GUIDE-seq Decoded: The Ultimate Protocol for Genome-Wide CRISPR Off-Target Detection
This comprehensive guide explores GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing), the premier experimental method for unbiased, genome-wide detection of CRISPR-Cas nuclease off-target effects.
GUIDE-seq Decoded: The Ultimate Protocol for Genome-Wide CRISPR Off-Target Detection
Abstract
This comprehensive guide explores GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing), the premier experimental method for unbiased, genome-wide detection of CRISPR-Cas nuclease off-target effects. Tailored for researchers and drug development professionals, the article provides foundational knowledge on the necessity of off-target profiling in therapeutic applications, a step-by-step methodological deep dive, expert troubleshooting and optimization strategies, and a critical validation framework comparing GUIDE-seq to alternative techniques like CIRCLE-seq, Digenome-seq, and SITE-seq. The content synthesizes current best practices to empower scientists to implement robust off-target screening, a critical step for ensuring the safety and efficacy of genome editing technologies.
Why Off-Targets Matter: The Critical Foundation of GUIDE-seq for Safe Genome Editing
Application Notes: The GUIDE-seq Workflow in Therapeutic Development
Genome-wide off-target detection is a critical component in the safety assessment of therapeutic genome-editing agents. GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by sequencing) remains a foundational, highly sensitive method for the unbiased identification of off-target sites. Its application spans from initial tool characterization in basic research to mandatory safety packages for regulatory submissions in clinical trials.
Table 1: Comparative Analysis of Key Off-Target Detection Methods
| Method | Principle | Sensitivity (Theoretical) | Requires Pre-Defined Sites? | Primary Application Stage |
|---|---|---|---|---|
| GUIDE-seq | Capture of double-stranded oligodeoxynucleotide tags into DSBs | High (detects ~0.1% of sequencing reads) | No | Basic Research, Preclinical Safety |
| CIRCLE-seq | In vitro circularization and amplification of off-target cleaved genomic DNA | Very High (detects ~0.01% of events) | No | Preclinical Safety, Lead Optimization |
| Digenome-seq | In vitro digestion of genomic DNA and whole-genome sequencing | High | No | Preclinical Safety |
| SITE-seq | In vitro capture of Cas9-cleaved DNA ends | High | No | Preclinical Safety |
| Targeted NGS | Deep sequencing of predicted off-target loci | N/A (Targeted) | Yes | Clinical Lot Release, Patient Monitoring |
Key Insight: GUIDE-seq provides a critical in cellulo snapshot of nuclease activity within a native chromatin context, making its data indispensable for early-risk identification, even when followed by ultra-sensitive in vitro methods like CIRCLE-seq for comprehensive profiling.
Detailed Experimental Protocol: GUIDE-seq
A. Materials and Cell Preparation
- Cells: HeLa or relevant therapeutic cell line (e.g., iPSCs, primary T-cells).
- Transfection Components: Cas9 nuclease (or mRNA), sgRNA, and the GUIDE-seq oligonucleotide (dsODN).
- GUIDE-seq dsODN: A blunt, double-stranded, 34-bp phosphorothioate-modified oligo. Sequence example: 5′-/5Phos/GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′ (T = phosphorothioate).
B. Step-by-Step Workflow
- Co-transfection: Transfect 200,000-500,000 cells with a complex of Cas9:sgRNA RNP (e.g., 100 pmol Cas9, 120 pmol sgRNA) and 100 pmol of GUIDE-seq dsODN using an appropriate method (e.g., nucleofection for primary cells).
- Genomic DNA Extraction: Harvest cells 72 hours post-transfection. Extract high-molecular-weight gDNA using a silica-column or magnetic bead-based method.
- Shearing and Size Selection: Shear gDNA to ~400 bp fragments (e.g., via sonication). Perform size selection using SPRI beads to enrich fragments of 300-500 bp.
- End Repair & A-tailing: Use a commercial end repair/dA-tailing module to prepare fragments for adapter ligation.
- Adapter Ligation: Ligation of Illumina-compatible Y-shaped adapters.
- dsODN-Specific PCR Enrichment: Perform two sequential nested PCRs (12-15 cycles each) using primers specific to the dsODN sequence and adapter sequence to exclusively amplify fragments containing the integrated dsODN tag.
- Library Purification & QC: Purify PCR products with SPRI beads. Assess library quality via Bioanalyzer/TapeStation and quantify by qPCR.
- Sequencing: Pool and sequence on an Illumina platform (Minimum depth: 20-30 million paired-end 150 bp reads per sample).
- Data Analysis: Process reads using the publicly available GUIDE-seq software suite to map dsODN integration sites, identify off-target loci, and quantify read counts.
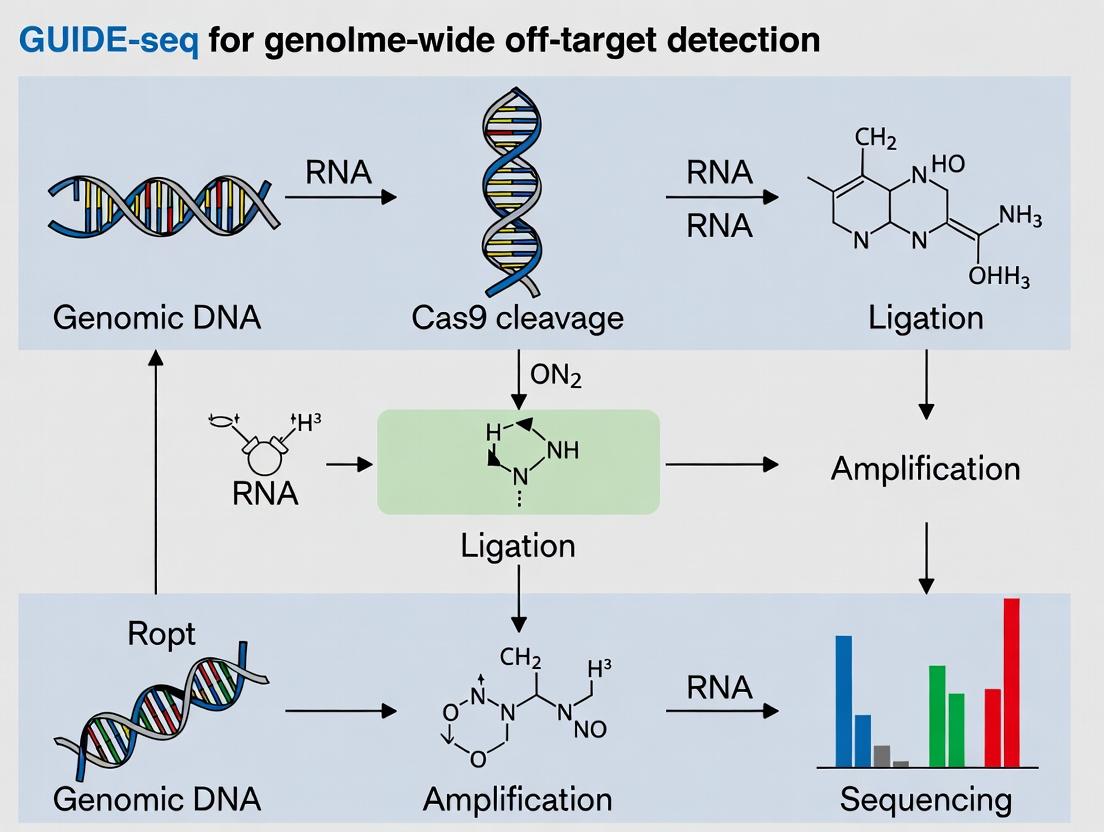
Visualization: The GUIDE-seq Pathway and Workflow
Title: GUIDE-seq Molecular Pathway and Experimental Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for GUIDE-seq and Off-Target Analysis
| Item | Function & Specification | Example Provider/Cat. No. |
|---|---|---|
| Recombinant Cas9 Nuclease | High-activity, endotoxin-free nuclease for RNP formation. | IDT, Thermo Fisher, Aldevron |
| Chemically Modified sgRNA | Enhanced stability and reduced immunogenicity; 2'-O-methyl 3' phosphorothioate modifications. | Synthego, IDT |
| GUIDE-seq dsODN | Blunt, 34-bp, phosphorothioate-modified double-stranded tag for NHEJ capture. | Custom synthesis (IDT, Eurofins). |
| Next-Generation Sequencing Kit | High-fidelity library preparation for Illumina platforms. | Illumina DNA Prep, NEB Next Ultra II |
| dsODN-Specific PCR Primers | Nested primer sets for specific amplification of tagged genomic fragments. | Custom oligonucleotides. |
| Cell Transfection/Nucleofection Kit | For efficient delivery of RNP + dsODN into difficult cell types. | Lonza Nucleofector, NEON Transfection System |
| Genomic DNA Extraction Kit | For high-integrity, high-molecular-weight gDNA from low cell inputs. | Qiagen DNeasy, Zymo Quick-DNA |
| Bioinformatics Pipeline | Software for mapping dsODN integration sites and identifying off-targets. | GUIDE-seq (open-source), CRISPResso2. |
Within the broader thesis investigating genome-wide off-target effects of CRISPR-Cas9 and other programmable nucleases, GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by sequencing) serves as a foundational, high-sensitivity method for unbiased off-target detection. This application note details its core principle of tagging double-strand breaks (DSBs) with oligonucleotides, providing the protocols necessary to integrate this technique into comprehensive genome-editing safety assessments critical for therapeutic drug development.
Core Principle
The GUIDE-seq method leverages the cell's endogenous DNA repair machinery to integrate a double-stranded, blunt-ended oligonucleotide (the "GUIDE-seq tag") directly into genomic DSBs generated by a programmable nuclease. This tag then serves as a unique molecular handle for PCR amplification and next-generation sequencing (NGS) to map the precise genomic locations of both on-target and off-target cleavage events genome-wide.
Key Research Reagent Solutions
| Item | Function in GUIDE-seq |
|---|---|
| dsODN Tag | A blunt-ended, 5'-phosphorylated, 34-bp double-stranded oligonucleotide with a central degeneracy. Serves as the repair template integrated into DSBs. The critical reagent for break tagging. |
| Transfection Reagent | For co-delivery of the nuclease components (e.g., Cas9/gRNA RNP or plasmid) and the dsODN tag into target cells. Lipofectamine-based or electroporation systems are common. |
| PCR Primers (Tag-Specific) | Oligonucleotides designed to bind the constant region of the integrated dsODN tag, enabling specific amplification of tagged genomic loci. |
| High-Fidelity Polymerase | Used for the primary and nested PCRs to amplify tag-integrated sites with high accuracy and yield for NGS library preparation. |
| NGS Library Prep Kit | A kit for constructing Illumina-compatible sequencing libraries from the amplified GUIDE-seq products. |
| Genomic DNA Isolation Kit | For clean, high-molecular-weight genomic DNA extraction post-transfection. |
| Surveyor/T7 Endonuclease I | (Optional) For initial validation of nuclease activity at the intended on-target site prior to GUIDE-seq. |
Detailed Protocols
Protocol 1: dsODN Tag Design and Preparation
- Design: Synthesize two oligonucleotides: GUIDE-seq-Oligo-A: 5'-/5Phos/-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCNNNNNNNNNNNNNNNNNNNN-3' and GUIDE-seq-Oligo-B: 5'-/5Phos/-/iSp18//iSp18/NAGATCGGAAGAGCACACGTCTGAACTCCAGTCAC-3'. The "N" denotes random degenerate bases, and /iSp18/ is a hexa-ethylene glycol spacer.
- Annealing:
- Resuspend oligos in annealing buffer (10 mM Tris, pH 8.0, 50 mM NaCl, 1 mM EDTA) to 100 µM.
- Mix equimolar amounts of Oligo-A and Oligo-B.
- Heat to 95°C for 5 minutes in a thermal cycler, then ramp down to 25°C at 0.1°C/sec.
- The resulting dsODN is 5'-phosphorylated, blunt-ended, and ready for use.
Protocol 2: Cell Transfection and Tag Integration
- Cell Seeding: Seed 2e5 - 4e5 HEK293T or other target cells per well in a 24-well plate 18-24 hours prior.
- Transfection Mix:
- For RNP delivery: Complex 2 µg of purified Cas9 protein, 200 pmol of synthetic gRNA, and 100 pmol of annealed dsODN tag.
- Use an optimized transfection reagent (e.g., Lipofectamine CRISPRMAX) according to the manufacturer's protocol.
- Delivery: Add complexes to cells. Include controls: nuclease only (no tag) and tag only (no nuclease).
- Incubation: Culture cells for 48-72 hours to allow for DSB generation, tag integration, and repair.
Protocol 3: Genomic DNA Isolation and PCR Amplification
- gDNA Extraction: Harvest cells and extract genomic DNA using a silica-column based kit. Elute in 50-100 µL of elution buffer. Quantify by spectrophotometry.
- Primary PCR:
- Set up 50 µL reactions: 100 ng gDNA, 0.5 µM Tag-specific Primer 1, 0.5 µM Tag-specific Primer 2, 1x High-Fidelity PCR Master Mix.
- Cycling: 98°C 30s; (98°C 10s, 65°C 30s, 72°C 30s) x 25 cycles; 72°C 5 min.
- Nested PCR:
- Dilute primary PCR product 1:50.
- Use 2 µL as template with nested tag-specific primers containing full Illumina adapter sequences.
- Cycling: 98°C 30s; (98°C 10s, 65°C 30s, 72°C 30s) x 15 cycles; 72°C 5 min.
- Clean-up: Purify the nested PCR product using SPRI beads. Quantify by qPCR or bioanalyzer.
Protocol 4: Sequencing and Data Analysis
- Sequencing: Pool libraries and sequence on an Illumina MiSeq or HiSeq platform (2x 150 bp or 2x 250 bp recommended).
- Bioinformatics:
- Use the original GUIDE-seq software or pipelines like GUIDE-seqScan or CRISPRseek.
- Steps: Demultiplex, align reads to reference genome, identify reads containing tag sequence, cluster tag integration sites, and score off-target sites.
Table 1: Typical GUIDE-seq Experimental Parameters and Outcomes
| Parameter | Typical Value / Outcome | Notes |
|---|---|---|
| dsODN Tag Concentration | 100 - 500 pmol per transfection (24-well) | Critical for sensitivity; too high can increase background. |
| Tag Integration Efficiency | ~1-5% of total reads contain tag | Measured by percentage of NGS reads with tag sequence. |
| Background (No Nuclease Control) | < 0.5% of on-target reads | Defines the detection threshold for true off-targets. |
| Detection Sensitivity | Can identify sites with < 0.1% modification frequency | More sensitive than in silico prediction or ChIP-based methods. |
| Typical Off-Targets per gRNA | 0 - 15+ | Varies greatly with gRNA sequence and nuclease specificity. |
| Sequencing Depth Required | 10 - 30 million reads per sample | Ensures coverage for low-frequency off-target detection. |
Visualizations
Title: GUIDE-seq Workflow: From DSB to Detection
Title: Molecular Detail of dsODN Tag Integration via NHEJ
Within the broader thesis of advancing CRISPR-Cas therapeutic safety, GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) represents a pivotal methodology for the comprehensive profiling of nuclease off-target effects. Its core advantages make it indispensable for rigorous preclinical assessment in drug development.
