL sensitivity [59]. |
| c-FLIP siRNA / Rocaglamide | Tools to downregulate c-FLIP, a key inhibitor of TRAIL-induced apoptosis. | Rocaglamide is a translational inhibitor of c-FLIP used to sensitize resistant cells [61]. |
| IAP Antagonists (e.g., AT406) | Small-molecule Smac mimetics that inhibit IAP proteins. | Used in combination to overcome TRAIL resistance in solid tumors [61]. |
FAQs on Addressing Off-Target Effects in RNAi Experiments
What are the primary causes of off-target effects in RNAi experiments?
Off-target effects occur when a short interfering RNA (siRNA) unintentionally reduces the expression of genes other than the intended target. The primary mechanism is through "miRNA-like" effects [3]. An siRNA can function like a microRNA (miRNA) when it exhibits partial complementarity, particularly in its seed region (nucleotides 2-7 or 2-8 from the 5' end of the guide strand), to sequences in the 3' untranslated regions (UTRs) of off-target mRNAs [3] [52]. This can lead to translational inhibition or degradation of those mRNAs, even with only 6-7 nucleotides of complementarity [52].
How can I confirm that my observed phenotype is due to on-target silencing?
Confirming on-target effects requires a multi-pronged approach. Best practices include [52]:
- Using Multiple siRNAs: Test at least two, and ideally three, independent siRNAs targeting the same gene. A phenotype observed with multiple siRNAs targeting different regions of the same mRNA is more likely to be on-target.
- Correlating Knockdown with Phenotype: Demonstrate that the siRNAs which produce the phenotypic effect also significantly reduce the target mRNA levels (e.g., by >70%). Conversely, siRNAs that do not effectively knock down the mRNA should not produce the phenotype [52].
- Rescuing the Phenotype: Expressing a modified version of the target gene that is resistant to the siRNA (e.g., through silent mutations) can confirm specificity if the phenotype is reversed.
What are the best negative controls for an RNAi experiment?
Proper negative controls are essential to isolate the effect of your specific siRNA from non-specific effects of the transfection process and the presence of any foreign RNA or DNA [62].
- Non-Targeting siRNA (Scrambled Control): A siRNA with a sequence that has no significant complementarity to any gene in the organism's transcriptome. This is the most common negative control.
- Mutant Plasmid Control: When using plasmid-based systems (e.g., for shRNA), a superior negative control is a plasmid that is identical to the experimental one but contains a mutated version of the gene of interest that lacks functionality. A modern solution is the eZ-stop peptide, a sequence inserted to halt translation early, ensuring the control plasmid has a similar length and metabolic burden as the experimental plasmid [62].
The table below summarizes the key types of controls for RNAi experiments.
| Control Type | Purpose | Description & Best Practices |
|---|---|---|
| Positive Control | Verify transfection efficiency and the functionality of the RNAi machinery [19] [62]. | Use a well-validated siRNA that knocks down a ubiquitous, non-essential gene (e.g., GAPDH) or a reporter gene (e.g., GFP). Run in parallel with your experiment. |
| Negative Control | Account for effects of the transfection process and the presence of foreign nucleic acids [19] [62]. | Use a non-targeting (scrambled) siRNA sequence. For plasmids, use a mutant control like the eZ-stop peptide to match the metabolic burden of the experimental plasmid [62]. |
| Transfection Control | Monitor and optimize the delivery of siRNA into cells [19]. | Use a fluorescently-labeled non-targeting siRNA to visualize uptake efficiency under a microscope. |
Troubleshooting Guides
Problem: No or Insufficient Knockdown of Target mRNA
Recommendations:
- Check mRNA levels properly: Use quantitative real-time PCR (qRT-PCR). Ensure the RNA is not degraded during isolation [19].
- Verify transfection efficiency: Use a positive control siRNA and a fluorescent transfection control to ensure the siRNA is entering the cells effectively [19].
- Optimize experimental conditions:
- Test multiple siRNAs: If one siRNA fails, others targeting different sites on the same mRNA may be effective [19].
Problem: High Cytotoxicity or Cell Death in siRNA-Treated Cells
Recommendations:
- Rule out transfection reagent toxicity: Run a control with the transfection reagent only (no siRNA). If toxicity is high, try different cell densities or alternative transfection reagents [19].
- Titrate down siRNA concentration: High siRNA concentrations (e.g., >100 nM) can trigger stress responses and cytotoxicity. Use the lowest effective concentration [19].
- Investigate seed sequence: The siRNA's seed sequence may be inadvertently targeting genes essential for cell survival. Re-design the siRNA or try an alternative sequence with a different seed region [52].
Problem: Observed Phenotype is Due to Off-Target Effects
Recommendations:
- Analyze the seed sequence: Systematically check if the top-performing siRNAs in your screen share common hexamer or heptamer seed sequences. Enrichment of specific seeds is a strong indicator of prevalent off-target effects [52].
- Use chemical modifications: Incorporate specific chemical modifications into the siRNA backbone to reduce off-target potential.
- Employ pooled siRNA designs: Instead of a single siRNA, use a pool of several siRNAs targeting the same gene, each at a lower concentration. This can dilute out individual seed-mediated off-target effects while maintaining on-target efficacy [3].
- Perform rescue experiments: The most definitive way to confirm an off-target effect is to rescue the phenotype by expressing the suspected off-target gene in a manner resistant to the siRNA.
Optimization Strategies: A Data-Driven Approach
Concentration Optimization
Systematically testing siRNA concentration is fundamental. The table below outlines a sample titration experiment to optimize for efficacy and minimize toxicity.
| siRNA Concentration | Expected mRNA Knockdown (qRT-PCR) | Expected Protein Knockdown (Western Blot) | Notes on Cytotoxicity |
|---|---|---|---|
| 1-5 nM | Low to Moderate (30-60%) | May be minimal | Typically minimal cytotoxicity. |
| 10-20 nM | Good (60-80%) | Observable (50-70%) | Low cytotoxicity in most cell lines. |
| 50 nM | High (>80%) | High (>70%) | Potential for cytotoxicity; monitor cell health. |
| 100 nM | Very High (>90%) | Very High (>80%) | High risk of non-specific and cytotoxic effects. |
Chemical Modifications to Reduce Off-Targets
Specific chemical modifications to the siRNA molecule can enhance stability and reduce immunogenicity and off-target effects [63]. The following table lists common modifications and their functions.
| Modification Type | Description | Function |
|---|---|---|
| Phosphorothioate (PS) Backbone | Replacement of a non-bridging oxygen atom in the phosphate backbone with sulfur [63]. | Increases nuclease resistance and improves pharmacokinetics. |
| 2'-Sugar Modifications | Substitution of the 2'-OH group with -O-Me, -O-Et, or -F [63]. | Reduces immunogenicity and protects against nuclease degradation. |
| Locked Nucleic Acid (LNA) | Introduction of a methylene bridge that "locks" the ribose ring [63]. | Dramatically increases affinity for the target mRNA (thermostability) and improves specificity. |
| GalNAc Conjugation | Covalent linkage of a triantennary N-acetylgalactosamine molecule to the siRNA [63]. | Enables targeted delivery to hepatocytes by binding to the asialoglycoprotein receptor. |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in RNAi Experiments |
|---|---|
| Pre-designed siRNA Libraries | Commercially available libraries (e.g., Silencer Select, Stealth RNAi) provide multiple, validated siRNAs per gene, often with guarantees of >70% knockdown [19]. |
| Bioinformatics Algorithms | Tools like BLOCK-iT RNAi Designer and algorithms from IDT use machine learning to select siRNA sequences for high efficiency and reduced off-target potential [63]. |
| Validated Positive Control siRNAs | siRNAs targeting common genes like GAPDH to standardize and verify the performance of your RNAi workflow in every experiment [19]. |
| Fluorescent Transfection Controls | Labeled, non-targeting RNAs that allow researchers to visually confirm and quantify delivery efficiency into their specific cell type under the microscope [19]. |
| Specialized Transfection Reagents | Formulations optimized for different nucleic acid types (siRNA, plasmid) and for challenging cell lines, including primary and suspension cells. |
Experimental Workflows and Mechanisms
The following diagram illustrates the core mechanism of RNAi and the primary source of off-target effects, providing a visual reference for troubleshooting.