The following table quantifies the key performance metrics of GUIDE-seq against earlier off-target detection methods.
Table 1: Comparison of Genome-Wide Off-Target Detection Methods
| Method | Principle | Sensitivity (Theoretical) | Specificity | Requires Prior Target Prediction? | Experimental Bias |
|---|---|---|---|---|---|
| GUIDE-seq | Capture of double-stranded oligodeoxynucleotide (dsODN) integration at DSB sites. | Can detect sites with <0.1% frequency of indels (Tsai et al., 2015). | High (relies on physical tag integration). | No (Unbiased) | Minimal; tag integration efficiency can vary. |
| CIRCLE-seq | In vitro circularization and amplification of nuclease-cleaved genomic DNA. | Extremely high on purified genomic DNA. | High in vitro, but may identify sites not cleaved in cells. | No (Unbiased) | Bias from in vitro digestion and amplification. |
| BLISS | Direct ligation of adapters to DSB ends in situ. | Detects individual DSB events; quantitative. | High. | No (Unbiased) | Requires optimized in situ ligation. |
| Digenome-seq | In vitro digestion of genomic DNA followed by whole-genome sequencing. | High on purified DNA. | High in vitro, similar to CIRCLE-seq. | No (Unbiased) | Bias from in vitro digestion conditions. |
| CHAMP | Computational prediction based on sequence homology. | Varies with algorithm. | Low to moderate; high false-positive rate. | Yes (Biased) | Purely computational, no experimental data. |
Detailed Application Notes & Protocols
Application Note 1: Validating Therapeutic gRNA Specificity
- Context: Prior to IND (Investigational New Drug) application for a CRISPR-based therapy.
- Procedure: Transfert target cells (e.g., iPSC-derived cardiomyocytes, primary T-cells) with Cas9/gRNA RNP complexes alongside the GUIDE-seq dsODN tag. Harvest genomic DNA 72 hours post-transfection. Enrich for tag-integrated sites via PCR and prepare sequencing libraries.
- Outcome: A genome-wide map of on- and off-target sites with indel frequencies. Confirmation of <3-5 high-confidence off-target sites with frequencies >0.1% typically triggers gRNA re-design or protein engineering (e.g., high-fidelity Cas9 variants).
Application Note 2: Profiling Novel Nuclease Platforms
- Context: Characterizing the specificity profile of a newly engineered Cas nuclease (e.g., Cas12a variant, base editor).
- Procedure: Adapt the dsODN tag sequence if necessary (some nucleases have distinct end-chemistry). Co-deliver nuclease, gRNA, and tag. Analysis must account for the nuclease's characteristic DSB or nick profile (e.g., staggered vs. blunt ends).
- Outcome: A specificity "fingerprint" for the novel enzyme, enabling direct comparison to SpCas9 or other benchmarks, crucial for platform selection in drug development.
Experimental Protocol: GUIDE-seq Workflow
Key Research Reagent Solutions:
- GUIDE-seq dsODN: A blunt, double-stranded, 5'-phosphorylated 34-bp oligodeoxynucleotide with a 5' biotin modification. Function: Serves as the exogenous tag integrated into genomic DSBs.
- Streptavidin C1 Dynabeads: Magnetic beads. Function: To capture biotinylated dsODN-integrated genomic fragments.
- Next-Generation Sequencing (NGS) Library Prep Kit (e.g., Illumina): Function: To prepare amplified tag-integrated sites for high-throughput sequencing.
- Cas9 Nuclease (WT or HiFi): Function: Creates the DSBs at target and off-target sites.
- Transfection Reagent (e.g., Lipofectamine CRISPRMAX): Function: For efficient delivery of RNP and dsODN into target cells.
- GUIDE-seq Analysis Software (e.g., GUIDE-seq software, CRIS.py): Function: Bioinformatics pipeline to align sequencing reads and identify significant off-target integration sites from background.
Protocol:
- dsODN Design & Preparation: Resuspend the HPLC-purified dsODN duplex at 100 µM in nuclease-free buffer.
- Cell Transfection: For a 24-well plate, complex 100-200 pmol of Cas9 protein, 100-200 pmol of synthetic gRNA, and 100 pmol of dsODN to form the RNP. Transfect into 70-80% confluent cells using a lipid-based transfection reagent optimized for RNP delivery.
- Genomic DNA (gDNA) Extraction: Harvest cells 72 hours post-transfection. Isolate high-molecular-weight gDNA using a silica-column or magnetic bead-based kit.
- Shearing & Size Selection: Shear 1-3 µg of gDNA to an average fragment size of 300-400 bp using a focused-ultrasonicator. Perform a double-sided size selection (e.g., with SPRI beads) to retain fragments between 200-600 bp.
- Biotinylated Fragment Enrichment: Incubate sheared, size-selected DNA with Streptavidin C1 beads overnight at room temperature with rotation. Wash beads stringently.
- On-Bead Library Preparation: Perform end-repair, A-tailing, and adapter ligation directly on the beads. Elute the library via heat denaturation.
- Amplification & Sequencing: Amplify the eluted library with indexed primers for 14-18 PCR cycles. Purify the final library and sequence on an Illumina platform (2x150 bp recommended).
- Bioinformatic Analysis: Process fastq files using the standard GUIDE-seq computational pipeline (alignment with BWA, peak calling, and off-target site identification).
Visualizations
Diagram 1: GUIDE-seq Experimental Workflow
Diagram 2: GUIDE-seq Data Analysis Logic Pathway
Introduction Within the broader thesis on comprehensive GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by sequencing) methodology for genome-wide off-target detection in therapeutic genome editing, this document details the core functional components. The reliable detection of nuclease-induced double-strand breaks (DSBs) hinges on the precise interplay of a defined double-stranded oligodeoxynucleotide (dsODN) tag, the nuclease of interest, and a robust sequencing workflow.
Core Components
- dsODN Tag: A short, blunt-ended, double-stranded DNA molecule with a known, non-genomic sequence. Its 5' ends are phosphorylated to facilitate direct ligation into nuclease-induced DSBs.
- Nuclease: The genome-editing agent (e.g., Cas9-gRNA RNP, TALEN, ZFN) whose off-target cleavage profile is being characterized.
- Sequencing Workflow: The multi-step molecular biology and computational pipeline to capture, amplify, and analyze dsODN-tagged genomic sites.
Research Reagent Solutions Toolkit
| Item | Function in GUIDE-seq |
|---|---|
| Defined dsODN Tag | Serves as the molecular marker for DSB capture. Its known sequence allows for specific PCR enrichment of tagged genomic loci. |
| Cationic Lipid Transfection Reagent | Enables efficient co-delivery of the dsODN tag and nuclease (as RNP or plasmid) into target cells. |
| High-Fidelity DNA Polymerase | Used in the primary nested PCR to specifically amplify dsODN-tagged genomic fragments with minimal errors. |
| Magnetic Beads for Size Selection | Critical for purifying and size-selecting adapter-ligated libraries prior to final PCR to ensure appropriate fragment length for sequencing. |
| Paired-End High-Throughput Sequencing Platform | Enables precise mapping of junction reads containing both genomic sequence and the integrated dsODN tag. |
Detailed Protocol: GUIDE-seq Workflow
1. Cell Transfection & dsODN Integration
- Materials: Cultured mammalian cells, nuclease (e.g., pre-complexed Cas9-gRNA RNP), defined dsODN tag (e.g., 100 µM stock), transfection reagent, appropriate cell culture medium.
- Procedure:
- Co-deliver the nuclease (e.g., 100 pmol RNP) and dsODN tag (e.g., 100 fmol) into 1-2x10⁵ cells per condition using a cationic lipid transfection system according to the manufacturer's protocol.
- Incubate cells for 48-72 hours to allow for nuclease activity, DSB generation, and cellular repair with dsODN integration.
- Harvest genomic DNA using a silica-column or magnetic bead-based purification method. Elute in 50-100 µL of elution buffer.
2. Primary Nested PCR Enrichment of Tagged Sites
- Materials: Purified genomic DNA, dsODN-specific primer (Primer_SSODN), gene-specific primer 1 (GSP1), high-fidelity PCR master mix.
- Procedure (First Round):
- Set up a 50 µL reaction: 100 ng genomic DNA, 0.5 µM Primer_SSODN, 0.5 µM GSP1, 1x high-fidelity polymerase mix.
- Cycling: 98°C for 30s; 15 cycles of (98°C for 10s, 69°C for 30s, 72°C for 30s); 72°C for 1 min.
- Procedure (Second Round - Nested):
- Use 1 µL of the first-round product as template.
- Perform a second PCR using a nested dsODN-specific primer (PrimerSSODNnested) and a nested gene-specific primer (GSP2). Use the same cycling conditions but increase cycles to 20.
3. Library Preparation & Sequencing
- Materials: Purified nested PCR product, library preparation kit (end-repair/A-tailing, adapter ligation), size selection beads, indexing primers.
- Procedure:
- Purify the nested PCR product using magnetic beads.
- Perform end-repair/A-tailing and adapter ligation using a commercial library prep kit.
- Perform a second bead-based size selection (e.g., 0.55x left-side, 0.85x right-side) to isolate fragments ~200-500 bp.
- Amplify the final library with indexed primers for 12-15 cycles.
- Quantify by qPCR, validate fragment size by capillary electrophoresis, and sequence on a paired-end 150-300 bp platform (Illumina).
Data Presentation: Key Experimental Parameters
Table 1: Typical Quantitative Parameters for GUIDE-seq Experiments
| Parameter | Typical Range / Value | Notes |
|---|---|---|
| dsODN Concentration | 50 - 200 fmol per transfection | Higher amounts increase background. |
| Nuclease Amount (RNP) | 50 - 200 pmol per transfection | Must be titrated for optimal on-target activity. |
| Cell Number | 1x10⁵ - 2x10⁵ per condition | Sufficient for genomic DNA yield. |
| Primary PCR Cycles | 12 - 18 cycles | Minimize to reduce PCR bias. |
| Nested PCR Cycles | 15 - 20 cycles | Optimize for sufficient library yield. |
| Sequencing Depth | 20 - 50 million paired-end reads | Ensures saturation for off-target detection. |
Visualization: GUIDE-seq Experimental Workflow
dsODN Integration & Library Construction
dsODN Tag Capture of a DSB Site
Within the broader thesis of GUIDE-seq genome-wide off-target detection research, the evolution of methodologies represents a critical narrative. This thesis posits that the transition from early, labor-intensive techniques to current streamlined, high-sensitivity standards has been the primary enabler for the therapeutic application of CRISPR-Cas systems. This application note details the key methodologies in this evolution, providing protocols and analytical frameworks essential for researchers and drug development professionals.
Table 1: Evolution of Genome-Wide Off-Target Detection Methods
| Method (Year) | Core Principle | Detection Sensitivity (Theoretical) | Key Advantages | Key Limitations | Typical Experimental Duration |
|---|---|---|---|---|---|
| GUIDE-seq (2015) | Integration of oligonucleotide duplexes into DSBs followed by amplification and sequencing. | ~0.1% of total reads | Unbiased, sensitive, works in cells. | Requires oligonucleotide delivery, background in high DSB contexts. | 10-14 days |
| BLISS (2016) | Direct in situ ligation of adapters to DSB ends followed by sequencing. | Single-cell; quantitative. | Direct DSB labeling, single-cell resolution. | Lower complexity, requires high sequencing depth. | 7-10 days |
| BLISS (2017) | Digested genome ligation to hairpin adapters and sequencing. | Highly sensitive (can detect rare sites). | In vitro, no transfection bias, very sensitive. | Does not capture cellular repair context. | 5-7 days |
| SITE-Seq (2017) | In vitro Cas9 digestion of genomic DNA, hairpin adapter ligation, and sequencing. | Highly sensitive (can detect rare sites). | In vitro, no transfection bias, very sensitive. | Does not capture cellular repair context. | 5-7 days |
| DISCOVER-Seq (2019) | Immunoprecipitation of MRE11-bound DSB ends. | Endogenous marker; works in vivo. | Utilizes endogenous repair machinery, applicable in vivo. | Relies on MRE11 recruitment kinetics. | 10-12 days |
| CIRCLE-Seq (2019) | Circularization of sheared genomic DNA, in vitro digestion, and linearization of cut circles. | Extremely sensitive (<0.01%). | Ultra-high sensitivity, low background. | In vitro, complex library prep. | 7-9 days |
| LEDGAR-seq (2024) | CRISPR-Cas12-based amplification of GUIDE-seq tags. | ~10-100x more sensitive than GUIDE-seq. | Exceptional sensitivity, retains cellular context. | Newer method, less community validation. | 12-15 days |
Detailed Experimental Protocols
Protocol A: Original GUIDE-seq Workflow
Title: Detailed Protocol for Genome-wide, Unbiased Identification of DNA Double-Strand Breaks Induced by Engineered Nucleases.
Key Research Reagent Solutions:
- GUIDE-seq Oligoduplex: A 34-bp double-stranded oligonucleotide with phosphorothioate modifications. Function: Integrates into nuclease-induced DSBs via NHEJ, serving as a tag for amplification.
- 5´-Phosphorylated adapters (ILL-INI-5P, ILL-INI-3P): Function: For Illumina library preparation from amplified genomic DNA containing the integrated oligo.
- PCR Primers (GUIDE-seq Primer 1 & 2): Function: Specifically amplify genomic regions flanking the integrated oligoduplex tag.
- High-Fidelity DNA Polymerase (e.g., Q5): Function: Ensures accurate amplification of target regions for sequencing.
- Magnetic Beads for Size Selection (e.g., SPRI beads): Function: Clean and size-select DNA fragments post-amplification and adapter ligation.
Procedure:
- Cell Transfection: Co-transfect cultured cells (e.g., HEK293T) with plasmid(s) encoding the CRISPR-Cas nuclease and the GUIDE-seq oligoduplex using a preferred method (lipofection, electroporation). Critical: Optimize oligoduplex concentration (typical range 50-200 pmol).
- Genomic DNA (gDNA) Harvest: 72 hours post-transfection, harvest cells and extract high-molecular-weight gDNA using a silica-column or phenol-chloroform method.
- Fragmentation & Size Selection: Shear 1-3 µg of gDNA to an average fragment size of 400-500 bp using a focused-ultrasonicator. Perform size selection using SPRI beads to enrich fragments of 300-600 bp.
- End Repair, A-tailing, and Adapter Ligation: Perform standard Illumina library prep steps on the sheared DNA: end repair, 3´ adenylation, and ligation of phosphoryated Illumina adapters.
- Primary GUIDE-seq PCR: Perform the first PCR (15-18 cycles) using the GUIDE-seq Primer 1 and Primer 2, which bind the integrated oligo and the Illumina adapter, respectively.
- Secondary Indexing PCR: Perform a second, limited-cycle PCR (4-8 cycles) to add full Illumina P5/P7 flow cell binding sites and unique dual indices (UDIs) for sample multiplexing.
- Library Purification & QC: Purify the final library using SPRI beads. Quantify by Qubit and analyze fragment distribution by Bioanalyzer/TapeStation.
- Sequencing & Analysis: Sequence on an Illumina platform (2x150 bp or 2x250 bp recommended). Process data using the original GUIDE-seq analysis pipeline (alignment, tag identification, off-target site calling).
Protocol B: CIRCLE-Seq for Ultra-Sensitive In Vitro Detection
Title: Protocol for Circularization for In Vitro Reporting of Cleavage Effects by Sequencing.
Key Research Reagent Solutions:
- Cas9 RNP Complex: Pre-complexed recombinant Cas9 protein and synthetic sgRNA. Function: Provides highly active nuclease for in vitro digestions.
- Circligase ssDNA Ligase: Function: Catalyzes the intramolecular circularization of single-stranded DNA, a key step to eliminate background.
- Hairpin Adapter (Splint Oligo): Function: Bridges the ends of a Cas9-cut site during ligation to create a PCR-amplifiable template.
- Phi29 DNA Polymerase: Function: Performs rolling circle amplification (RCA) of circularized DNA, linearly amplifying cleaved sites.
Procedure:
- gDNA Isolation & Shearing: Extract gDNA from untreated cells. Mechanically shear 3 µg of gDNA to ~300 bp fragments.
- End Repair & 3´ Adenylation: Perform blunt-end repair and add a single 3´-A overhang to the sheared fragments.
- Circularization: Treat the A-tailed DNA with Circligase to promote self-circularization of fragments. This step eliminates free ends from shearing.
- Cas9 RNP Cleavage In Vitro: Incubate the circularized DNA library with pre-assembled Cas9 ribonucleoprotein (RNP) targeting the locus of interest. Only circles containing the target site will be linearized by cleavage.
- Hairpin Adapter Ligation: Ligate a biotinylated hairpin adapter to the newly created DSB ends in the linearized circles. This adapter contains a sequencing primer binding site.
- Rolling Circle Amplification (RCA): Treat the ligated product with exonuclease to remove non-circular DNA, then amplify using Phi29 polymerase, which extends from the hairpin primer.
- Library Generation & Sequencing: Fragment the RCA product, perform Illumina adapter ligation and PCR. Sequence and analyze using the CIRCLE-seq bioinformatics tool to identify cleaved sites.
Visualized Workflows and Relationships
Title: Comparison of GUIDE-seq and CIRCLE-seq Workflows.
Title: Evolution Timeline of Off-Target Detection Methods.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Off-Target Detection Experiments
| Reagent Category | Specific Example/Name | Function in Protocol | Critical Considerations |
|---|---|---|---|
| Tagging Molecule | GUIDE-seq Oligoduplex (dsODN) | Integrates into DSBs in cells to tag cleavage sites. | Phosphorothioate modifications essential for stability. Optimal concentration must be titrated. |
| Nuclease Delivery | Cas9 Ribonucleoprotein (RNP) | Provides immediate nuclease activity; used in vitro (CIRCLE-seq) or for editing. | Reduces time for off-target activity vs. plasmid expression. High purity required. |
| Adapter/Linker | Phosphorylated Illumina Adapters / Hairpin Adapters | Allows for PCR amplification and sequencing of tagged or cleaved fragments. | Ligation efficiency is critical for library complexity. Hairpin adapters reduce adapter-dimer formation. |
| Specialized Enzymes | Circligase ssDNA Ligase (CIRCLE-seq) | Circularizes sheared DNA to eliminate background from free ends. | High enzyme fidelity is required to maintain sequence integrity. |
| Specialized Enzymes | Phi29 DNA Polymerase (CIRCLE-seq) | Performs Rolling Circle Amplification (RCA) of circularized, cleaved DNA. | Provides high-fidelity, linear amplification from minimal input. |
| DNA Repair Factor Binder | MRE11 Antibody (DISCOVER-Seq) | Immunoprecipitates DNA bound by the endogenous MRE11 repair complex at DSBs. | Antibody specificity and affinity are paramount for clean signal. |
| Library Prep Beads | SPRI (Solid Phase Reversible Immobilization) Beads | Size selection and purification of DNA fragments during library preparation. | Bead-to-sample ratio dictates size cut-off; critical for reproducibility. |
| High-Fidelity Polymerase | Q5 or KAPA HiFi Polymerase | Amplifies target regions with minimal error during library PCR steps. | Essential for accurate representation of sequences, especially for low-frequency sites. |
Mastering the GUIDE-seq Protocol: A Step-by-Step Application Guide
Within the broader thesis investigating GUIDE-seq as a genome-wide off-target detection method for therapeutic CRISPR-Cas applications, the initial experimental design phase is critical. The design and incorporation of the double-stranded Oligodeoxynucleotide (dsODN) donor tag fundamentally enable the sensitive detection of double-strand breaks (DSBs). This protocol details best practices for this foundational phase.
Key Design Parameters for dsODNs
The dsODN serves as the molecular tag that integrates into CRISPR-Cas-induced DSBs. Its design directly influences tagging efficiency and detection sensitivity. Current literature and experimental data emphasize the following optimized parameters:
Table 1: Optimized dsODN Design Specifications
| Parameter | Recommended Specification | Rationale & Impact |
|---|---|---|
| Structure | Double-stranded, blunt-ended | Facilitates direct ligation into blunt-ended DSBs generated by SpCas9. |
| Length | 34-36 bp double-stranded region | Optimal for stable integration and PCR amplification; longer sequences may reduce efficiency. |
| Overhangs | None (blunt) or 5'-phosphorylated | Blunt ends mimic natural DSB ends; 5'-phosphorylation may enhance ligation. |
| Core Sequence | Non-homologous to target genome | Prevents homologous recombination, ensuring tag integration primarily via NHEJ at DSB sites. |
| Asymmetric PCR Handles | Unique 20-24 bp sequences on each strand | Enables specific, nested PCR amplification of tagged genomic loci for library preparation. |
| Purification | HPLC or PAGE-purified | Critical for high yield and to remove truncated oligos that can reduce signal-to-noise. |
Detailed Protocol: dsODN Design, Preparation, and Validation
Protocol 1:In SilicoDesign of the dsODN Tag
- Generate Core Sequence: Use a random sequence generator to create a 34 bp sequence (e.g., 5'-NNNN...-3'). Analyze this sequence in silico using BLAST against the reference genome of your experimental model to ensure it lacks significant homology (>15 bp contiguous match).
- Append Asymmetric Handles: To the 5' end of one strand of the core, append a unique 24 bp sequence (Handle A). To the 5' end of the complementary strand, append a different unique 24 bp sequence (Handle B). The final single-stranded oligos will be 58-60 nt.
- Order Oligos: Specify synthesis scale (typically 100 nmole, PAGE-purified) and 5' phosphorylation for both oligos to facilitate ligation.
Protocol 2: dsODN Annealing and Purification
Materials:
- Complementary HPLC/PAGE-purified single-stranded oligos.
- Annealing Buffer (10x: 100 mM Tris, 500 mM NaCl, 10 mM EDTA, pH 8.0).
- Nuclease-free water.
- Thermal cycler.
Method:
- Resuspend each oligo in nuclease-free water to 100 µM.
- In a PCR tube, mix:
- 10 µL Oligo 1 (100 µM)
- 10 µL Oligo 2 (100 µM)
- 25 µL 10x Annealing Buffer
- 205 µL Nuclease-free water
- Total Volume: 250 µL (final 4 µM dsODN).
- Anneal in a thermal cycler: Heat to 95°C for 5 min, then ramp down to 25°C at a rate of 0.1°C/sec.
- Verify annealing and purity via non-denaturing PAGE (4-20% gel) or by analyzing a 1:1000 dilution on a high-sensitivity DNA Bioanalyzer chip. A single, sharp band at ~68 bp (34 bp core + handles) is expected.
- Store the annealed dsODN at -20°C.
Protocol 3: Cell Transfection and dsODN Tag Integration
Materials:
- Cultured cells (e.g., HEK293T, primary T-cells).
- SpCas9 ribonucleoprotein (RNP) complex: pre-complexed Cas9 protein and target-specific sgRNA.
- Annealed dsODN from Protocol 2.
- Appropriate transfection reagent (e.g., Lipofectamine CRISPRMAX for adherent cells, Neon/Nucleofector for primary cells).
Method:
- Complex Formation: Pre-complex the SpCas9 protein with sgRNA at a molar ratio of 1:1.2 to 1:2 (Cas9:sgRNA) in Opti-MEM. Incubate at room temperature for 10-20 minutes.
- Transfection Mixture: For a 24-well format, prepare a master mix containing:
- 50-200 nM final concentration of Cas9 RNP.
- 50-250 nM final concentration of annealed dsODN.
- Appropriate volume of transfection reagent per manufacturer's instructions.
- Transfection: Add the mixture to cells. Include critical controls:
- No RNP, +dsODN: Controls for non-specific dsODN integration.
- +RNP, -dsODN: Controls for background signal in later steps.
- Incubation: Culture cells for 48-72 hours post-transfection to allow for DSB generation, dsODN integration, and repair via NHEJ.
Visualizing the GUIDE-seq Experimental Workflow
Diagram 1: GUIDE-seq workflow for off-target detection.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for GUIDE-seq Phase 1
| Item | Function & Rationale | Example/Specification |
|---|---|---|
| High-Purity ssODNs | Template for dsODN. PAGE/HPLC purification minimizes failed sequences, increasing tagging specificity. | 100 nmole scale, 5'-phosphorylated, desalted. |
| Recombinant SpCas9 Protein | For RNP formation. Offers faster kinetics, reduced off-target effects, and higher efficiency vs. plasmid delivery. | Nuclease-grade, endotoxin-free. |
| Chemically Modified sgRNA | Incorporation of 2'-O-methyl 3' phosphorothioate analogs enhances stability and reduces immune responses in cells. | CRISPR-modified synthetic sgRNA. |
| Cell-Type Specific Transfection Reagent | Enables efficient co-delivery of RNP (large, charged) and dsODN (small) into diverse cell types. | e.g., Lipofectamine CRISPRMAX for adherent lines; Nucleofector kits for primary/immune cells. |
| High-Sensitivity DNA Analysis Kit | Critical for quality control of dsODN annealing and final library quantification. | e.g., Agilent High Sensitivity DNA Kit for Bioanalyzer/TapeStation. |
| dsODN-Specific PCR Primers | Unique primers targeting Handle A/B allow specific amplification of tagged genomic loci, minimizing background. | HPLC-purified primers for nested PCR. |
| PCR Enzyme for High-GC/Complex Templates | Robust polymerase is needed for efficient amplification of GC-rich genomic regions captured during GUIDE-seq. | e.g., Q5 High-Fidelity or KAPA HiFi HotStart polymerase. |
Within the comprehensive thesis on "Genome-wide Off-Target Detection in CRISPR-Cas9 Therapeutics Using GUIDE-seq," Phase 2 represents the critical experimental step of introducing the CRISPR-Cas9 ribonucleoprotein (RNP) complex and the double-stranded Oligodeoxynucleotide (dsODN) GUIDE-seq tag into the target cell population. Successful transfection and integration of the dsODN tag into nuclease-induced double-strand breaks (DSBs) are prerequisites for the subsequent genome-wide sequencing and analysis phases that identify off-target cleavage events. This protocol details optimized methodologies for this phase.
Research Reagent Solutions Toolkit
| Reagent/Material | Function in Phase 2 |
|---|---|
| CRISPR-Cas9 RNP Complex | Pre-assembled complex of recombinant Cas9 protein and target-specific sgRNA. Direct delivery increases editing efficiency and reduces off-target effects compared to plasmid DNA. |
| GUIDE-seq dsODN | 34-36 bp double-stranded Oligodeoxynucleotide with 5' phosphorothioate linkages. Serves as the tag integrated into DSBs, providing a universal primer binding site for later PCR amplification. |
| Electroporation System (e.g., Neon, Nucleofector) | Device for delivering macromolecules into hard-to-transfect cells (e.g., primary T cells, stem cells) via electrical pulses that create transient pores in the cell membrane. |
| Cell Culture Media (Full & Electroporation-specific) | Full media for cell health pre/post electroporation. Electroporation-specific buffers (R-buffer, Supplement) are optimized for low conductivity and high cell viability. |
| Viability Stain (e.g., Trypan Blue) | For assessing post-electroporation cell viability prior to plating. |
| Control dsODN (Fluorophore-labeled) | Used in optimization experiments to quantify transfection efficiency via flow cytometry. |
Table 1: Optimized Electroporation Parameters for Common Cell Lines in GUIDE-seq
| Cell Line | System/Kit | Voltage (V) | Pulse Width (ms) | # of Pulses | Cell Density | RNP (pmol) | dsODN (pmol) | Approx. Viability (%) | Editing Efficiency (%)* |
|---|---|---|---|---|---|---|---|---|---|
| HEK293T | Neon (100 µL) | 1,100 | 20 | 2 | 1.2e6 | 100 | 100 | 75-85 | >60 |
| U2OS | 4D-Nucleofector (SE) | 1,350 | 10 | 1 | 1.0e5 | 50 | 50 | 70-80 | ~40 |
| Primary Human T Cells | Neon (100 µL) | 1,600 | 10 | 3 | 2.0e6 | 200 | 200 | 60-70 | 40-50 |
| K562 | 4D-Nucleofector (SF) | 1,300 | 10 | 1 | 5.0e5 | 100 | 100 | 80-90 | >50 |
*Editing efficiency is measured at the on-target locus via NGS or T7E1 assay 72 hours post-transfection.
Table 2: Key dsODN Design and Stoichiometry
| Parameter | Specification | Rationale |
|---|---|---|
| Length | 34-36 bp | Maximizes tag retention and PCR efficiency. |
| 5' Modification | Phosphorothioate (3 linkages each end) | Protects against exonuclease degradation, enhancing tag stability. |
| Strand Polarity | Blunt ends | Promotes integration into Cas9-generated DSBs. |
| Molar Ratio (RNP:dsODN) | 1:1 to 1:2 | Ensures sufficient tag for integration without excessive cellular toxicity. |
| Final Concentration in Reaction | 100 - 200 pmol per 1e5 - 1e6 cells | Optimized for detection sensitivity and cell viability. |
Detailed Experimental Protocols
Protocol A: RNP-dsODN Electroporation for Adherent Cells (HEK293T)
Objective: To co-deliver Cas9 RNP and GUIDE-seq dsODN into HEK293T cells via electroporation.
Materials:
- Prepared Cas9 RNP complex (100 pmol per reaction).
- GUIDE-seq dsODN (100 pmol per reaction).
- HEK293T cells at 70-80% confluence.