Diagram 1: RNAi Mechanism and Off-Target Effects Pathway.
This workflow outlines a systematic approach for optimizing an RNAi experiment and validating its results.
Diagram 2: RNAi Experimental Optimization and Validation Workflow.
Validation and Comparative Analysis: Confirming On-Target Activity in RNAi Workflows
Why is using multiple siRNAs considered the gold standard?
In RNAi screening, a primary challenge is distinguishing genuine on-target gene silencing from false positives caused by off-target effects (OTEs). Off-target effects occur when an siRNA directly affects the expression of genes other than the one it is designed to target, often because the siRNA acts like a microRNA (miRNA). This can happen when a short sequence within the siRNA (6-8 nucleotides long, called the "seed" sequence) binds to partially complementary sites in the 3' untranslated regions (UTRs) of non-target mRNAs, leading to their degradation or translational repression [59] [23].
Using multiple, independent siRNAs targeting the same gene is the most reliable strategy to confirm that an observed phenotypic change is due to the knockdown of the intended gene, rather than an off-target artifact. The core principle is that different siRNAs against the same gene, which have distinct seed sequences and thus different off-target profiles, should produce the same phenotypic outcome if it is a true on-target effect. If only one siRNA produces a phenotype, it is likely due to its unique off-target activity [51] [44].
Experimental Protocol for Hit Confirmation
The following workflow outlines a robust, multi-step process for confirming hits from a primary RNAi screen.
Detailed Methodology
Selection of Multiple siRNAs:
- Number: Use at least two, but ideally three or more, siRNAs per target gene [51] [64].
- Design: Ensure siRNAs are independent, meaning they target different, non-overlapping sequences (exons) within the same target mRNA. This minimizes the chance they share the same off-target genes.
- Specificity: Utilize pre-designed siRNAs that have undergone stringent computational checks to minimize sequence homology with non-target genes (e.g., ensuring at least 2-3 nucleotide mismatches against all closely related non-target genes) [51].
Confirmatory Transfection and mRNA Knockdown Assessment:
- Transfection: Transfect each siRNA into the relevant cell line under optimized conditions. Use a non-targeting scrambled siRNA as a negative control and a siRNA targeting a known pathway component as a positive control.
- mRNA Quantification: 48 hours post-transfection, isolate total RNA and perform quantitative real-time PCR (qRT-PCR) to measure the mRNA levels of the target gene.
- Acceptance Criterion: Successful knockdown is typically defined as ≥70% reduction in target mRNA levels compared to the negative control [64].
Phenotypic Validation in a Secondary Assay:
- Assay Type: Do not rely solely on the primary screening assay. Develop a secondary, orthogonal assay that also measures the biological phenotype of interest. For example, if the primary screen measured cell death, a secondary assay could measure Caspase-3/7 activity as a more specific marker of apoptosis [59].
- Acceptance Criterion: At least two siRNAs that effectively knock down the target mRNA must also reproduce the significant phenotypic effect in this secondary assay.
Correlation of Knockdown and Phenotype:
- Analyze the data to ensure that the magnitude of the phenotypic effect correlates with the efficiency of target mRNA knockdown. An siRNA that produces a strong phenotype but has poor knockdown is likely working through an off-target mechanism [59].
Rescue Experiment with an siRNA-Resistant Construct (Gold Standard Validation):
- Construct Design: A powerful control is to express a version of the target gene that is resistant to the siRNA. This is often a codon-optimized gene where the siRNA target sequence has been altered without changing the amino acid sequence of the encoded protein [51].
- Experiment: Co-transfect the cells with the confirmed siRNA and the siRNA-resistant gene construct.
- Acceptance Criterion: Expression of the resistant construct should "rescue" or reverse the phenotypic effect caused by the siRNA, providing the strongest evidence that the phenotype is due to the specific knockdown of the intended target [51].
Troubleshooting Guide & FAQs
Q: I have two siRNAs for my target gene. One reduces mRNA well and gives a strong phenotype, but the second reduces mRNA without a phenotype. What does this mean? A: This is a classic sign of an off-target effect. The phenotypic outcome from the first siRNA is likely not due to the knockdown of your intended target but rather to the silencing of other genes via its unique seed sequence. The hit should not be confirmed. You should test additional siRNAs against the target [59].
Q: All of my siRNAs against a target gene show strong mRNA knockdown, but none of them produce a phenotype. Why? A: This suggests the gene is not involved in the biological pathway you are studying. The knockdown is on-target, but the gene is not essential for your specific phenotype. Your screening assay is working correctly, but this is a negative result.
Q: How can I be sure that my multiple siRNAs aren't all hitting the same off-target? A: While possible, it is statistically unlikely if you use independent siRNAs with different seed sequences. Their off-target profiles will be different. The consistency of the phenotype across these different siRNAs is what gives confidence in the on-target effect. Using siRNAs from different commercial designs (e.g., Silencer vs. Silencer Select) can further diversify risk.
Q: What concentration of siRNA should I use for confirmation studies? A: Use the lowest effective concentration of siRNA (often <30 nM) to minimize off-target effects, which are more prevalent at high concentrations (≥100 nM). Titrate your siRNAs to find this optimal concentration [51].
Q: My siRNA knocks down mRNA, but I don't see a reduction in the target protein. What should I do? A: Check the protein's half-life (turnover rate). If the protein is long-lived, you may need to extend the time course of your experiment and analyze protein levels at 72 or 96 hours post-transfection [64].
RNAscope Scoring Guidelines for Validation
For researchers using RNA in situ hybridization (like RNAscope) as a secondary phenotypic or validation assay, the following semi-quantitative scoring guidelines should be applied. This helps in consistently evaluating the level of target RNA expression in tissue samples.
| Score | Criteria | Interpretation |
|---|---|---|
| 0 | No staining or <1 dot/10 cells | Negative |
| 1 | 1-3 dots/cell | Low/very low expression |
| 2 | 4-9 dots/cell; very few dot clusters | Moderate expression |
| 3 | 10-15 dots/cell; <10% dots in clusters | High expression |
| 4 | >15 dots/cell; >10% dots in clusters | Very high expression |
Note: When validating knockdown, you would expect a significant reduction in this score in samples treated with effective siRNAs compared to controls. Always run positive and negative control probes to qualify your sample and assay performance [35].
The Scientist's Toolkit: Key Research Reagents
The table below lists essential materials and reagents for conducting rigorous siRNA hit confirmation experiments.
| Item | Function/Description | Examples & Notes |
|---|---|---|
| Validated siRNAs | Algorithm-designed siRNAs experimentally verified to knock down mRNA by ≥70%. Using pre-validated reagents increases success. | Silencer Select, ON-TARGETplus [64] |
| Scrambled siRNA | A negative control siRNA with a sequence that does not target any known gene in the organism. | Critical for establishing a baseline for phenotype and mRNA levels [51] |
| siRNA-Resistant Gene | A codon-optimized version of the target gene used in rescue experiments to confirm on-target activity. | Can be synthesized via services like GeneArt [51] |
| Positive Control siRNA | An siRNA targeting a gene known to elicit a strong phenotypic response in your assay system. | e.g., siRNA against CASP8 in a death assay [59] [64] |
| qRT-PCR Assay Kits | For quantitative measurement of target mRNA knockdown. | Ensure primers do not detect the siRNA-resistant rescue construct [51] [64] |
In the development of RNA interference (RNAi) therapies, identifying unintended, or "off-target," effects is a critical safety challenge. Off-target effects occur when an RNAi drug, such as a small interfering RNA (siRNA), silences genes other than its intended target, potentially leading to adverse consequences [23]. Transcriptomic profiling technologies are essential tools for unbiased detection of these effects, as they can measure the expression of thousands of genes simultaneously. This guide details the use of Microarrays and RNA-Sequencing (RNA-Seq) for this purpose, providing troubleshooting and methodological support to ensure the reliability of your off-target assessments.