- Trypsin-EDTA, full growth medium (DMEM + 10% FBS).
- Neon Transfection System (Thermo Fisher), Neon Kit (100 µL tips).
- DPBS, without Ca2+/Mg2+.
Methodology:
- Cell Preparation: Harvest HEK293T cells using trypsin. Neutralize with full medium, count, and centrifuge at 300 x g for 5 min. Wash cell pellet once with DPBS.
- Resuspension: Resuspend cell pellet in Neon Resuspension Buffer R to a density of 1.2 x 10^7 cells/mL.
- Complex Assembly: In a sterile tube, mix 100 pmol of pre-assembled Cas9 RNP with 100 pmol of dsODN. Incubate at room temperature for 5-10 min.
- Electroporation Mix: Combine 10 µL of cell suspension (1.2e5 cells) with the RNP-dsODN mixture. Gently mix.
- Electroporation: Aspirate the mix into a Neon 100 µL tip. Electroporate using pre-optimized parameters: 1100V, 20ms, 2 pulses.
- Recovery: Immediately transfer electroporated cells into a pre-warmed 24-well plate containing 500 µL of full medium.
- Incubation: Place plate in a 37°C, 5% CO2 incubator. Refresh medium after 24 hours.
- Harvest: Harvest cells 72 hours post-electroporation for genomic DNA extraction and GUIDE-seq library preparation (Phase 3).
Protocol B: Optimization for Transfection Efficiency & Viability
Objective: To determine optimal RNP:dsODN ratios and cell densities using a fluorophore-labeled control dsODN.
Methodology:
- Prepare a master cell suspension as in Protocol A, Step 1-2.
- Set up a matrix of electroporation reactions with varying RNP (50-200 pmol) and fluorescent dsODN (50-400 pmol) amounts.
- Perform electroporation in triplicate for each condition.
- At 24 hours post-transfection, harvest an aliquot of cells, wash with DPBS, and analyze via flow cytometry to measure the percentage of fluorescent-positive cells (transfection efficiency).
- In parallel, use a viability stain (e.g., Trypan Blue) to assess cell viability.
- Plot viability (%) vs. transfection efficiency (%) to identify the optimal condition that maximizes both parameters for your specific cell line.
Visualized Workflows and Pathways
Diagram Title: Phase 2 Experimental Workflow
Diagram Title: GUIDE-seq Tag Integration Mechanism
Application Notes
Within GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) workflows for genome-wide off-target detection of CRISPR-Cas nucleases, Phase 3 is critical for converting in situ double-strand breaks (DSBs) into a sequencedable library. This phase isolates and processes genomic DNA to specifically enrich for fragments containing the integrated GUIDE-seq oligonucleotide tag, which marks nuclease cut sites. The efficiency of this phase directly impacts the sensitivity and specificity of off-target detection. Current methodologies emphasize balancing high DNA yield with preservation of tag integrity, utilizing optimized shearing techniques to achieve ideal fragment sizes for Illumina sequencing, and employing robust enrichment strategies to reduce background noise.
Protocols
Protocol 3.1: Genomic DNA (gDNA) Extraction from Nucleofected Cells
Principle: High-molecular-weight gDNA is extracted while ensuring the preservation of the integrated double-stranded oligonucleotide tag.
- Cell Lysis: At 72 hours post-nucleofection, pellet ~1-2 million cells. Resuspend in 500 µL of lysis buffer (10 mM Tris-HCl pH 8.0, 25 mM EDTA, 100 mM NaCl, 0.5% SDS) with 1 µL of RNase A (20 mg/mL). Incubate at 37°C for 30 minutes.
- Protein Degradation: Add 2.5 µL of Proteinase K (20 mg/mL). Mix and incubate at 56°C overnight with gentle agitation.
- DNA Precipitation: Add 500 µL of room-temperature isopropanol. Invert tube gently until DNA threads form. Spool out DNA using a sealed Pasteur pipette.
- Wash & Hydration: Dip spooled DNA into 1 mL of 70% ethanol. Transfer to a microcentrifuge tube with 200 µL of low-EDTA TE buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0). Hydrate at 4°C overnight. Gently vortex to mix.
- Quantification: Measure DNA concentration using a fluorometric assay (e.g., Qubit dsDNA HS Assay). Expected yield: 5-20 µg from 1 million mammalian cells.
Protocol 3.2: Ultrasonication-Based DNA Shearing
Principle: gDNA is fragmented to a target size optimal for library construction and sequencing (~500 bp).
- Instrument Setup: Use a focused-ultrasonicator (e.g., Covaris S2/S220). Fill the water bath with degassed, chilled water (4-8°C).
- Sample Preparation: Dilute 1-3 µg of gDNA in 130 µL of low-EDTA TE in a microTUBE. Ensure no bubbles are present.
- Shearing Parameters: Set the following program:
- Peak Incident Power: 175 W
- Duty Factor: 10%
- Cycles per Burst: 200
- Treatment Time: 60 seconds
- Mode: Frequency Sweeping
- Verification: Analyze 50 ng of sheared DNA on a 2% agarose gel or Bioanalyzer. A tight distribution centered at 500 bp is ideal.
Protocol 3.3: Enrichment for Tagged DNA Fragments
Principle: Biotinylated probes complementary to the integrated GUIDE-seq tag are used to pull down tagged fragments via streptavidin beads.
- End Repair & A-Tailing: Perform using a commercial library preparation kit (e.g., NEBNext Ultra II). Follow manufacturer instructions for 50-200 ng of sheared DNA.
- Adapter Ligation: Ligate pre-methylated, hairpin-loop ("barcoded") adapters to the A-tailed DNA. These adapters contain sequencing primer sites and sample indexes.
- Denaturation & Hybridization: Denature the adapter-ligated DNA at 95°C for 5 minutes and immediately snap-cool. Add 5 µL of biotinylated GUIDE-seq capture probe (100 µM stock) in 6x SSC, 0.1% SDS hybridization buffer. Incubate at 65°C for 16-20 hours.
- Streptavidin Pull-down: Pre-wash 50 µL of Streptavidin C1 Dynabeads twice with 1x B&W buffer (5 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 1 M NaCl). Add the hybridization reaction to the beads and incubate at room temperature for 30 minutes with rotation.
- Stringency Washes: Wash beads sequentially with:
- Wash I: 2x SSC, 0.1% SDS at room temperature (2x, 5 min).
- Wash II: 0.2x SSC, 0.1% SDS at 65°C (3x, 10 min).
- Wash III: 0.2x SSC at room temperature (1x, 2 min).
- Elution: Resuspend beads in 25 µL of low-EDTE TE. Heat at 95°C for 5 minutes. Immediately place on a magnetic rack and transfer supernatant containing enriched, tagged DNA to a new tube.
- PCR Amplification: Amplify the eluted DNA for 12-18 cycles using primers complementary to the hairpin adapters. Purify the final library using SPRI beads. Quantify by qPCR and profile on a Bioanalyzer.
Protocol 3.4: Quality Control and Quantification
- Fragment Analysis: Verify final library size distribution (expected peak ~550-650 bp) using a High Sensitivity DNA chip on a Bioanalyzer or TapeStation.
- Quantitative PCR: Use a library quantification kit (e.g., KAPA SYBR FAST qPCR) against known standards to determine the molar concentration of amplifiable fragments. Enriched libraries typically yield 1-10 nM in 20 µL.
Table 1: Expected Yield and Size Metrics for Phase 3 Procedures
| Procedure | Input Amount | Expected Output | Optimal Size Range | Key QC Metric |
|---|---|---|---|---|
| gDNA Extraction | 1-2 x 10^6 cells | 5-20 µg | >20 kb | A260/A280 = 1.8-2.0 |
| Ultrasonic Shearing | 1-3 µg gDNA | >90% recovery | 450-550 bp | Tight peak on Bioanalyzer |
| Adapter Ligation | 50-200 ng sheared DNA | 30-70% efficiency | N/A | Successful shift on gel |
| Streptavidin Enrichment | Adapter-ligated DNA | 5-20% recovery* | N/A | >1000-fold enrichment of tagged sites |
| Final Library Amplification | Eluted DNA | 20-100 ng/µL | 550-650 bp | Distinct peak, no adapter dimer |
*Recovery is highly dependent on cutting efficiency and tag integration.
Table 2: Recommended Reagent and Instrument Parameters
| Reagent/Instrument | Key Component/Parameter | Purpose/Setting |
|---|---|---|
| Lysis Buffer | SDS (0.5%), EDTA (25 mM) | Efficient cell lysis, inhibit nucleases |
| Proteinase K | 20 mg/mL, 56°C incubation | Digest proteins, inactivate nucleases |
| Covaris S2 | Duty Factor: 10%, 200 cycles/burst | Controlled, reproducible acoustic shearing |
| Streptavidin C1 Beads | High binding capacity (~10 µg/mg) | Efficient capture of biotinylated probe-DNA complexes |
| Biotinylated Capture Probe | 5' Biotin, 3' C3 Spacer, 50 bp | High-specificity hybridization to integrated tag |
| Stringency Wash Buffer | 0.2x SSC, 0.1% SDS at 65°C | Remove non-specifically bound DNA |
Diagrams
Title: GUIDE-seq Phase 3: DNA Processing & Enrichment Workflow
Title: Molecular Basis of Tagged Fragment Enrichment
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Phase 3 of GUIDE-seq
| Item | Function in Protocol | Key Consideration |
|---|---|---|
| Proteinase K | Digests nucleases and cellular proteins during gDNA extraction, preventing DNA degradation. | Quality affects yield; must be RNase-free. |
| Covaris microTUBE | A specialized vessel for holding DNA sample during focused-ultrasonication. | Ensures consistent acoustic coupling and shearing efficiency. |
| NEBNext Ultra II DNA Library Prep Kit | Provides optimized enzymes and buffers for end repair, A-tailing, and adapter ligation. | Streamlines workflow and improves reproducibility. |
| Hairpin (Loop) Adapters | Double-stranded adapters with a hairpin loop that blocks self-ligation and reduces adapter dimer formation. | Critical for maintaining library complexity. |
| Biotinylated GUIDE-seq Capture Probe | A single-stranded DNA oligonucleotide complementary to the integrated tag, with 5' biotin for pull-down. | Specificity and melting temperature (Tm) are crucial for low background. |
| Streptavidin C1 Dynabeads | Magnetic beads coated with streptavidin for capturing biotinylated probe-DNA complexes. | High binding capacity minimizes probe saturation. |
| KAPA Library Quantification Kit | qPCR-based kit for accurate molar quantification of sequencing-ready libraries. | Essential for proper pooling and loading on sequencer. |
| AMPure XP/SPRI Beads | Magnetic beads for size-selective purification and cleanup of DNA between enzymatic steps. | Ratios determine size cut-off; critical for removing primers and adapter dimers. |
Within GUIDE-seq genome-wide off-target detection research, the Phase 4 library preparation step is critical for converting the isolated, adapter-ligated DNA fragments into a sequencer-compatible format. This process involves PCR amplification with indexing primers to introduce unique sample barcodes and platform-specific sequences, followed by purification and quantification to ensure high-quality NGS data for the identification of CRISPR-Cas nuclease off-target sites.
Table 1: Typical Library Preparation Reagent Volumes and Specifications
| Component | Volume/Amount (for 50 µL rxn) | Purpose | Notes for GUIDE-seq |
|---|---|---|---|
| Adapter-Ligated DNA | 10-25 µL (up to 50 ng) | Template | Contains GUIDE-seq oligo-integrated fragments. |
| High-Fidelity PCR Master Mix | 25 µL | Amplification enzyme & dNTPs | Use low-cycle number (≤12) to minimize bias. |
| Universal PCR Primer (i5) | 2.5 µL (10 µM) | Attaches flow cell binding site, P5 | Standard Illumina primer sequence. |
| Indexed PCR Primer (i7) | 2.5 µL (10 µM) | Attaches index & flow cell site, P7 | Unique index per sample for multiplexing. |
| Nuclease-Free Water | To 50 µL total | Reaction volume adjustment | Ensure no contamination. |
| Post-PCR Purification (SPR Beads) | 1.0x - 1.2x sample volume | Size selection & cleanup | Removes primers, dimers; retains >150 bp fragments. |
Table 2: Critical Library QC Metrics for NGS
| QC Metric | Target Range (Illumina) | Method | Impact on GUIDE-seq Data |
|---|---|---|---|
| Library Concentration | ≥ 2 nM (post-PCR) | Qubit dsDNA HS Assay | Ensures sufficient cluster density. |
| Fragment Size Distribution | Peak ~250-350 bp | Bioanalyzer/TapeStation | Confirms successful adapter ligation & PCR. |
| Molarity (for pooling) | 4-10 nM (final) | qPCR with Library Quant Kit | Accurate equimolar pooling for multiplexing. |
| Adapter Dimer | < 5% of total signal | Bioanalyzer/TapeStation | High dimer % reduces usable sequencing reads. |
Detailed Experimental Protocols
Protocol 1: PCR Amplification and Indexing of GUIDE-seq Libraries
Objective: To amplify adapter-ligated DNA fragments and add sample-specific indices and full Illumina adapters. Materials: Purified adapter-ligated DNA, high-fidelity DNA polymerase master mix (e.g., KAPA HiFi, NEBNext Ultra II Q5), universal and index PCR primers (Illumina-compatible), nuclease-free water, thermal cycler. Procedure:
- Reaction Setup: On ice, assemble the following in a 0.2 mL PCR tube:
- Nuclease-free water: X µL (to 50 µL total)
- High-fidelity PCR Master Mix (2X): 25 µL
- Universal PCR Primer (10 µM): 2.5 µL
- Indexed PCR Primer (10 µM): 2.5 µL
- Purified Adapter-Ligated DNA: 10-25 µL (not exceeding 50 ng input)
- Thermocycling: Place tubes in a pre-heated thermal cycler and run the following program:
- Initial Denaturation: 98°C for 45 seconds.
- Cycling (10-12 cycles):
- Denature: 98°C for 15 seconds.
- Anneal: 65°C for 30 seconds.
- Extend: 72°C for 30 seconds.
- Final Extension: 72°C for 1 minute.
- Hold: 4°C.
- Post-PCR Hold: Proceed immediately to purification or store at -20°C.
Protocol 2: Purification and Size Selection with Solid-Phase Reversible Immobilization (SPRI) Beads
Objective: To remove PCR reagents, primers, and primer dimers while selectively retaining correctly sized library fragments. Materials: AMPure XP or SPRIselect beads, fresh 80% ethanol, nuclease-free water, magnetic stand, 1.5 mL low-binding tubes. Procedure:
- Bring PCR product to room temperature. Vortex SPRI beads thoroughly.
- Bind: Add a calculated volume of SPRI beads (typically 1.0X to 1.2X the sample volume, e.g., 55 µL beads to 50 µL PCR product) directly to the PCR reaction. Mix thoroughly by pipetting 10 times. Incubate at room temperature for 5 minutes.