FAQ: Technology Selection and Core Concepts
1. Why is transcriptomic profiling necessary for detecting off-target effects in RNAi experiments? RNAi off-target effects often arise when the short "guide strand" of an siRNA exhibits partial complementarity to non-target mRNAs, leading to their unintended silencing via the miRNA-like pathway [23] [1]. Standard assays like qRT-PCR are targeted and would miss these unexpected events. Profiling technologies like Microarrays and RNA-Seq provide an unbiased, genome-wide view of the transcriptome, enabling the discovery of these unforeseen changes in gene expression [23].
2. How do I choose between Microarrays and RNA-Seq for off-target detection? The choice involves a trade-off between the scope of discovery, budget, and computational resources. The following table summarizes the key differences:
Table 1: Comparison of Microarrays and RNA-Seq for Off-Target Detection
| Feature | RNA-Seq | Microarrays |
|---|---|---|
| Principle | Direct sequencing of cDNA fragments [65] | Hybridization to pre-defined DNA probes [66] |
| Discovery Power | High. Can detect novel transcripts, isoforms, and genes not previously annotated [67] [68] | Limited. Restricted to detecting only the genes for which probes are on the array [68] [66] |
| Dynamic Range | Very wide (>10⁵). Superior for quantifying both very high and very low abundance transcripts [67] [66] | Narrower (~10³). Signal saturation can limit accurate quantification of highly expressed genes [67] [66] |
| Sensitivity | High. More effective at detecting differentially expressed genes, especially those with low expression [67] [68] | Lower. Can miss low-abundance transcripts [66] |
| Data Complexity | High. Requires significant bioinformatics expertise and computing power for data storage and analysis [69] [66] | Low. Data analysis is more straightforward and requires less computational resources [66] |
| Cost | Higher per sample for sequencing and analysis [68] [66] | Lower upfront cost, particularly for large studies on known genes [66] |
For a maximal, unbiased discovery of off-target effects—including in non-coding regions or for novel transcripts—RNA-Seq is the superior choice [67]. For targeted, cost-effective studies where the gene set of interest is well-defined, microarrays remain a viable option.
3. What are the common sources of off-target effects in RNAi, and how does this guide profiling? The primary source is sequence-dependent off-targeting, where the siRNA seed region (nucleotides 2-8) binds to partially complementary sites in the 3'UTR of non-target mRNAs [23] [1]. Your experimental design should ensure sufficient sequencing depth or array coverage to detect subtle changes in gene expression that may result from this imperfect pairing.
Troubleshooting Guides
Issue 1: High Technical Variation Obscures Biological Signals
Problem: Principal Component Analysis (PCA) plots show that samples cluster more by batch (e.g., date of library prep) than by experimental group (e.g., treated vs. control).
Solutions:
- At the experimental design stage: Minimize batch effects by processing all samples (controls and treated) simultaneously for RNA isolation, library preparation, and sequencing [70]. Use biological replicates (ideally 3 or more per group) to reliably capture biological over technical variability [69].
- At the analysis stage: Include batch as a covariate in your statistical model for differential expression. Use normalization methods designed to remove unwanted variation (RUV) or correct for batch effects [69].
Issue 2: Suspected RNA Degradation Affecting Data Quality
Problem: RNA Integrity Number (RIN) is low, or Bioanalyzer plots show degraded RNA. This leads to poor 3' coverage in poly(A)-selected RNA-Seq libraries or high background on microarrays.
Solutions:
- Prevention: Use fresh cells/tissues and RNA stabilization reagents. Handle RNA with RNase-free techniques.
- Mitigation: If RNA quality is suboptimal but usable, switch to an rRNA depletion protocol for RNA-Seq instead of poly(A) selection, as it is less biased towards the 3' end of transcripts [69]. For microarrays, ensure the RNA input meets the manufacturer's specified quality thresholds.
Issue 3: High Background or Low Signal-to-Noise Ratio
Problem: Microarray data shows high background fluorescence, or a large percentage of RNA-Seq reads do not map to the reference genome.
Solutions:
- For Microarrays: This can indicate non-specific hybridization or probe-related issues. Ensure stringent washing protocols are followed according to the array manufacturer's guidelines. Verify that the sample label and hybridization conditions are optimal [71].
- For RNA-Seq: A low mapping rate can suggest DNA contamination or poor library quality. Re-assess the raw read quality with FastQC [69]. Trim adapters and low-quality bases with tools like Trimmomatic [69]. Ensure you are using an appropriate and well-indexed reference genome for alignment.
Experimental Protocols for Off-Target Detection
Protocol 1: RNA-Seq-Based Off-Target Screening
This protocol is designed for comprehensive, unbiased detection of transcriptional changes.
1. Sample Preparation and RNA Extraction:
- Treat cells with the RNAi therapeutic (e.g., siRNA) and a negative control (e.g., non-targeting siRNA) in biological triplicate.
- Extract total RNA using a column-based or phenol-chloroform method. Assess RNA quality and integrity using an instrument such as a Bioanalyzer or TapeStation. An RNA Integrity Number (RIN) > 8.0 is generally recommended [70].
2. Library Preparation and Sequencing:
- RNA Selection: For mRNA sequencing, use poly(A) selection to enrich for messenger RNA. If working with degraded samples or non-polyadenylated RNAs, use ribosomal RNA depletion [69].
- Library Construction: Convert RNA to a cDNA library using a strand-specific protocol. This preserves information about which DNA strand was transcribed, which is crucial for accurate annotation and detecting antisense transcripts [69]. Fragment the cDNA to an appropriate size (e.g., 200-500 bp).
- Sequencing: Use a platform like Illumina for high-throughput sequencing. Aim for a minimum of 20-30 million paired-end reads per sample to ensure sufficient depth for quantifying mid-to-low abundance transcripts [69].
3. Data Analysis Workflow:
- Quality Control: Use FastQC to evaluate raw read quality. Trim adapters and low-quality bases with Trimmomatic [69].
- Alignment: Map the cleaned reads to the reference genome using a splice-aware aligner such as STAR.
- Quantification: Generate a count matrix of reads mapped to each gene using tools like HTSeq or featureCounts [70].
- Differential Expression: Identify genes with statistically significant expression changes between treatment and control groups using tools such as DESeq2 or edgeR [70]. These genes represent the putative off-target hits.
The following diagram illustrates the core workflow for an RNA-Seq experiment:
Protocol 2: Microarray-Based Off-Target Screening
This protocol is suitable for targeted profiling where the relevant pathways are well-annotated.
1. Sample Preparation and Labeling:
- Prepare RNA from treated and control cells as described in the RNA-Seq protocol.
- Convert the purified RNA to cDNA and then to cRNA, incorporating a fluorescent label (e.g., Cy3 or Cy5) during an in vitro transcription reaction.
2. Hybridization and Scanning:
- Hybridize the labeled cRNA to the microarray (e.g., Affymetrix Clariom D or similar) according to the manufacturer's instructions [71].
- After hybridization and washing, scan the array using a dedicated scanner to generate fluorescent intensity data for each probe.
3. Data Analysis:
- Normalization: Use the Robust Multi-array Average (RMA) algorithm to perform background correction, quantile normalization, and summarization of probe-level data into expression values [68].
- Differential Expression: Use a linear models approach (e.g., in Limma package in R/Bioconductor) to identify probe sets with statistically significant changes in intensity between groups.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Transcriptomic Profiling Experiments
| Reagent / Material | Function in Experiment |
|---|---|
| RNA Stabilization Reagents | Preserves RNA integrity immediately after cell lysis or tissue collection, preventing degradation that confounds results. |
| Poly(A) Selection Beads | Enriches for messenger RNA from total RNA by binding to the poly-adenylated tail, reducing ribosomal RNA background [70]. |
| rRNA Depletion Kits | Removes abundant ribosomal RNA sequences, crucial for sequencing non-poly(A) transcripts or degraded samples [69]. |
| Stranded cDNA Library Prep Kit | Creates sequencing-ready libraries while preserving strand-of-origin information, critical for accurate transcript annotation [69]. |
| Microarray Platform | Pre-fabricated slide or chip containing hundreds of thousands of DNA probes for specific gene targets [71]. |
| Fluorescent Labeling Dyes | Tags cRNA for detection during microarray scanning, allowing quantification of hybridization intensity [71]. |
Workflow and Pathway Diagrams
The following diagram outlines the logical decision-making process for designing a robust off-target detection study:
Within RNA interference (RNAi) research, a persistent challenge is the occurrence of off-target effects, where gene silencing reagents suppress unintended transcripts alongside the intended target [3] [1]. This phenomenon can lead to misleading phenotypic readouts, confounding experimental results and potentially derailing downstream applications like drug target validation. Phenotypic cross-checking—the practice of systematically correlating gene knockdown efficiency with functional outcomes—provides a critical framework for identifying and mitigating these false positives. This guide offers troubleshooting protocols and FAQs to help researchers ensure their observed phenotypes are a direct consequence of on-target knockdown.
Troubleshooting Guides and FAQs
My negative control shows a phenotypic effect. What could be wrong?
This is a classic sign of non-specific or off-target effects.
- Potential Cause 1: Sequence-Dependent Off-Targeting. Your siRNA may have partial complementarity to non-targeted mRNAs, causing miRNA-like translational repression or mRNA degradation [3] [72].
- Solution: Redesign your siRNA with stricter bioinformatic tools. Use tools that account for seed region homology (nucleotides 2-8 of the guide strand) to minimize miRNA-like off-target effects. Consider using pooled siRNAs, where a mixture of several siRNAs against the same target can dilute out individual off-target effects [3].
- Potential Cause 2: Activation of the Immune Response. The siRNA may be triggering a sequence-independent interferon response [1].
- Solution: Check the sequence for known immunostimulatory motifs. Utilize siRNAs with chemical modifications, such as 2'-O-methyl groups, which can reduce immunogenicity and improve stability [3] [72].
I have strong mRNA knockdown but no expected phenotype. What should I do?
This discrepancy suggests the knockdown may not be sufficient to elicit a functional change.
- Potential Cause 1: Incomplete Protein Knockdown. Residual protein levels may be sufficient to maintain cellular function. mRNA degradation does not always correlate perfectly with a reduction in protein levels, especially for proteins with a long half-life [72].
- Solution: Always measure protein levels (e.g., by western blot or immunofluorescence) in addition to mRNA levels. Extend the time point of your analysis to allow for pre-existing protein turnover.
- Potential Cause 2: Compensatory Mechanisms. The cell may be activating alternative pathways to compensate for the loss of your target gene.
- Solution: Perform a time-course experiment to analyze phenotypic effects at multiple time points post-knockdown. Early phenotypes may be masked by later compensatory adaptations.
How can I be confident that my phenotype is on-target?
The gold standard is to demonstrate phenotype reversal with a rescue experiment.
- Protocol: Rescue Experiment
- Clone a Rescue Construct: Create an expression vector for your target gene that is resistant to the siRNA. This is achieved by introducing silent mutations in the siRNA target site so that the mRNA sequence is altered, but the amino acid sequence of the protein remains the same.
- Transfect: Co-transfect cells with both the siRNA and the rescue construct.
- Analyze: Measure the phenotype in the presence of the siRNA alone and in the presence of both the siRNA and the rescue construct.
- Interpretation: If the phenotype is specifically due to the knockdown of your target gene, expression of the rescue construct should revert the phenotype back to wild-type conditions.
My shRNA vector shows no silencing activity. What are the common issues?
- Potential Cause 1: Mutated Inserts. shRNA sequences, particularly hairpins, can be unstable in bacteria, leading to mutations during plasmid propagation [8].
- Solution: Sequence-verify your final plasmid construct. It is recommended to screen multiple bacterial clones, as up to 20% may contain mutated inserts [8].
- Potential Cause 2: Low Transfection Efficiency or Improper Expression. The shRNA may not be being delivered or expressed effectively.
- Solution: Optimize transfection conditions and ensure your cells are at the correct confluency. Use a positive control shRNA known to be effective. For inducible systems, ensure the cell line expresses the required repressor protein (e.g., Tet repressor) and that the inducing agent (e.g., doxycycline) is active and present in the correct concentration [8] [73].
Experimental Protocols for Validation
Protocol 1: A Multi-Method Approach to Assess Knockdown Efficiency
A thorough evaluation of knockdown requires measuring its effect at multiple levels.
1. mRNA Quantification (qRT-PCR)
- Methodology: Extract total RNA from treated and control cells. Perform reverse transcription followed by quantitative PCR (qRT-PCR) using primers specific for your target gene.
- Key Controls: Include a housekeeping gene (e.g., GAPDH, β-actin) for normalization. Use non-targeting siRNA and a positive control siRNA.
2. Protein Level Analysis (Western Blotting)
- Methodology: Lyse cells and separate proteins by SDS-PAGE. Transfer to a membrane and probe with an antibody against your target protein.
- Key Controls: Use an antibody for a loading control protein (e.g., tubulin, GAPDH). This is crucial, as mRNA knockdown does not always reflect protein knockdown.
3. Functional/Phenotypic Readout
- Methodology: This is assay-specific (e.g., cell viability assay using a tetrazolium dye like MTT, apoptosis assay by flow cytometry, migration assay using a Boyden chamber).
- Key Controls: Always run the functional assay in parallel with your mRNA and protein analysis on the same batch of treated cells to enable direct correlation.
The table below summarizes the key methods for a comprehensive knockdown validation.
| Method | Target | Readout | Key Advantage |
|---|---|---|---|
| qRT-PCR | mRNA | mRNA levels | Highly sensitive, quantitative [74] |
| Western Blot | Protein | Protein levels | Confirms functional knockdown; accounts for protein half-life [72] |
| Viability/Phenotypic Assay | Cellular Function | Phenotype (e.g., proliferation, death) | Directly links gene function to biological outcome |
Protocol 2: Sensor Assay for Functional shRNA Identification
This protocol uses a single-vector reporter system to functionally identify highly potent shRNAs in a high-throughput manner, as detailed in [73].
Principle: An shRNA and its cognate target site ("Sensor") are cloned into a single vector. The Sensor is placed in the 3'UTR of a fluorescent reporter gene (e.g., Venus). When the shRNA is induced and is potent, it silences the reporter, leading to a loss of fluorescence.
Workflow:
- Clone: Generate a library of pSENSOR vectors, each containing a unique shRNA and its 50nt target sensor sequence.
- Infect & Induce: Transduce the pooled pSENSOR library into reporter cells at a low MOI to ensure single-copy integration. Induce shRNA expression (e.g., with doxycycline for a Tet-on system).