- Capture: Place the tube on a magnetic stand for 5 minutes or until the supernatant is clear.
- Wash (2x): With the tube on the magnet, carefully remove and discard the supernatant. Add 200 µL of freshly prepared 80% ethanol without disturbing the bead pellet. Incubate at room temperature for 30 seconds, then remove and discard the ethanol. Repeat this wash a second time.
- Dry: Briefly spin the tube, place it back on the magnet, and use a low-volume pipette to remove any residual ethanol. Air-dry the bead pellet for 2-3 minutes (do not over-dry).
- Elute: Remove the tube from the magnet. Resuspend the dried beads in 20-30 µL of nuclease-free water or 10 mM Tris-HCl (pH 8.0-8.5). Mix thoroughly. Incubate at room temperature for 2 minutes.
- Recapture: Place the tube back on the magnet for 2 minutes or until the supernatant is clear.
- Recover: Carefully transfer the purified library supernatant (eluent) to a new, labeled 1.5 mL tube.
Protocol 3: Library Quantification and Quality Control
Objective: To accurately quantify the final library concentration and assess fragment size distribution prior to sequencing. Materials: Qubit fluorometer and dsDNA HS Assay Kit, Agilent Bioanalyzer with High Sensitivity DNA kit or Agilent TapeStation with D1000/High Sensitivity D1000 ScreenTape, library quantification standard and qPCR mix (e.g., KAPA Library Quant Kit). Procedure (Multi-Step QC):
- Fluorometric Quantification (Qubit):
- Perform the Qubit dsDNA HS Assay according to the manufacturer's protocol.
- Use 1-2 µL of the purified library. This gives a precise DNA concentration (ng/µL) but does not discriminate between adapter-ligated fragments and primer dimers.
- Fragment Size Analysis (Bioanalyzer/TapeStation):
- Run 1 µL of the purified library on a High Sensitivity DNA chip or TapeStation.
- The profile should show a peak in the 250-400 bp range (inclusive of adapters, ~130-180 bp insert). Note the percentage of the peak area in the adapter dimer region (~100-150 bp).
- qPCR Quantification for Pooling (Recommended):
- Perform the qPCR-based library quantification kit according to the protocol.
- This method quantifies only fragments containing full adapters, providing the most accurate molarity (nM) for equimolar pooling of multiple GUIDE-seq libraries.
Visualizations
Title: NGS Library Prep Workflow for GUIDE-seq
Title: Final NGS Library Structure with Dual Indices
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for NGS Library Preparation
| Item | Function in GUIDE-seq Library Prep | Example Product(s) |
|---|---|---|
| High-Fidelity PCR Master Mix | Provides robust, low-bias amplification of adapter-ligated fragments with high fidelity, critical for maintaining sequence integrity of off-target sites. | KAPA HiFi HotStart ReadyMix, NEBNext Ultra II Q5 Master Mix. |
| Indexed PCR Primers (i5/i7) | Dual-indexed primers that add full Illumina P5/P7 flow cell adapters and unique combinatorial barcodes for multiplexing multiple samples in one sequencing run. | Illumina TruSeq CD Indexes, IDT for Illumina UD Indexes. |
| SPRI Magnetic Beads | Enable rapid, size-selective cleanup and concentration of PCR-amplified libraries, removing primers, dimers, and salts. Crucial for clean size profiles. | Beckman Coulter AMPure XP, SPRIselect Reagent. |
| dsDNA High-Sensitivity Assay Kit | Fluorometrically quantifies double-stranded library DNA concentration with high accuracy in dilute samples, essential for normalization. | Qubit dsDNA HS Assay Kit. |
| Library Quantification Kit (qPCR) | Quantifies only library fragments containing complete adapters via qPCR, providing the most accurate molarity for equitable pooling prior to sequencing. | KAPA Library Quantification Kit (Illumina), NEBNext Library Quant Kit. |
| High Sensitivity DNA Analysis Kit | Provides precise electrophoretic analysis of library fragment size distribution and detects adapter dimer contamination. | Agilent High Sensitivity DNA Kit (Bioanalyzer), D1000 HS ScreenTape (TapeStation). |
| Nuclease-Free Water & Tubes | Ensures reactions are free of RNase, DNase, and PCR inhibitors. Low-bind tubes minimize sample loss during purification steps. | Various molecular biology grade suppliers (Ambion, Sigma). |
This protocol details the computational phase of a GUIDE-seq experiment, a critical component of genome-wide off-target detection research. The objective is to process raw sequencing reads, identify integration events of the GUIDE-seq Oligonucleotide Tag, and generate a high-confidence list of off-target sites for a given CRISPR-Cas guide RNA. This pipeline is designed for reproducibility and accuracy, minimizing false positives while capturing genuine off-target events.
Core Computational Workflow
Table 1: Key Performance Metrics and Benchmarks for the GUIDE-seq Computational Pipeline
| Pipeline Stage | Typical Input Data | Key Output Metric | Expected Range/Threshold | Common Software/Tool |
|---|---|---|---|---|
| Read Preprocessing & Alignment | Paired-end FASTQ files (~50-100M reads) | Percentage of reads aligned to reference genome | >85% | BWA-MEM, Bowtie2 |
| Tag Extraction & Site Identification | Aligned BAM file | Unique genomic loci with tag integrations | 50 - 5,000+ sites | GUIDE-seq specific scripts (e.g., GUIDE-seq R package) |
| Peak Calling & Scoring | Candidate integration sites | High-confidence off-target sites | 10 - 200 sites (guide-dependent) | MAGeCK, custom peak-calling |
| False Positive Filtering | Scored off-target sites | Final validated off-targets | Signal-to-noise ratio > 5; Read count > background median + 5 MAD | BEDTools, SAMtools |
| Annotation & Prioritization | Final off-target list | Sites in coding/exonic regions | Variable | ANNOVAR, ChIPseeker |
Detailed Experimental Protocol: Computational Analysis
Protocol 5.1: Raw Read Processing and Alignment
Objective: To align paired-end sequencing reads to the reference genome and extract properly paired reads. Materials: High-performance computing cluster, reference genome (e.g., hg38), sequencing reads in FASTQ format. Procedure:
- Quality Control: Run FastQC on raw FASTQ files. Use Trimmomatic or Cutadapt to remove adapter sequences and low-quality bases (Phred score < 20).
- Genome Alignment: Index the reference genome using BWA (
bwa index). Align trimmed reads using BWA-MEM with default parameters:bwa mem -M -t [threads] [reference_genome] [R1.fastq] [R2.fastq] > aligned.sam. - File Conversion and Sorting: Convert SAM to BAM, sort, and index using SAMtools:
- Remove Duplicates: Use Picard Tools to mark and remove PCR duplicates:
java -jar picard.jar MarkDuplicates I=aligned.sorted.bam O=dedup.bam M=metrics.txt.
Protocol 5.2: GUIDE-seq Tag Identification and Off-Target Locus Calling
Objective: To identify genomic loci flanking the integrated double-stranded oligodeoxynucleotide (dsODN) tag. Materials: Deduplicated BAM file, dsODN tag sequence, reference genome. Procedure:
- Extract Read Pairs Containing Tag: Use a custom script (e.g.,
guideseq.pyfrom the original GUIDE-seq publication) or theGUIDEseqBioconductor package to scan read pairs for the dsODN tag sequence. Retain read pairs where one read contains the tag and its mate is mapped to the genome. - Generate Integration Site Bed File: For each tag-containing read, extract the genomic coordinate of its mate (the putative cleavage site). Create a BED file of these unique sites, with read count per site.
- Peak Calling and Background Subtraction: Use a sliding window (e.g., 50 bp) to merge nearby sites. Calculate a local background read density from regions flanking the candidate peaks. Sites with a read count significantly above background (e.g., p-value < 0.01 by Fisher's exact test) are retained.
- Annotate Sites with Mismatches: Align the guide RNA sequence to each candidate genomic locus using a local aligner (e.g., Bowtie2 in local mode). Record the number and position of mismatches and bulges.
Protocol 5.3: Generation of High-Confidence Off-Target List
Objective: To filter and prioritize candidate off-target sites into a final high-confidence list. Materials: Candidate site BED file, mismatch annotations, blacklist regions (e.g., ENCODE DAC Blacklisted Regions). Procedure:
- Apply Read Count and Ratio Thresholds: Filter sites requiring a minimum number of unique supporting reads (e.g., ≥ 3 reads) and a minimum ratio of tag-containing reads to total reads in the region.
- Remove Artifactual Sites: Subtract sites falling within known genome blacklist regions (using BEDTools
intersect -v). Filter out sites with high read counts in negative control samples (untreated or non-targeting guide). - Prioritize by Genomic Context and Mismatch Pattern: Annotate remaining sites with genomic features (promoter, exon, intron, intergenic) using ANNOVAR. Prioritize sites in transcriptionally active regions or with specific mismatch patterns known to be permissive (e.g., mismatches in distal PAM region).
- Generate Final Report: Create a final table (CSV/TSV) containing columns for genomic coordinates, strand, read count, mismatch pattern, and genomic annotation.
Visualization of the Computational Pipeline
Title: GUIDE-seq Computational Pipeline Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools and Resources for the GUIDE-seq Pipeline
| Item Name | Category | Supplier/Provider | Critical Function in Pipeline |
|---|---|---|---|
| BWA (Burrows-Wheeler Aligner) | Alignment Software | Open Source (GitHub) | Aligns sequencing reads to the reference genome with high speed and accuracy. |
| SAMtools/BCFtools | File Processing Suite | Open Source (htslib.org) | Manipulates and analyzes aligned sequencing data in SAM/BAM format (sorting, indexing, filtering). |
| GUIDE-seq Bioconductor Package | Specialized Analysis Tool | Bioconductor | Core software for identifying tag integration sites and performing initial peak calling. |
| UCSC Genome Browser/Table Browser | Genomic Database & Visualization | UCSC | Provides reference genomes, annotation tracks, and data retrieval for analysis and verification. |
| ENCODE DAC Blacklist Regions | Genomic Filter File | ENCODE Consortium | Lists artifact-prone regions to filter out false-positive off-target calls. |
| ANNOVAR | Annotation Tool | Open Source | Annotates genomic variants/positions with functional context (genes, conserved regions, etc.). |
| High-Performance Computing (HPC) Cluster | Infrastructure | Institutional IT/Cloud (AWS, Google Cloud) | Provides the necessary computational power for memory-intensive alignment and processing steps. |
| R/Bioconductor & Python Environments | Programming Frameworks | Open Source | Flexible platforms for running specialized packages and custom analysis scripts. |
Within the broader thesis of GUIDE-seq (Genome-wide Unbiased Identification of DSBs Enabled by sequencing) genome-wide off-target detection research, this application note details its critical adaptation for profiling unintended edits by therapeutic CRISPR-Cas9 guide RNAs (gRNAs) and base editors. As these technologies advance toward the clinic, comprehensive off-target characterization is non-negotiable for regulatory approval and patient safety. GUIDE-seq remains a gold-standard, unbiased method for identifying double-strand break (DSB) locations genome-wide, which can be extended to map the off-target profiles of base editors by capturing their nicking or DNA mismatch repair intermediates.
Table 1: Comparison of Off-Target Detection Methods for gRNAs and Base Editors
| Method | Detection Principle | Suitable for Cas9 Nuclease? | Suitable for Base Editors? | Sensitivity (Theoretical) | Key Limitation |
|---|---|---|---|---|---|
| GUIDE-seq | Integration of dsODN tag at DSB sites | Yes | Yes (via nicking or repair) | ~0.1% of reads | Requires DSB or nick; lower sensitivity for very rare events. |
| CIRCLE-seq | In vitro circularization & sequencing of genomic DNA | Yes | Limited | High (in vitro) | Purely in vitro; lacks cellular context. |
| SITE-Seq | In vitro Cas9 digestion & fragment capture | Yes | No | High (in vitro) | In vitro only; biased by cleavage efficiency. |
| ONE-seq | Captures nicked DNA strands | Yes (nickase) | Yes (ABEs, CBEs) | Comparable to GUIDE-seq | Optimized for nickase-derived edits. |
| Digenome-seq | In vitro Cas9 digestion & whole-genome sequencing | Yes | Limited | High (in vitro) | High false-positive rate; in vitro context. |
Table 2: Representative Off-Target Counts from Recent Studies
| Study (Year) | Target Gene | Editor (gRNA) | Detection Method | Total Off-Targets Identified | Highest Frequency Off-Target (% Indel or Edit) |
|---|---|---|---|---|---|
| Tsai et al. (2023) | VEGFA | SpCas9 (gRNA1) | GUIDE-seq | 12 | 4.2% |
| Lee et al. (2024) | HEK3 | ABE8e (High-fidelity) | GUIDE-seq adaptation | 3 | 0.8% |
| Kim et al. (2023) | FANCF | BE4max (High-fidelity) | ONE-seq | 7 | 1.5% |
| Clinical Trial (2022) | CEP290 | AsCas12a (ABE) | GUIDE-seq & CIRCLE-seq | 2 | <0.1% |
Detailed Experimental Protocols
Protocol 1: Standard GUIDE-seq for Cas9 Nuclease Off-Target Profiling
- Principle: A double-stranded oligodeoxynucleotide (dsODN) tag is integrated into CRISPR-Cas9-induced DSBs during repair. Tagged sites are then enriched and sequenced.
- Detailed Steps:
- Cell Transfection: Co-transfect 2e5 HEK293T cells (or target therapeutic cell line) with 1.5 µg SpCas9 expression plasmid, 500 ng gRNA expression plasmid, and 100 pmol of annealed GUIDE-seq dsODN tag using a nucleofection system (e.g., Lonza 4D-Nucleofector).
- Genomic DNA Extraction: 72 hours post-transfection, harvest cells and extract high-molecular-weight genomic DNA using the QIAamp DNA Mini Kit.
- Library Preparation: Shear 2 µg gDNA to ~400 bp using a Covaris S2. Perform end-repair, A-tailing, and ligation of sequencing adapters. Enrich for dsODN-tagged fragments via PCR using one primer specific to the dsODN and another to the adapter.
- Sequencing & Analysis: Sequence on an Illumina MiSeq (2x150 bp). Process reads using the GUIDE-seq computational pipeline (available on GitHub) to align tags, identify unique integration sites, and call significant off-target loci (requiring ≥2 unique tag integrations and p-value < 0.05).
Protocol 2: Modified GUIDE-seq for Adenine Base Editor (ABE) Off-Target Profiling
- Principle: ABEs create an A•T to G•C conversion via a nick in the non-edited strand. The GUIDE-seq dsODN can be integrated during the repair of this nick or subsequent mismatch repair.
- Detailed Modifications to Protocol 1:
- Transfection: Replace SpCas9 plasmid with an ABE8e expression plasmid. Use a nickase version of Cas9 (D10A) fused to the deaminase. The gRNA remains the same.
- Timing: Extend incubation time to 96-120 hours post-transfection to allow for sufficient base editing and repair processes.