- Sort & Sequence: Use Fluorescence-Activated Cell Sorting (FACS) to separate cells with low fluorescence (indicating potent shRNA activity) from cells with high fluorescence (ineffective shRNAs).
- Identify: Isolate genomic DNA from sorted populations and sequence the integrated shRNA cassettes to identify the shRNAs enriched in the low-fluorescence population.
Functional shRNA Identification Workflow
Data Presentation: Strategies to Minimize Off-Target Effects
Various strategies have been developed to minimize the impact of off-target effects in RNAi experiments. The following table summarizes the key approaches, their mechanisms, and limitations.
| Strategy | Mechanism | Advantages | Limitations/Notes |
|---|---|---|---|
| Chemical Modifications (e.g., 2'-O-methyl) [3] [72] | Modifies siRNA sugar phosphate backbone to enhance stability and reduce immunogenicity. | Improves pharmacokinetics; reduces immune activation. | Requires optimization to maintain RISC loading and activity. |
| Pooled siRNAs [3] | Uses a mixture of several siRNAs targeting the same mRNA. | Dilutes individual off-target effects; improves on-target confidence. | Can make rescue experiments more complex; higher cost. |
| Bioinformatic Design [74] [73] | Algorithms select siRNAs with low seed homology to other transcripts. | Proactive reduction of sequence-based off-targets. | Not a guarantee; empirical validation is still required. |
| Appropriate Controls [74] | Includes non-targeting (scrambled) siRNA and positive control siRNA. | Essential for distinguishing specific from non-specific effects. | A fundamental requirement for any rigorous RNAi experiment. |
| CRISPR Knockout [1] | Creates permanent DNA breaks and gene knockouts, acting at the DNA level. | Higher specificity; fewer documented off-target effects than RNAi. | Not suitable for essential genes (lethal); knockout is permanent. |
| CRISPR Interference (CRISPRi) [1] | Uses a catalytically "dead" Cas9 to block transcription without cutting DNA. | Reversible knockdown; high specificity of DNA targeting. | Silencing may not be as complete as with catalytic Cas9. |
The Scientist's Toolkit: Research Reagent Solutions
| Reagent / Tool | Function / Description | Application Note |
|---|---|---|
| Validated siRNA | Pre-designed and experimentally tested for efficacy and specificity [74]. | Saves time and resources; ideal for initial experiments. |
| Non-Targeting siRNA | A scrambled sequence with no known genomic target [74]. | The essential negative control for measuring background and off-target effects. |
| Positive Control siRNA | An siRNA known to effectively knockdown a specific, often ubiquitous, gene [74]. | Validates that your transfection and assay conditions are working. |
| Lipid-Based Transfection Reagent | Forms liposomes that facilitate cellular uptake of nucleic acids like siRNA [74]. | Common for standard cell lines; efficiency varies by cell type. |
| Nucleofector System | Electroporation-based technology for delivering siRNA into hard-to-transfect cells [74]. | Useful for primary cells or non-dividing cells. |
| Sensor Assay Vector (pSENSOR) | Single-vector system linking shRNA expression to fluorescent reporter silencing [73]. | High-throughput functional identification of potent shRNAs. |
| Tet-On Inducible System | Allows precise, dose-dependent control of shRNA expression using doxycycline [73]. | Enables study of temporal gene function and reduces toxicity from constitutive expression. |
Troubleshooting Guides and FAQs
Frequently Asked Questions (FAQs)
Q1: What is the primary cause of off-target effects in RNAi experiments? The primary cause of off-target effects is seed sequence-mediated targeting, where the siRNA functions similarly to a microRNA (miRNA). A 6-8 nucleotide "seed" sequence (nucleotides 2-7 or 2-8 from the 5' end of the guide strand) can bind to partially complementary sites in the 3' untranslated regions (UTRs) of non-target mRNAs, leading to their unintended degradation or translational inhibition [75] [52].
Q2: When should I use long dsRNA versus siRNA? The choice depends on your experimental system:
- Long dsRNA: Use in organisms and systems that tolerate it without triggering a potent interferon response, such as plants, nematodes (C. elegans), and insects [75] [29].
- siRNA: Essential for use in mammalian systems, where long dsRNA activates the innate immune response, leading to global translational shutdown and apoptosis [75] [76].
Q3: My siRNA shows strong mRNA knockdown, but I see no phenotype. What could be wrong? This is a common issue. Please check the following:
- Protein Turnover Rate: The target protein may have a long half-life. A longer time course may be needed to see an effect on protein levels after mRNA knockdown [19].
- Assay Sensitivity: Ensure your phenotypic assay is sensitive enough to detect the level of knockdown achieved.
- Functional Redundancy: Other genes or pathways may be compensating for the loss of your target gene's function.
Q4: My positive control siRNA works, but my experimental siRNAs do not. What should I do?
- Test Multiple siRNAs: Always design and test at least 2-3 different siRNAs targeting non-overlapping regions of the same gene to confirm on-target effects [77] [19].
- Check siRNA Design: Verify that your siRNAs follow standard design rules (e.g., optimal GC content, 5'-end stability) and use updated algorithms that minimize off-target potential [29].
- Confirm mRNA Knockdown: Use a different qRT-PCR assay to ensure you are accurately measuring the mRNA levels, and check that the assay target site is not located too far from the siRNA cut site [19].
Troubleshooting Common Experimental Issues
Problem: High Cytotoxicity in Transfected Cells
- Potential Cause: The transfection reagent or the siRNA itself can be toxic to cells, especially at high concentrations.
- Solutions:
- Run a transfection reagent-only control to determine baseline toxicity.
- Titrate the siRNA concentration. Test a range of concentrations (e.g., 5-100 nM) to find the lowest effective dose [19].
- Try using different cell densities or alternative transfection reagents.
Problem: Inconsistent Knockdown Efficiency Between Replicates
- Potential Cause: Low transfection efficiency or variability in experimental handling.
- Solutions:
- Always include a validated positive control siRNA (e.g., targeting a housekeeping gene like GAPDH) to monitor transfection efficiency across experiments [19].
- Use a fluorescently labeled negative control siRNA to visually confirm and quantify the percentage of transfected cells using microscopy or flow cytometry.
- Standardize cell passage number, confluence at transfection, and reagent handling procedures.
The tables below summarize the key differences between long dsRNA and siRNA, and strategies to mitigate off-target effects.
Table 1: Specificity and Efficiency Profile of RNAi Triggers
| Feature | Long dsRNA | synthetic siRNA |
|---|---|---|
| Mechanism of Processing | Processed by Dicer into a complex pool of many siRNAs [75] | Precisely defined 21-23 nt duplex; loaded directly into RISC [75] |
| Primary Specificity Concern | Lower specificity; can silence multiple genes sharing short homologous regions due to diverse siRNA pool [29] | miRNA-like off-target effects due to seed sequence complementarity [75] [52] |
| Immune Response | Triggers strong interferon response in mammalian cells [76] | Bypasses interferon response in mammals [75] |
| RNAi Signal Amplification | Yes, in systems with RNA-dependent RNA polymerases (RdRPs) like plants and worms [75] | Typically no |
| Ideal Application | Organisms with efficient systemic RNAi (plants, insects, nematodes); pest control [75] [29] | Mammalian cell culture, therapeutic development [75] [78] |
Table 2: Strategies to Minimize Off-Target Effects in RNAi Experiments
| Strategy | Description | Key Benefit |
|---|---|---|
| Chemical Modifications | Incorporating 2'-O-methyl [76] or unlocked nucleic acid (UNA) [76] modifications in the seed region of the guide strand. | Significantly reduces miRNA-like off-targeting without compromising on-target activity. |
| siRNA Pooling | Using a pool of several siRNAs targeting the same gene at low concentrations. | Dilutes seed sequence-specific effects, reducing off-target signatures [75]. |
| Bioinformatic Design | Using algorithms that select siRNAs with minimal seed sequence matches to off-target transcripts. | Proactively avoids siRNAs with high predicted off-target potential [52]. |
| Modified siRNA Structures | Using novel designs like small internally segmented siRNAs (sisiRNAs) or paperclip RNA (pcRNA) [76] [29]. | Can alter strand selection and improve specificity. |
| Proper Controls | Using multiple siRNAs per target and scrambled negative controls. | Helps distinguish on-target from sequence-specific off-target effects [77]. |
Experimental Protocols for Specificity Assessment
Protocol 1: Microarray Analysis for Genome-Wide Off-Target Detection
This protocol is used to identify transcriptome-wide changes due to off-target effects [77] [52].