- Analysis Pipeline: Use the GUIDE-seq pipeline with relaxed parameters for nick integration, followed by targeted amplicon sequencing of predicted and identified off-target loci to confirm A-to-G edits at each site.
Visualized Workflows and Pathways
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for GUIDE-seq Based Off-Target Profiling
| Reagent / Solution | Function & Importance in the Protocol | Example Product / Component |
|---|---|---|
| GUIDE-seq dsODN Duplex | The double-stranded oligo tag that integrates at DSB/nick sites, enabling amplification and identification. Must be HPLC-purified. | Custom synthesized (e.g., IDT): 5′-[Phos]GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNN...-3′ |
| High-Efficiency Transfection Reagent/System | Critical for delivering multiple components (editor, gRNA, dsODN) into relevant therapeutic cell types (e.g., primary T cells, stem cells). | Lonza 4D-Nucleofector X Kit; Lipofectamine CRISPRMAX. |
| High-Fidelity PCR Master Mix | Essential for the specific, low-bias amplification of dsODN-tagged genomic fragments during library prep. | NEB Q5 Hot Start HiFi PCR Master Mix; KAPA HiFi HotStart ReadyMix. |
| Cas9/Base Editor Expression Plasmid | Source of the nuclease or editor protein. High-fidelity variants (e.g., SpCas9-HF1, ABE8e) are recommended to reduce off-target background. | Addgene plasmids #108100 (ABE8e), #108106 (SpCas9-HF1). |
| Targeted Amplicon Sequencing Kit | Required for orthogonal validation of candidate off-target sites identified by GUIDE-seq. | Illumina TruSeq Custom Amplicon; Twist Target Enrichment. |
| Bioinformatics Pipeline | Software to process sequencing data, map tag integrations, and call off-target sites with statistical confidence. | GUIDE-seq (R package, GitHub); CRISPResso2 for amplicon analysis. |
Solving GUIDE-seq Challenges: Expert Troubleshooting and Optimization Strategies
Within GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) research for profiling CRISPR-Cas nuclease off-target effects, the successful integration of a double-stranded oligodeoxynucleotide (dsODN) tag is foundational. This dsODN serves as a marker for double-strand break (DSB) sites, enabling their subsequent enrichment and sequencing. Low dsODN integration efficiency is a critical pitfall that leads to poor tag recovery, low sequencing library complexity, and ultimately, failure to detect bona fide off-target sites. This application note details the causes of low efficiency and provides optimized protocols to overcome this challenge.
The following table summarizes key variables and their impact on integration efficiency, as reported in recent literature (2023-2024).
Table 1: Factors Influencing dsODN Integration Efficiency in GUIDE-seq
| Factor | Low-Efficiency Condition | High-Efficiency Condition | Typical Impact on Read Counts |
|---|---|---|---|
| dsODN Design | Single-stranded, short (< 30 bp), no phosphorothioate (PS) bonds | Double-stranded, 34-36 bp, PS bonds on 3-5 terminal nucleotides | Up to 100-fold increase |
| dsODN Concentration | < 50 nM | 50 - 250 nM (optimal ~100 nM) | Plateau above 250 nM |
| Cell Transfection Method | Lipofection of difficult cells (e.g., primary, stem cells) | Nucleofection / Electroporation | 10- to 50-fold increase in hard-to-transfect cells |
| Cell Confluence | >90% or <40% | 70-80% | ~2- to 5-fold variation |
| dsODN:Cas9-gRNA Molar Ratio | dsODN in excess (>10:1) | Balanced ratio (1:1 to 3:1 dsODN:Cas9 RNP) | Optimal signal-to-noise ratio |
| Harvest Timing | < 48 hours post-transfection | 72 hours post-transfection | ~3-fold increase in tag recovery |
Optimized Experimental Protocols
Protocol 3.1: Synthesis and Preparation of High-Efficiency dsODN Tag
Materials:
- HPLC-purified single-stranded oligos (ssODNs).
- Annealing Buffer: 10 mM Tris, 50 mM NaCl, 1 mM EDTA, pH 8.0.
- Thermal cycler or heat block.
Procedure:
- Design: Order two complementary oligos. The "tag strand" should be 5'-phosphorylated. The "PCR handle strand" should have a 5' phosphate and 3-5 phosphorothioate linkages at its 5' end. Total length: 34-36 bp.
- Example:
5'-P-GGTCACCTCTGAGTTCCATAGACTGGATAGTGG-3'(Tag) - Example:
5'-[PS]TCCACTATCCAGTCTATGGAACTCAGAGGTGACC-3'(Handle, PS bonds on first 3-5 nucleotides).
- Example:
- Resuspend oligos to 100 µM in nuclease-free water.
- Mix 10 µL of each oligo (100 µM) with 80 µL of annealing buffer in a PCR tube.
- Anneal using a thermal cycler: 95°C for 5 minutes, ramp down to 25°C at 0.1°C/sec.
- Dilute annealed dsODN to 5 µM working stock in annealing buffer. Store at -20°C.
Protocol 3.2: Co-Delivery of RNP and dsODN via Nucleofection for Adherent Cells (e.g., HEK293T, U2OS)
Materials:
- Lonza Nucleofector System (2b or 4D) with appropriate kit (e.g., SF Cell Line Kit).
- Cas9 nuclease (IDT Alt-R S.p., TrueCut Cas9 Protein v2).
- Chemically synthesized crRNA and tracrRNA (or sgRNA).
- Optimized dsODN from Protocol 3.1.
- Complete growth medium.
Procedure:
- Cell Preparation: Harvest cells at 70-80% confluence. Count and pellet 2e5 - 1e6 cells.
- RNP Complex Formation: For 1 reaction, complex 30 pmol of Cas9 protein with 36 pmol of sgRNA (or equimolar crRNA:tracrRNA duplex) in 10 µL total volume of the provided Nucleofector buffer. Incubate at RT for 10-20 min.
- dsODN Addition: Add 3 µL of 5 µM dsODN stock (15 fmol, final ~1:1 molar ratio with RNP) directly to the RNP complex. Mix gently.
- Nucleofection: Resuspend cell pellet in the RNP-dsODN mixture. Transfer to a Nucleocuvette and run the recommended program (e.g., CM-130 for HEK293T).
- Recovery: Immediately add pre-warmed medium and transfer cells to a culture plate.
- Harvest: Incubate for 72 hours. Harvest genomic DNA using a silica-membrane column kit with RNAse A treatment.
Protocol 3.3: GUIDE-seq Library Preparation and Enrichment (Adapted)
Key Modification: Use two sequential PCRs to minimize bias and maximize complexity.
- Primary PCR (1-4 cycles): Amplify integrated tags using a tag-specific primer and a primer binding to the ligated adapter. Use a high-fidelity polymerase. Keep cycles minimal.
- Purification: Clean up primary PCR product with a size-selection bead-based method (0.6x-0.8x ratio).
- Secondary Indexing PCR (8-12 cycles): Add full Illumina adapters and sample indices.
- Sequencing: Pool libraries and sequence on an Illumina platform (≥ 2x150 bp recommended).
Visualizations
Diagram 1: dsODN Integration and GUIDE-seq Workflow
Diagram 2: Causes & Solutions for Low Integration
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Reagents for High-Efficiency GUIDE-seq
| Reagent / Material | Function & Rationale | Recommended Example / Specification |
|---|---|---|
| Phosphorothioate-Modified dsODN | Protects dsODN from exonuclease degradation, dramatically increasing stability and integration frequency. | 3-5 phosphorothioate bonds at each 5' end of the duplex. HPLC purification. |
| Alt-R S.p. Cas9 V3/V4 Protein | High-activity, recombinant Cas9 nuclease for RNP formation. Reduces off-target effects vs. plasmid delivery. | IDT Alt-R S.p. Cas9 Nuclease V3 or similar. |
| Chemically Modified Synthetic gRNA | Increased stability and potency, leading to higher cleavage efficiency and improved dsODN capture. | IDT Alt-R crRNA & tracrRNA (or sgRNA) with 2'-O-methyl modifications. |
| Nucleofector System & Kit | Electroporation-based delivery for maximal co-transfection efficiency of RNP and dsODN into difficult cells. | Lonza 4D-Nucleofector with Cell Line Specific Kit (e.g., SF Kit). |
| High-Fidelity PCR Master Mix | For minimal-bias amplification of tagged genomic loci during library construction. | KAPA HiFi HotStart ReadyMix or NEB Next Ultra II Q5. |
| SPRIselect Beads | For precise size selection and clean-up during library prep, removing primer dimers and large fragments. | Beckman Coulter SPRIselect or equivalent. |
This application note, framed within a thesis on comprehensive GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) methodology for CRISPR-Cas off-target detection, details the optimization of three critical experimental parameters. Successful GUIDE-seq relies on efficient integration of a double-stranded oligodeoxynucleotide (dsODN) tag into nuclease-induced double-strand breaks (DSBs). We present optimized protocols and quantitative data for determining the ideal cell number, transfection reagent parameters, and dsODN concentration to maximize tag integration and sequencing library diversity while minimizing cytotoxicity, specifically in HEK293T and U2OS cell lines.
Within genome-editing research, identifying off-target sites is paramount for therapeutic safety. GUIDE-seq is a sensitive, unbiased method for detecting CRISPR-Cas9 off-target cleavages in vivo. Its efficacy hinges on the co-delivery and capture of a designed dsODN into Cas9-induced DSBs. Inconsistent dsODN integration, often due to suboptimal experimental conditions, leads to poor sequencing library complexity and false-negative results. This document provides a systematic approach to optimizing the foundational variables of cell number, transfection efficiency, and dsODN dosage to establish a robust and reproducible GUIDE-seq workflow.
Core Optimization Data Tables
Table 1: Optimization of Seeding Cell Number for GUIDE-seq (24-well plate)
| Cell Line | Recommended Seeding Density (cells/well) | Seeding Time Pre-Transfection | Rationale & Outcome |
|---|---|---|---|
| HEK293T | 1.0 - 1.2 x 10^5 | 18-24 hours | Ensures ~80-90% confluence at transfection, critical for lipofection efficiency. Lower density reduces transfection efficacy; higher density increases cytotoxicity post-transfection. |
| U2OS (osteosarcoma) | 0.8 - 1.0 x 10^5 | 18-24 hours | U2OS cells adhere more firmly and divide slower. This range achieves optimal confluence without overgrowth, balancing transfection and cell health. |
Table 2: Transfection Condition Optimization for RNP + dsODN Co-delivery
| Transfection Reagent | Recommended Ratio (RNP:dsODN:Reagent) | Complex Formation Time | Media Change Post-Transfection | Key Consideration |
|---|---|---|---|---|
| Lipofectamine CRISPRMAX | 1 µg: 100 fmol: 1.5 µL (per well of 24-well) | 10-20 min at RT | At 4-6 hours | Minimizes cytotoxicity. For Cas9 RNP. Form complexes in separate tubes for RNP and dsODN before combining. |
| Lipofectamine 2000 | 1 µg: 100 fmol: 2.0 µL (per well of 24-well) | 20 min at RT | At 4-6 hours | Higher potential cytotoxicity; requires precise timing for media change. Proven high efficiency for plasmid + dsODN co-transfection. |
Table 3: dsODN Concentration Titration Impact on GUIDE-seq Outcomes
| dsODN Concentration (fmol per well, 24-well) | Relative Tag Integration Efficiency* | Library Complexity & Off-target Sites Detected | Cytotoxicity & Observed Cell Health |
|---|---|---|---|
| 25 fmol | Low (Baseline = 1.0) | Low diversity; only canonical on-target and top off-targets detected. | Minimal impact. |
| 100 fmol | High (Optimal, 3.5 - 4.2x) | High library complexity; maximal number of confident off-target sites identified. | Mild, manageable. |
| 200 fmol | Moderate (2.1 - 2.5x) | Saturated integration, potential for increased PCR duplicates. Library complexity may plateau or drop. | Noticeable cytotoxicity observed. |
| 400 fmol | Low to Moderate (1.5 - 2.0x) | High background noise; significant cytotoxicity reduces viable cell yield for genomic DNA extraction. | Severe, reduces final gDNA yield. |
*Tag integration efficiency measured via qPCR amplification of dsODN junctions relative to a genomic control locus.
Detailed Experimental Protocols
Protocol 3.1: Seeding Cells for GUIDE-seq Optimization
- Trypsinize and count cells using an automated cell counter or hemocytometer.
- Dilute cells in complete growth medium (e.g., DMEM + 10% FBS, no antibiotics) to the densities specified in Table 1.
- Seed cells into a tissue-culture treated 24-well plate, adding 500 µL of cell suspension per well. Gently rock the plate to ensure even distribution.
- Incubate the plate at 37°C, 5% CO2 for 18-24 hours until cells are 80-90% confluent at the time of transfection.
Protocol 3.2: Co-transfection of Cas9 RNP and dsODN using Lipofectamine CRISPRMAX Materials: Cas9 protein, sgRNA, GUIDE-seq dsODN (HPLC-purified), Lipofectamine CRISPRMAX, Opti-MEM I Reduced Serum Medium.
- Prepare RNP Complex: Anneal sgRNA to Cas9 protein (3:1 molar ratio) by incubating at room temperature (RT) for 10 minutes.
- Dilute Components: In Tube A, dilute 1 µg of pre-complexed RNP in 25 µL Opti-MEM. In Tube B, dilute 100 fmol of dsODN in 25 µL Opti-MEM.
- Prepare Lipid Mix: In Tube C, dilute 1.5 µL of CRISPRMAX reagent in 25 µL Opti-MEM. Incubate 5 minutes at RT.
- Combine Complexes: Add Tube A (RNP) to Tube C (lipid). Mix gently. Immediately add Tube B (dsODN) to the combined A+C mixture. Mix by pipetting gently.
- Incubate and Transfect: Incubate the final mixture for 10-15 minutes at RT. Add the entire 75 µL complex drop-wise to the pre-seeded well (from Protocol 3.1). Gently swirl the plate.
- Media Change: After 4-6 hours incubation (37°C, 5% CO2), carefully aspirate the transfection mixture and replace with 500 µL of fresh, pre-warmed complete growth medium.
Protocol 3.3: Genomic DNA Harvest for GUIDE-seq Library Preparation
- Incubate: Post-transfection, incubate cells for 48-72 hours to allow for dsODN integration and DNA repair.
- Wash and Lyse: Aspirate medium, wash wells with 1x PBS. Aspirate PBS and add 200 µL of direct lysis buffer (e.g., 50mM NaOH) per well. Incubate at 95°C for 10 minutes.
- Neutralize: Add 200 µL of neutralization buffer (e.g., 1M Tris-HCl, pH 8.0). Mix thoroughly by pipetting.
- Purify gDNA: Transfer lysate to a 1.5 mL tube. Perform a standard ethanol precipitation or use a commercial gDNA clean-up kit. Elute in 50-100 µL of TE buffer or nuclease-free water.
- Quantify: Measure gDNA concentration using a fluorescence-based assay (e.g., Qubit dsDNA HS Assay).
Visualizations
Diagram 1: Core GUIDE-seq experimental workflow.
Diagram 2: Three interdependent optimization parameters.
The Scientist's Toolkit: Key Reagents & Materials
| Item | Function in GUIDE-seq Optimization |
|---|---|
| HPLC-purified dsODN | Defined sequence oligo for tagging DSBs. High purity is critical to prevent side reactions and ensure precise integration. |
| Lipofectamine CRISPRMAX | A lipid-based transfection reagent specifically formulated for RNP delivery, offering high efficiency with lower cytotoxicity. |
| Opti-MEM I Reduced Serum Medium | A low-serum medium used for diluting lipids and forming transfection complexes, minimizing interference. |
| Recombinant Cas9 Nuclease | High-activity, endotoxin-free protein for forming RNP complexes with in vitro transcribed or synthetic sgRNA. |
| Synthetic sgRNA | Chemically synthesized, modified sgRNA for high stability and reduced immunogenicity compared to in vitro transcribed RNA. |
| Fluorometric DNA Quantification Kit (e.g., Qubit) | Essential for accurate measurement of low-concentration dsODN stocks and harvested genomic DNA prior to library prep. |
| Direct Lysis Buffer (e.g., NaOH/Tris) | Enables rapid, in-well genomic DNA harvest from 24/96-well plates, streamlining parallel processing of optimization samples. |
Addressing High Background Noise and Improving Signal-to-Noise Ratio
In GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by sequencing), a critical challenge is distinguishing true nuclease-induced off-target double-strand breaks (DSBs) from the immense background of random genomic breaks and sequencing artifacts. High background noise directly obscures low-frequency editing events, compromising the sensitivity and accuracy of off-target detection. This document outlines application notes and protocols for mitigating noise and enhancing the signal-to-noise ratio (SNR) within the context of a broader thesis on comprehensive genome-wide off-target profiling.
The primary noise sources in GUIDE-seq and related assays are categorized below.
Table 1: Primary Noise Sources in GUIDE-seq Assays
| Noise Source | Description | Typical Impact on Background |
|---|---|---|
| Endogenous DSBs | Naturally occurring breaks from replication stress, oxidative damage, or V(D)J recombination. | Constitutes >60% of non-insert background in untreated controls. |
| Tagmentation & PCR Bias | Unefficient tag integration and amplification artifacts during library prep. | Can cause >50% variance in background read distribution between replicates. |
| Oligonucleotide Uptake | Random, non-DSB-associated integration of the dsODN tag into transient nicks or gaps. | Accounts for ~15-25% of total tag integration events in negative controls. |
| Sequencing Errors | Base-calling errors and mis-mapping of reads, especially in repetitive regions. | Varies by platform; can obscure ~5-10% of legitimate sites. |
Research Reagent Solutions Toolkit
Table 2: Essential Reagents for Noise Reduction in GUIDE-seq
| Reagent / Material | Function & Rationale |
|---|---|
| Phosphorothioate-modified dsODN | Enhances cellular stability and reduces non-specific degradation, improving legitimate DSB tagging efficiency. |
| Cas9 D10A nickase (for paired nicking) | Used in negative control experiments to distinguish DSB-dependent tagging from nick-associated background integration. |
| ATM/DNA-PK inhibitors (e.g., KU-55933, NU7441) | Suppresses end-joining repair of endogenous DSBs, reducing competing background tag integration sites. |
| High-fidelity DNA polymerases (e.g., Q5, KAPA HiFi) | Minimizes PCR errors and chimeric amplicon formation during library amplification. |
| Magnetic streptavidin beads (e.g., Dynabeads) | Efficient pull-down of biotinylated PCR products reduces non-specific carryover in library purification. |
| Duplex-specific nuclease (DSN) | Normalizes library complexity by degrading over-amplified common sequences, improving rare target detection. |
Optimized Experimental Protocols
Protocol 1: dsODN Tag Design and Transfection for Maximum SNR
Objective: Maximize specific integration at true off-target DSBs. Detailed Steps:
- Design: Synthesize a 34-bp double-stranded oligonucleotide (dsODN) with 5´ phosphates and 3´ dideoxy-C modifications. Include internal phosphorothioate linkages at the 5´-most three nucleotides on both strands.
- Complex Formation: Pre-complex 100 pmol of purified Cas9 protein with 120 pmol of sgRNA in nucleofection buffer for 15 min at 25°C.
- Co-transfection: Mix the RNP complex with 100 pmol of dsODN tag. Transfect into 1x10⁵ HEK293T cells via nucleofection (Lonza 4D-Nucleofector, program CA-137).
- Control: Include a "dsODN-only" transfection (no RNP) to profile background, non-DSB-associated tag integration.
Protocol 2: GUIDE-seq Library Prep with DSN Normalization
Objective: Generate sequencing libraries while suppressing amplification bias. Detailed Steps:
- Genomic DNA Extraction: Harvest cells 72h post-transfection. Extract gDNA using a silica-column based kit with RNAse A treatment.
- Tag-Specific Enrichment: Perform primary PCR (12 cycles) using a biotinylated primer specific to the dsODN tag and a second primer binding to a common adapter ligated to sheared genomic DNA.
- Purification: Bind PCR products to streptavidin magnetic beads. Wash stringently (2x SSC, 0.1% SDS at 65°C) to remove non-specific amplicons.
- DSN Normalization: Elute DNA from beads. Add 2U of Duplex-Specific Nuclease (DSN) in 1x DSN buffer. Incubate at 55°C for 25 min. Stop reaction with EDTA.
- Final Library Amplification: Perform a limited-cycle (8-10 cycles) secondary PCR to add full Illumina adapters and sample indices. Purify with size-selection beads.
Data Analysis & Signal Thresholding
True off-target sites are distinguished from background using statistical modeling. The core metric is the GUIDE-seq Read Count (GRC) and the Noise-adjusted Confidence Score.
Table 3: Key Analysis Parameters for SNR Improvement
| Parameter | Formula/Definition | Recommended Threshold |
|---|---|---|
| GRC | Raw sequencing reads aligning uniquely to a locus. | > Median background + 3 SD |
| Uniquely Mapped Reads (%) | (Uniquely mapped reads / Total reads) * 100 | > 70% |
| R² (Replicate Correlation) | Pearson correlation of GRC between biological replicates. | > 0.85 |
| Background Subtraction | Adjusted GRC = Sample GRC - Mean(Control GRC at matched locus) |
Apply to all candidate sites |
Visualizations
Diagram 1: GUIDE-seq Signal vs Noise Workflow
Diagram 2: Noise Mitigation Strategy Map
Within GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) research for profiling CRISPR-Cas nuclease off-target effects, establishing critical controls is paramount. This document details application notes and protocols for validating on-target editing and specificity, ensuring data fidelity for therapeutic development.
Application Notes: Essential Controls for GUIDE-seq Studies
Positive Control for On-Target Cleavage
A validated, high-efficiency single guide RNA (sgRNA) must be included in every experiment to confirm nuclease activity. Failure of this control invalidates the entire run.
Negative Controls for Specificity
- Nuclease-deficient Control (dCas9): Distinguishes true cleavage events from background sequencing noise.
- Untransfected/Uninjected Control: Identifies baseline genomic DNA breaks.
- Non-targeting sgRNA Control: Accounts for non-specific cellular responses to RNP transfection.
Replicate and Sequencing Depth Requirements
Recent studies (2023-2024) indicate that robust off-target identification requires:
- Minimum Biological Replicates: 3
- Minimum Sequencing Depth at On-target Site: 100,000x coverage
- Recommended Whole-Genome Coverage: 0.5x - 1x
Table 1: Quantitative Benchmarks for Control Read Analysis
| Control Type | Acceptable On-target Efficiency Range | Maximum Allowed Background DSB Reads (per million) | Typical sgRNA Example |
|---|---|---|---|
| Experimental sgRNA | >40% INDELs (NGS) | N/A | VEGFA site 2 |
| Positive Control sgRNA | >60% INDELs (NGS) | N/A | EMX1 site 1 |
| dCas9 + sgRNA | <0.5% INDELs | < 10 | Any target |
| Non-targeting sgRNA | <0.1% INDELs | < 5 | Scrambled sequence |
| Untransfected Cells | 0% INDELs | < 2 | N/A |
Orthogonal Validation
All putative off-target sites from GUIDE-seq require confirmation via an orthogonal method (e.g., targeted amplicon sequencing, T7E1 assay) before being reported.
Detailed Protocols
Protocol 1: GUIDE-seq with Integrated Controls
Goal: Perform GUIDE-seq with mandatory positive, negative, and specificity controls.
Materials:
- GUIDE-seq Oligo: 5’-Phosphorylated, blunt-ended, double-stranded oligodeoxynucleotide (dsODN) tag.
- Cas9 Nuclease: Wild-type SpyCas9 or high-fidelity variant.
- sgRNAs: Experimental, Positive Control (EMX1), Non-targeting Control.
- Cells: HEK293T or other target cell line (≥ 80% viability).
- PCR Reagents: For tag-specific amplification.
- NGS Platform: Illumina HiSeq/MiSeq.
Procedure:
- Day 1: Cell Seeding
- Seed 2e5 HEK293T cells per well in a 24-well plate. Prepare wells for: Experimental sgRNA, Positive Control sgRNA, dCas9 + Experimental sgRNA, Non-targeting sgRNA, Untransfected.
Day 2: Transfection
- Complex 1.5 pmol Cas9 protein, 1.5 pmol sgRNA, and 100 pmol GUIDE-seq dsODN tag to form RNP.
- Transfect using lipid-based transfection reagent per manufacturer's protocol.
- For dCas9 control, use nuclease-deficient protein.
Day 5: Genomic DNA (gDNA) Harvest
- Extract gDNA using silica-membrane columns. Elute in 50 µL TE buffer.
- Quantify by fluorometry. Yield should be > 1 µg.
Tag Integration Enrichment & Library Prep
- First PCR (Tag-specific): Use 500 ng gDNA with primers specific to GUIDE-seq dsODN and a common linker sequence. Use 18-20 cycles.
- Purify PCR product (50-200 bp smear expected).
- Second PCR (Indexing): Add Illumina adapters and barcodes using 12-15 cycles.
- Pool libraries equimolarly.
Sequencing & Analysis
- Sequence on a 150 bp paired-end run.
- Process using the GUIDE-seq analysis software (PMID: 25513782) or updated pipelines (e.g., crispresso2 GUIDE-seq mode).
- Critical Analysis Step: Subtract sites found in dCas9 and non-targeting controls from the experimental sgRNA hit list.
Protocol 2: Orthogonal Validation by Amplicon Sequencing
Goal: Validate candidate off-target sites from GUIDE-seq.
Procedure:
- Design primers flanking each candidate off-target locus (including the positive on-target site). Amplicon size: 250-350 bp.
- Perform PCR on original gDNA from the experimental condition.
- Purify amplicons, construct an NGS library, and sequence at high depth (>100,000x per amplicon).
- Analyze INDEL frequencies using CRISPResso2 or similar.
Table 2: Criteria for Validating a True Off-Target Site
| Metric | Threshold for Validation | Notes |
|---|---|---|
| GUIDE-seq Read Count | ≥ 10 unique tag integrations | After background subtraction |
| Amplicon-Seq INDEL % | ≥ 0.5% | Must be above noise floor |
| Sequence Homology | Mismatch/bulge pattern consistent with Cas9 tolerance | Central mismatches usually disruptive |
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions
| Item | Function | Example/Product |
|---|---|---|
| High-Fidelity Cas9 Variant | Reduces off-target cleavage; critical for therapeutic profiling. | Alt-R S.p. HiFi Cas9 Nuclease V3 |
| Synthetic, Modified sgRNA | Enhances stability and reduces immune response in cells. | TruGuide sgRNA with 2'-O-methyl modifications |
| GUIDE-seq dsODN Tag | Blunt, phosphorylated double-stranded tag for DSB capture. | Alt-R GUIDE-seq Oligo (IDT) |
| Nuclease-Deficient dCas9 | Essential negative control for background DSB identification. | Alt-R S.p. dCas9 Protein |
| Non-targeting sgRNA Control | Control for non-specific cellular effects of sgRNA delivery. | Alt-R CRISPR-Cas9 Negative Control crRNA |
| Multiplexed NGS Validation Kit | For efficient orthogonal validation of multiple off-target loci. | xGEN Amplicon NGS workflow |
Visualizations
Title: Control-Integrated GUIDE-seq Experimental Workflow
Title: GUIDE-seq Core Principle: Tag Capture of DSBs
Within a broader thesis on GUIDE-seq genome-wide off-target detection research, a critical and often underappreciated hurdle is the precise tuning of parameters within bioinformatics analysis pipelines. The accuracy and reproducibility of off-target site identification hinge entirely on these settings. This document provides application notes and detailed protocols for navigating this complex step, focusing on the empirical tuning of key software parameters to minimize false positives and negatives.
Key Parameter Analysis and Comparison
The performance of GUIDE-seq analysis software (e.g., GUIDE-seq, MAGeCK, CRISPResso2, and custom pipelines) is governed by several interdependent parameters. Incorrect settings can lead to significant data misinterpretation.
Table 1: Core Parameters in GUIDE-seq Analysis Pipelines
| Software/Tool | Key Tunable Parameter | Typical Default | Impact of Low Value | Impact of High Value | Recommended Tuning Approach |
|---|---|---|---|---|---|
| GUIDE-seq (Original) | max.mismatch |
6 | Misses bona fide off-targets with more mismatches. | Increases false positives from genomic noise. | Titrate from 4 to 7 using positive/negative control sites. |
min.reads |
3-5 | Increases false positives from sequencing/PCR errors. | Misses low-efficiency, genuine off-target sites. | Scale with sequencing depth; use min.reads = sqrt(total reads)/N. |
|
| MAGeCK | read-count-threshold |
0 | Includes low-confidence, noisy integration sites. | May remove true, low-frequency integrations. | Set based on distribution of read counts per unique site. |
mapping-quality |
20 | Allows poorly mapped reads to contribute. | Excessively stringent, discards valid alignments. | Use aligned control samples to assess mapping error rate. | |
| CRISPResso2 | MIN_READS_ALIGNMENT |
100 | Processes loci with insufficient data for statistics. | Excludes valid loci with lower capture efficiency. | Align with min.reads from GUIDE-seq; keep consistent. |
| Alignment (Bowtie2/BWA) | -N (Mismatches in seed) |
0 | Fails to align gapped or highly mismatched integrations. | Spurious alignments increase noise dramatically. | Rarely increase above 1. Control via --score-min instead. |
--score-min (Stringency) |
Function-based | Similar to high max.mismatch—too permissive. |
Similar to low max.mismatch—too stringent. |
Use L,-0.6,-0.6 for GUIDE-seq; requires empirical validation. |
Experimental Protocol: Empirical Parameter Optimization
This protocol describes a systematic method for tuning the max.mismatch and min.reads parameters using control datasets.