- Cell Transfection: Transfert cells with your target siRNA, a non-targeting control siRNA, and a mock transfection control. It is critical to use at least two different siRNAs against your target gene.
- RNA Isolation: Harvest cells 48 hours post-transfection (earlier time points are preferable to minimize secondary effects). Isolate total RNA using a standard method (e.g., TRIzol). Check RNA quality using an instrument like a Bioanalyzer.
- Microarray Processing: Label the RNA and hybridize it to a whole-genome expression microarray according to the manufacturer's instructions.
- Data Analysis:
- Identify genes significantly deregulated in the target siRNA sample compared to both control samples.
- Use bioinformatic tools to search for enrichment of the 6-7 nt seed sequence of your siRNA in the 3' UTRs of the deregulated genes. Tools like the Significance Analysis of Microarrays (SAM) can be used [77] [52].
Protocol 2: Validating Seed Sequence-Driven Off-Target Effects
This protocol confirms that an observed phenotype is due to a specific seed-mediated off-target effect [52].
- Identify Potential Off-Target Gene:
- From your microarray data or using prediction software, identify a candidate off-target gene with a 7-8 nt perfect match to the siRNA's seed sequence in its 3' UTR.
- Design a Rescue Experiment:
- Create a reporter construct (e.g., luciferase) where the 3' UTR of the candidate off-target gene is cloned downstream of the reporter gene.
- Reporter Assay:
- Co-transfect cells with the reporter construct and the original siRNA.
- If the siRNA directly targets the 3' UTR via its seed sequence, you will observe a dose-dependent reduction in reporter activity.
- As a control, use a reporter with a mutated seed match sequence in the 3' UTR; this should not be silenced.
- Phenotypic Rescue:
- To confirm the phenotype is off-target, attempt to rescue it by overexpressing the candidate off-target gene (lacking its 3' UTR) in the presence of the siRNA.
RNA Interference Mechanisms and Workflows
The following diagrams illustrate the core mechanisms of RNAi and a standard experimental workflow for comparing RNAi triggers.
RNAi Triggers and Specificity Pathways
RNAi Specificity Experimental Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for RNAi Specificity Research
| Reagent / Material | Function in Experiment | Key Considerations |
|---|---|---|
| Pre-designed siRNAs | Target gene knockdown. | Select vendors that guarantee >70% knockdown and provide multiple, chemically modified options (e.g., Silencer Select, Stealth RNAi) [19]. |
| Chemically Modified Nucleotides | Reduce off-target effects and improve stability. | 2'-O-methyl, UNA, or LNA modifications, particularly in the seed region (guide strand positions 2-8) [76]. |
| Positive Control siRNA | Monitor transfection efficiency and experimental conditions. | Typically targets a ubiquitous, essential gene (e.g., GAPDH, Polo-like kinase 1). |
| Fluorescent Negative Control | Determine transfection efficiency and cell viability. | A non-targeting siRNA with a fluorescent tag (e.g., Cy3, FITC). |
| Transfection Reagent | Deliver nucleic acids into cells. | Must be optimized for specific cell line and nucleic acid type (siRNA vs. long dsRNA). |
| RNA Isolation Kit | Extract high-quality total RNA for downstream analysis. | Ensure RNA is free of genomic DNA and nucleases for accurate qRT-PCR and microarray results. |
| qRT-PCR Assays | Quantify mRNA knockdown of target and potential off-target genes. | Design assays close to the siRNA cut site and validate primer efficiency [19]. |
Welcome to the Technical Support Center for RNAi Research. A primary challenge in RNA interference (RNAi) experimentation is the occurrence of off-target effects, which can lead to the misidentification of false positives and erroneous biological conclusions [23] [79]. This resource provides a structured framework and practical protocols to help you validate screening results, ensuring that reported phenotypes are due to specific on-target gene silencing.
FAQ: Understanding the Core Issue
What are the primary mechanisms of RNAi off-target effects?
Off-target effects in RNAi are primarily mediated through two mechanisms:
- Sequence-Dependent Off-Targets: This is the most common cause. The guide strand of the siRNA can behave like a microRNA (miRNA) by binding to partially complementary sequences, primarily within the 3' untranslated regions (UTRs) of non-target mRNAs [23] [79] [3]. This interaction is largely driven by perfect complementarity to the "seed region" (nucleotides 2-8, and especially 2-5, of the guide strand's 5' end) and leads to the degradation or translational repression of those mRNAs [36] [80].
- Sequence-Independent Off-Targets: The introduction of exogenous siRNA can trigger innate immune responses, such as the interferon pathway, in a sequence-independent manner [79] [1]. Furthermore, high levels of siRNA or shRNA can saturate the endogenous RNAi machinery, displacing natural miRNAs and disrupting cellular physiology [79].
How do I know if my observed phenotype is real?
A true on-target phenotype should be reproducible using multiple, independent RNAi triggers (e.g., siRNAs with different sequences) targeting the same gene. If disparate siRNAs against the same gene produce the same phenotypic outcome, it strongly suggests an on-target effect. Conversely, if different siRNAs yield conflicting results, the phenotype is likely caused by an off-target artifact [79] [36].
Troubleshooting Guide: Validating Your RNAi Hits
Problem: Inconsistent Phenotypes from Different siRNAs Targeting the Same Gene
Diagnosis: This is a classic indicator of off-target effects. The variable regions outside the seed sequence of each siRNA are likely silencing different sets of off-target genes, producing conflicting phenotypes [79].
Solution:
- Employ siRNA Pooling: Use a pooled library of 3-4 siRNAs, each targeting different regions of the same mRNA. This dilutes the concentration of any single seed sequence, thereby minimizing seed-driven off-target effects while maintaining strong on-target silencing [36].
- Validate with a Second Modality: Confirm the phenotype using a mechanistically independent technology, such as CRISPR-Cas9 knockout or CRISPR inhibition (CRISPRi). Since CRISPR acts at the DNA level, it is not susceptible to the same mRNA-level off-target effects as RNAi [1].
Problem: Widespread Transcriptomic Changes Unrelated to the Intended Target
Diagnosis: Your siRNA is likely causing extensive miRNA-like off-target silencing, often due to a highly stable seed sequence or high transfection concentrations [80].
Solution:
- Profile the Transcriptome: Use global gene expression analysis (e.g., RNA sequencing) to compare cells treated with your siRNA against a non-targeting control siRNA. This will reveal the full spectrum of genes being silenced [36].
- Refine siRNA Design: Utilize advanced design algorithms that prioritize siRNAs with low thermodynamic stability in the nucleotides 2-5 sub-region of the seed. This specific region has been shown to be critical for initiating off-target binding, and reducing its stability can minimize these effects without compromising on-target potency [80].
- Apply Chemical Modifications: Incorporate specific chemical modifications, such as 2'-O-methyl (2'-OMe) modifications, particularly in the seed region (nucleotides 2-5) of the guide strand. These modifications sterically hinder off-target binding while preserving on-target cleavage activity [36] [80].
Experimental Protocols for Robust Validation
The following workflow and detailed protocols provide a systematic approach for confirming on-target activity.
Protocol 1: Multi-siRNA and Rescue Validation
Objective: To confirm that an observed phenotype is specific to the knockdown of the target gene.
Materials:
- At least 3 independent siRNAs targeting different regions of your gene of interest.
- A non-targeting control siRNA (scramble).
- A plasmid vector for expressing a recombinant, RNAi-resistant version of your target gene (cDNA with silent mutations in the siRNA binding sites).
Method:
- Multi-siRNA Transfection: Transfect each individual siRNA and the control siRNA into your cell model in biological triplicate.
- Phenotypic Analysis: Quantify the phenotypic readout (e.g., cell viability, migration) 72-96 hours post-transfection.
- Efficiency Check: Confirm knockdown efficiency for each siRNA via qRT-PCR or immunoblotting.
- Rescue Experiment: Co-transfect the siRNA that produced the strongest phenotype along with the RNAi-resistant rescue plasmid. A control group should be co-transfected with an empty vector.
- Assessment: Measure if the expression of the recombinant protein reverses or "rescues" the phenotype.
Interpretation: A phenotype consistent across multiple siRNAs that is subsequently reversed by the rescue construct provides strong evidence for an on-target effect [79].
Protocol 2: Quantitative Luciferase Reporter Assay for Off-Target Potential
Objective: To directly measure the potential of a specific siRNA to cause seed-dependent off-target effects [80].
Materials:
- siRNA to be tested.
- psiCHECK-1 or psiCHECK-2 vector (Promega).
- Oligonucleotides containing three tandem repeats of the seed-matched (SM) sequence for your siRNA.
- Control plasmids (with perfectly matched and non-targeting sequences).
Method:
- Clone SM Sequence: Insert the synthesized SM oligonucleotide into the 3' UTR of the Renilla luciferase gene in the psiCHECK vector.
- Co-transfection: Co-transfect HeLa (or other relevant) cells with the siRNA and the constructed reporter plasmid. A firefly luciferase plasmid (e.g., pGL3-Control) serves as an internal control.
- Dual-Luciferase Assay: Perform the Dual-Luciferase assay 24 hours post-transfection.
- Calculate Activity: Normalize Renilla luciferase activity to firefly luciferase activity. Off-target activity is presented as the percentage of Renilla luminescence relative to a non-targeting control siRNA.
Interpretation: A significant reduction in Renilla luminescence indicates strong seed-mediated off-target activity for that siRNA, suggesting it should be avoided or redesigned [80].
Data Presentation: Strategies to Mitigate Off-Target Effects
The table below summarizes the primary strategies to reduce off-target effects, their mechanisms, and key considerations.
| Strategy | Mechanism of Action | Key Considerations | Experimental Tip |
|---|---|---|---|
| siRNA Pooling [36] | Dilutes concentration of any single seed sequence, reducing seed-driven off-targets. | Must ensure pools target different mRNA regions with distinct seeds. | Use commercially available pooled libraries or design in-house with bioinformatics tools. |
| Chemical Modifications [36] [80] | 2'-O-methyl modifications in seed region (nt 2-5) sterically hinder off-target binding. | Can improve nuclease resistance and reduce immunogenicity. | Focus modifications on guide strand nucleotides 2-5 to preserve on-target activity. |
| Asymmetric Design [36] | Destabilizing 5' end of passenger strand promotes RISC loading of the guide strand. | Prevents passenger strand from acting as a miRNA and silencing off-targets. | Use algorithms that consider thermodynamic asymmetry for RISC loading. |
| Bioinformatic Design [36] [80] | Machine learning algorithms predict and avoid siRNAs with high seed stability and complementarity to off-target transcripts. | Requires access to advanced software and comprehensive genomic databases. | Always BLAST siRNA sequence against the relevant transcriptome to check for homologies. |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Validation | Example Use Case |
|---|---|---|
| Non-Targeting Control siRNA | Distinguishes experimental effects from non-sequence-specific responses. | Serves as the baseline control in all experiments to account for transfection and cellular stress. |
| Multiple Independent siRNAs | Confirms phenotype is reproducible and not specific to a single siRNA's off-target profile. | Transfect 3-4 different siRNAs; consistent phenotype strengthens on-target claim [79]. |
| RNAi-Rescue Plasmid | Expresses target protein immune to siRNA, testing phenotypic causality. | Reversion of phenotype upon co-transfection confirms the target gene's role. |
| Dual-Luciferase Reporter System | Quantitatively measures the off-target potential of an siRNA via a seed-matched 3' UTR. | Used in Protocol 2 to screen and rank siRNAs for low off-target activity before full experiments [80]. |
| Chemically Modified siRNA | Reduces miRNA-like off-target binding and increases serum stability. | Applying 2'-O-methyl groups to the seed region is a precise strategy to enhance specificity [36] [80]. |
Conclusion
The successful application of RNAi, both as a research tool and a therapeutic modality, hinges on a thorough and proactive approach to managing off-target effects. As outlined, this requires a deep understanding of the underlying mechanisms, coupled with the rigorous application of strategic design, careful experimental optimization, and multi-layered validation. The convergence of advanced bioinformatic prediction tools, rational chemical modification, and precise dosing represents a powerful arsenal for enhancing specificity. For the field of drug development, where the first RNAi therapeutics have now been approved, these practices are not merely best practices but essential requirements for ensuring efficacy and patient safety. Future directions will likely involve the development of even more sophisticated prediction algorithms that account for seed accessibility in 3' UTRs, novel modification chemistries to further reduce immunostimulation, and standardized regulatory frameworks for the off-target risk assessment of RNAi-based pesticides and drugs. By systematically addressing off-target effects, researchers can unlock the full potential of RNAi to deliver precise and reliable genetic insights and life-changing therapies.
References
- 1. RNAi vs. CRISPR: Guide to Selecting the Best Gene ... [https://www.synthego.com/blog/rnai-vs-crispr-guide/]
- 2. Editorial focus: understanding off-target effects as the key to ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-019-0196-3]
- 3. RNAi Mechanisms and Strategies to Reduce Off-Target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7876455/]
- 4. CRISPR 101: Off-Target Effects [https://blog.addgene.org/crispr-101-off-target-effects]
- 5. Off-target effects in CRISPR/Cas9 gene editing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10034092/]
- 6. RNAi FAQs: siRNA Duplex and Oligo Ordering [https://www.thermofisher.com/us/en/home/life-science/rnai/rnai-misc/understand-how-rnai-works/rnai-faqs.html]
- 7. The Power of RNA Interference [https://www.numberanalytics.com/blog/harnessing-rna-interference-gene-silencing]
- 8. Vector-Based RNAi Support—Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/rnai-support-center/vector-based-rnai-support/vector-based-rnai-support-troubleshooting.html]
- 9. What is the "seed sequence" of a miRNA? [https://www.qiagen.com/us/resources/faq/2262]
- 10. Overview of MicroRNA Biogenesis, Mechanisms of Actions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6085463/]
- 11. Effective tools for RNA-derived therapeutics: siRNA ... [https://www.thno.org/v11p8771.htm]
- 12. SeedMatchR: identify off-target effects mediated by siRNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10799297/]
- 13. Resources - AUM Biotech [https://www.aumbiotech.com/resources]
- 14. Therapeutic potential of synthetic microRNA mimics based ... [https://www.nature.com/articles/s41417-025-00885-w]
- 15. Effective recognition of double-stranded RNA does not ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11980852/]
- 16. Toxicity in mice expressing short hairpin RNAs gives new ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1779583/]
- 17. Double-stranded RNA innate immune response activation ... [https://insight.jci.org/articles/view/120474]
- 18. RNA Interference (RNAi) | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/rnai.html]
- 19. RNAi Synthetics Support—Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/rnai-support-center/rnai-synthetics-support/rnai-synthetics-support-troubleshooting.html]
- 20. Endogenous spacing enables co-processing of microRNAs ... [https://www.sciencedirect.com/science/article/pii/S2667237522001084]
- 21. Classification of and detection techniques for RNAi ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1535384/full]
- 22. Off-target effects of RNAi correlate with the mismatch rate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8583100/]
- 23. Editorial focus: understanding off-target effects as the key to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6902517/]
- 24. Scientists Review RNAi-induced Effects in GM Plants [https://www.isaaa.org/kc/cropbiotechupdate/ged/article/default.asp?ID=21209]
- 25. Optimizing dsRNA sequences for RNAi in pest control and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12039203/]
- 26. Target-specific requirements for RNA interference can ... [https://elifesciences.org/articles/97487]
- 27. Risk assessment considerations for RNAi‐based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11926569/]
- 28. Guidelines for RNA Transfection [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/transfection-basics/guidelines-for-rna-transfection.html]
- 29. Assessment of RNA interference effectiveness mediated by ... [https://www.sciencedirect.com/science/article/abs/pii/S0965174825001626]
- 30. A versatile CRISPR/Cas9 system off-target prediction tool ... [https://www.nature.com/articles/s42003-025-08275-6]
- 31. CRISPR off-targets: a question of context - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7056574/]
- 32. Tools for experimental and computational analyses of off ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8049448/]
- 33. BLOG: Selecting the Right Gene Editing Off-Target Assay [https://seqwell.com/guide-to-selecting-right-gene-editing-off-target-assay/]
- 34. CRISPR Off-Target Editing: Prediction, Analysis, and More [https://www.synthego.com/blog/crispr-off-target-editing/]
- 35. RNAscope Troubleshooting Guide and FAQ [https://acdbio.com/technical-support/solutions]
- 36. Methods for reducing siRNA off-target binding [https://eclipsebio.com/eblogs/sirna-off-target-strategies/]
- 37. Position-specific chemical modification of siRNAs reduces “ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1484422/]
- 38. Therapeutic siRNA: state of the art [https://www.nature.com/articles/s41392-020-0207-x]
- 39. Asymmetric siRNA: New Strategy to Improve Specificity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3360511/]
- 40. RNAi: a revolution in functional genomics [https://www.sitoolsbiotech.com/products/sipools/rnai-introduction]
- 41. Asymmetry in siRNA design | Gene Therapy [https://www.nature.com/articles/gt200945]
- 42. Selection and Optimization of Asymmetric siRNA Targeting ... [https://www.sciencedirect.com/science/article/pii/S1016847823072539]
- 43. Are siRNA Pools Smart? [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rnai-sirna/tech-notes/are-sirna-pools-smart.html]
- 44. RNAi screening: tips and techniques - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3036573/]
- 45. Comprehensive analysis of lipid nanoparticle formulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11415597/]
- 46. A versatile antibody capture system drives specific in vivo ... [https://www.nature.com/articles/s41565-025-01954-9]
- 47. Targeted Delivery of RNAi Therapeutics With Endogenous ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2911264/]
- 48. Optimized lipid nanoparticles (LNPs) for organ-selective ... [https://www.sciencedirect.com/science/article/pii/S2589004224010265]
- 49. Antibody–Conjugated LNPs in the Spotlight [https://biocytogen.com/blogs/antibody-conjugated-lnps-precision-delivery-for-nucleic-acid-therapeutics]
- 50. Understanding the Impact of siRNA Off-Target Effects ... [https://www.linkedin.com/pulse/understanding-impact-sirna-off-target-effects-luke-mclaughlin-xouvf]
- 51. Top 10 Ways to Validate Your RNAi Data [https://www.thermofisher.com/us/en/home/references/ambion-tech-support/rnai-sirna/general-articles/top-ten-ways-to-ensure-valid-rnai-data.html]
- 52. Systematic analysis of off-target effects in an RNAi screen ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-11-175]
- 53. 10 tips on how to best optimize siRNA transfection [https://www.ptglab.com/news/blog/10-tips-on-how-to-best-optimize-sirna-transfection/?srsltid=AfmBOorxSVQB9FNziG6tGDXr2DXJ4MOcxRJSU6nwFd5zBXpn7Doa3WN7]
- 54. Protocol for Controlling the Strand Selectivity of siRNA ... [https://pubmed.ncbi.nlm.nih.gov/40110740/]
- 55. Optimizing siRNA Transfection [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/transfection-basics/guidelines-for-rna-transfection/optimizing-sirna-transfection.html]
- 56. Performing appropriate RNAi control experiments [https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/transfection/guidelines-for-transfection/performing-appropriate-rnai-control-experiments]
- 57. Seed sequences mediate off-target activity in the CRISPR- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11605690/]
- 58. Common Seed Analysis to Identify Off-Target Effects in ... [https://www.sciencedirect.com/science/article/pii/S2472555222076171]
- 59. Systematic analysis of off-target effects in an RNAi screen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2996961/]
- 60. Systematic analysis of off-target effects in an RNAi screen ... [https://pubmed.ncbi.nlm.nih.gov/20230625/]
- 61. Overcoming resistance to TRAIL-induced apoptosis in solid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4902065/]
- 62. Best Practices for Selecting Controls in Gene Expression ... [https://www.polyplus-sartorius.com/ezstop-gene-expression-experiments]
- 63. RNA Therapeutics: Delivery Problems and Solutions—A Review [https://pmc.ncbi.nlm.nih.gov/articles/PMC12567017/]
- 64. 4400003 - FAQs [https://www.thermofisher.com/order/catalog/product/4400003/faqs]
- 65. RNA-Seq: a revolutionary tool for transcriptomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2949280/]
- 66. Microarray vs. RNA-Seq: Which Is Better for Gene ... [https://rna.cd-genomics.com/resource/microarray-vs-rna-seq-gene-expression-profiling.html]
- 67. RNA-Seq vs Microarrays | Compare technologies [https://www.illumina.com/science/technology/next-generation-sequencing/beginners/advantages/rna-seq-vs-arrays.html]
- 68. Comparison of RNA-Seq and Microarray in Transcriptome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3894192/]
- 69. A survey of best practices for RNA-seq data analysis [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8]
- 70. A Beginner's Guide to Analysis of RNA Sequencing Data [https://pmc.ncbi.nlm.nih.gov/articles/PMC6096346/]
- 71. Transcriptome Profiling with Microarrays [https://www.thermofisher.com/es/en/home/life-science/microarray-analysis/transcriptome-profiling-microarrays.html]
- 72. RNA interference: Mechanism and applications [https://www.abcam.com/en-us/knowledge-center/dna-and-rna/rna-interference]
- 73. Functional identification of optimized RNAi triggers using a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3130540/]
- 74. RNA Interference to Knock Down Gene Expression - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6743327/]
- 75. siRNA Specificity: RNAi Mechanisms and Strategies to ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2020.526455/full]
- 76. RNA Interference Trigger Variants: Getting the Most Out of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3423874/]
- 77. Direct comparison of the specificity of gene silencing using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1138965/]
- 78. Advances in RNA-based therapeutics: current ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1675209/full]
- 79. Vigilance and Validation: Keys to Success in RNAi Screening [https://pmc.ncbi.nlm.nih.gov/articles/PMC3306249/]
- 80. The siRNA Off-Target Effect Is Determined by Base-Pairing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8872465/]