Objective: To determine the optimal parameter set that maximizes the recovery of known positive control off-target sites while minimizing the identification of false positive sites in a negative control.
Materials: See "Research Reagent Solutions" below.
Procedure:
- Prepare Control Data Sets:
- Positive Control: Generate or obtain a GUIDE-seq dataset for a well-characterized gRNA with known, validated off-target sites (e.g., from the literature using targeted sequencing confirmation).
- Negative Control: Use the experimental GUIDE-seq library prior to transfection (genomic DNA only) OR use a non-targeting gRNA GUIDE-seq dataset. This represents background noise.
Define Parameter Grid:
- Create a matrix of parameter combinations to test. Example:
min.reads: 2, 3, 5, 8, 10max.mismatch: 3, 4, 5, 6
- Create a matrix of parameter combinations to test. Example:
Parallel Pipeline Execution:
- Run the complete GUIDE-seq analysis pipeline (from raw FASTQ to peak calling) for each parameter combination on both the positive and negative control datasets.
- Scripting is essential. Use a cluster or local script to batch process (e.g., Snakemake, Nextflow).
Calculate Performance Metrics:
- For each run on the Positive Control, identify how many of the known validated off-target sites are recovered (True Positives, TP).
- For each run on the Negative Control, count the total number of peaks called (False Positives, FP).
- Note: True Negatives (TN) and False Negatives (FN) are difficult to define genome-wide. Therefore, focus on TP and FP.
Determine Optimal Set:
- For each parameter combination, calculate a simple F-score proxy:
F = (2 * TP) / (2*TP + FP). - Plot the F-score against the parameter combinations.
- The optimal parameter set is the one that yields the highest F-score, effectively maximizing recovery of known sites while minimizing background noise.
- This set should be used for analyzing novel, experimental GUIDE-seq data under identical experimental conditions.
- For each parameter combination, calculate a simple F-score proxy:
Visualization of Workflow and Logic
Diagram 1: Parameter tuning workflow for GUIDE-seq analysis.
Diagram 2: Parameter impact on GUIDE-seq results.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for GUIDE-seq Parameter Tuning Experiments
| Item | Function in Parameter Tuning | Example/Details |
|---|---|---|
| Validated Positive Control gRNA Plasmid | Provides the ground truth off-target sites required to calculate True Positives (TP) during optimization. | e.g., A gRNA targeting the EMX1 or VEGFA site with off-targets confirmed by targeted sequencing. |
| Non-targeting Control gRNA Plasmid | Generates the negative control dataset to assess background noise and calculate False Positives (FP). | A scrambled gRNA with no known genomic targets, processed identically to experimental samples. |
| GUIDE-seq Oligonucleotide (dsODN) | The double-stranded donor oligo that integrates at cleavage sites. Essential for generating both control and experimental datasets. | HPLC-purified, phosphorothioate-modified ends to enhance stability. |
| High-Fidelity PCR Kit | For amplification of integrated dsODN sites. Minimizes PCR errors that can manifest as false positive peaks if min.reads is set too low. |
e.g., KAPA HiFi or Q5 Hot Start. |
| High-Sensitivity DNA Assay | Accurate quantification of libraries for equitable sequencing, preventing sample over/under-representation that affects min.reads thresholding. |
e.g., Agilent Bioanalyzer, Fragment Analyzer, or Qubit dsDNA HS Assay. |
| Cluster & Sequencing Reagents | To generate sufficient sequencing depth (>30M reads per sample) so that low-abundance integration events can be statistically distinguished from noise. | Illumina SBS kits appropriate for your sequencer (NovaSeq, NextSeq, etc.). |
| Computational Resources | Parameter grid searches require multiple parallel pipeline runs. Access to a HPC cluster or cloud computing (AWS, GCP) is highly recommended. | Minimum 16-32 cores, 64GB RAM for efficient parallelization of alignment and peak calling. |
Adapting GUIDE-seq for Challenging Cell Types (e.g., Primary Cells, Neurons)
Within the broader thesis on genome-wide off-target detection for CRISPR-based therapeutics, a critical limitation persists: standard GUIDE-seq, optimized for immortalized cell lines, fails in challenging, therapeutically relevant primary and post-mitotic cells. This application note details the adaptations required to overcome low cell numbers, poor transfection efficiency, and cytotoxicity in these systems, thereby extending the thesis's core methodology to clinically essential models.
Table 1: Primary Challenges and Adapted Solutions for Challenging Cell Types
| Challenge | Standard GUIDE-seq Protocol | Adapted Protocol for Primary/Neuronal Cells | Key Quantitative Improvement |
|---|---|---|---|
| Transfection/Nucleofection | Lipofection of dsODN & RNP. | Cell-type specific Nucleofection (e.g., Amaxa kits). Pre-complex RNP at low conc. (37°C, 10 min). | Neuronal transfection efficiency increased from <5% to 40-70% (Primary T-cells: ~80%). |
| dsODN Toxicity & Integration | 50-100 pmol dsODN per 100k HEK293T cells. | Reduce dsODN to 5-20 pmol per 10^6 primary cells. Use gel-purified dsODN. | Reduces cytotoxicity by >50% while maintaining detectable integration events. |
| Low Cell Number | Requires 1-2 million transfected cells. | Miniaturized library prep (1/10th volume reactions). PCR amplification cycles increased cautiously. | Reliable libraries from 50,000-100,000 successfully transfected cells. |
| Nuclease Activity & Culture | 48-72 hr culture post-transfection. | Shorter culture (24-48 hr) with viability enhancers (e.g., ROCK inhibitors for neurons). | Maintains cell viability >70% while allowing dsODN integration. |
| Genomic DNA (gDNA) Yield | Standard phenol-chloroform extraction. | Column-based gDNA extraction from low cell numbers. Carrier RNA optional. | Yields sufficient gDNA (≥ 200 ng) from 50k cells for library construction. |
Table 2: Optimized Reagent Quantities for Primary Human T-Cells
| Reagent | Standard Amount (HEK293T) | Optimized Amount (Primary T-Cells) | Purpose |
|---|---|---|---|
| Cas9 RNP (SpCas9) | 100 pmol | 50-75 pmol per 10^6 cells | Balance efficiency & toxicity. |
| dsODN (GUIDE-seq) | 50-100 pmol | 10-15 pmol per 10^6 cells | Reduce cytotoxicity, maintain tag integration. |
| Cells per reaction | 1 x 10^5 | 0.5-1 x 10^6 | Account for lower transfection efficiency. |
| Recovery Media | Standard growth media | Media + 10% FBS, ± IL-2 (T-cells) | Enhance post-nucleofection viability. |
Detailed Adapted Protocol
Day 1: Nucleofection & Culture for Primary Human T-Cells
Materials: Isolated primary human T-cells, P3 Primary Cell 4D-Nucleofector X Kit (Lonza), SpCas9 protein, synthetic sgRNA, gel-purified dsODN.
- Prepare RNP Complex: Complex 50 pmol SpCas9 with 60 pmol sgRNA in duplex buffer. Incubate at 37°C for 10 minutes.
- Add dsODN: Add 12 pmol of gel-purified dsODN to the RNP complex. Mix gently. Incubate at room temperature for 2 minutes.
- Cell Preparation: Wash 1x10^6 T-cells in PBS. Resuspend cell pellet in 20 µL of room temperature P3 Primary Cell Solution.
- Nucleofection: Combine cell suspension with RNP+dsODN mix. Transfer to a nucleofection cuvette. Run the appropriate program (EO-115 for T-cells). Immediately add 80 µL of pre-warmed RPMI 1640 + 10% FBS.
- Recovery Culture: Transfer cells to a 24-well plate prefilled with 500 µL of complete media (RPMI 1640, 10% FBS, 100 U/mL IL-2). Culture at 37°C, 5% CO2 for 24-48 hours.
Day 2/3: gDNA Extraction & Library Preparation
- Harvest Cells: Collect cells, wash with PBS. Use a microcolumn gDNA extraction kit (e.g., QIAamp DNA Micro Kit). Elute in 20 µL.
- Shearing & End-Repair: Shear 200 ng gDNA to ~300 bp (Covaris). Perform end-repair and A-tailing (NEB Ultra II FS kit).
- Adaptor Ligation: Ligate methylated sequencing adaptors using a reduced-volume reaction (10 µL total).
- Size Selection & PCR Enrichment: Size-select for 300-500 bp fragments (SPRI beads). Perform dsODN-specific nested PCR.
- 1st PCR (10 cycles): Use primer complementary to adaptor and dsODN.
- 2nd PCR (12-15 cycles): Use primers adding full Illumina P5/P7 and sample indexes.
- Library QC: Quantify by qPCR (KAPA Library Quant Kit). Analyze fragment size (Bioanalyzer). Sequence on Illumina MiSeq (2x150 bp).
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Adapted GUIDE-seq
| Reagent / Kit | Supplier Example | Function in Adapted Protocol |
|---|---|---|
| P3 Primary Cell 4D-Nucleofector Kit | Lonza | Cell-type specific reagent for high-efficiency, low-toxicity RNP/dsODN delivery. |
| Alt-R S.p. Cas9 Nuclease V3 | IDT | High-activity, endotoxin-free Cas9 for robust RNP formation. |
| Alt-R CRISPR-Cas9 sgRNA | IDT | Chemically modified sgRNA for enhanced stability and reduced immune response in primary cells. |
| Gel-purified dsODN GUIDE-seq Tag | Custom Synthesis (IDT) | Purified to remove short fragments, reducing toxicity and background. |
| QIAamp DNA Micro Kit | Qiagen | Column-based extraction for high-yield gDNA from low cell numbers. |
| NEBNext Ultra II FS DNA Library Prep Kit | NEB | Efficient library prep for low-input, fragmented DNA. |
| KAPA Library Quantification Kit | Roche | Accurate qPCR-based quantification of sequencing libraries. |
| SMARTer Human scRNA-Seq Kit | Takara Bio | (Optional) For parallel transcriptomic analysis of transfected cell population. |
Visualization of Workflows and Pathways
Title: Adapted GUIDE-seq Workflow for Primary Cells
Title: Problem-Solution Logic for Adapted GUIDE-seq
Title: Core GUIDE-seq Molecular Mechanism
GUIDE-seq vs. The Field: Validation Frameworks and Comparative Analysis
Within the broader thesis of GUIDE-seq genome-wide off-target detection research, a critical question persists: which method establishes itself as the gold standard for unbiased, sensitive, and specific identification of CRISPR-Cas nuclease off-target effects? The translation of CRISPR-Cas systems into therapeutic applications demands rigorous genomic safety assessments. This application note provides a comparative analysis of GUIDE-seq against its primary alternatives, detailed protocols for its execution, and a toolkit for researchers in drug development.
Comparative Analysis of Genome-Wide Off-Target Detection Methods
The following table summarizes the core quantitative and qualitative characteristics of leading methods, based on current benchmarking studies.
Table 1: Comparison of Genome-Wide Off-Target Detection Methods
| Method | Acronym Expansion | Core Principle | Detection Sensitivity | Required Control | Time to Data (days) | Key Limitation |
|---|---|---|---|---|---|---|
| GUIDE-seq | Genome-wide, Unbiased Identification of DE-pendent seq | Integration of oligo double-stranded oligodeoxynucleotides (dsODNs) into double-strand breaks (DSBs) followed by sequencing. | High (Can detect sites with <0.1% indel frequency). | Untreated sample for background subtraction. | 5-7 | Requires nucleofection; dsODN capture efficiency variable. |
| CIRCLE-seq | CIRCLE-seq | In vitro circularization and amplification of genomic DNA, followed by in vitro Cas9 digestion and sequencing. | Very High (Detects sites with ~0.01% activity). | No cellular control needed (in vitro). | 7-10 | Purely in vitro; may identify biologically irrelevant sites. |
| Digenome-seq | Digested genome seq | In vitro Cas9 digestion of genomic DNA, whole-genome sequencing, and mapping of cleavage sites. | High. | Mock-digested genomic DNA. | 7-10 | In vitro method; requires high sequencing depth (>100x). |
| SITE-seq | Selective Identification of Target Enriched seq | In vitro Cas9 digestion of chromatinized DNA, capture of cleaved ends with streptavidin, and sequencing. | High. | No nuclease control. | 7-10 | In vitro; uses chromatinized nuclear extracts. |
| BLESS | Break Labeling, Enrichment on Streptavidin, and Sequencing | Direct in situ labeling of DSBs in fixed cells with biotinylated linkers. | Medium. | Multiple controls (no nuclease, no linker). | 5-7 | Captures all cellular DSBs, requiring careful background analysis. |
| HTGTS | High-Throughput Genome-wide Translocation Sequencing | Detection of translocations from a fixed "bait" DSB to "prey" off-target DSBs. | Medium-High for bait-proximal prey. | Requires engineered bait locus. | 10-14 | Primarily detects off-targets that translocate; not fully genome-wide. |
Table 2: Practical Implementation Metrics
| Method | Cellular Context | DNA Input Requirement | Approx. Cost per Sample (Reagents & Seq) | Bioinformatics Complexity |
|---|---|---|---|---|
| GUIDE-seq | Live cells (transfected) | ~1-2 µg genomic DNA | $$$ | Moderate (Specialized pipelines required) |
| CIRCLE-seq | In vitro (purified genomic DNA) | ~1-5 µg genomic DNA | $$ | Moderate |
| Digenome-seq | In vitro (purified genomic DNA) | High (> 3 µg for WGS) | $$$$ (due to WGS depth) | High |
| SITE-seq | In vitro (chromatinized DNA) | ~1-5 µg genomic DNA | $$ | Moderate |
| BLESS | Fixed cells/sections | ~1-2 µg enriched DNA | $$$ | High |
| HTGTS | Live or fixed cells | ~1-2 µg genomic DNA | $$$ | High |
Detailed Protocol for GUIDE-seq
This protocol is adapted for adherent human cell lines (e.g., HEK293T) and the SpCas9 nuclease.
Part A: Cell Transfection and Genomic DNA Harvesting
Key Reagents:
- Cultured cells of interest
- GUIDE-seq dsODN (e.g., 5’-/5Phos/ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTGTCAAGATCGGAAGAGCGTCGTGTAG-3’, where N is a random base)
- Cas9 protein or expression plasmid
- sgRNA (synthetic or expressed)
- Nucleofection kit (e.g., Lonza SF Cell Line Kit)
- DNA purification kit (e.g., DNeasy Blood & Tissue Kit, Qiagen)
Procedure:
- Prepare dsODN: Resuspend and anneal the GUIDE-seq oligos to form the blunt, phosphorylated dsODN.
- Form RNP Complex: For ribonucleoprotein (RNP) delivery, pre-complex 5 µg of purified SpCas9 protein with 200 pmol of synthetic sgRNA in nucleofection buffer. Incubate 10 min at 25°C. Add 100 pmol of GUIDE-seq dsODN to the RNP complex.
- Harvest and Nucleofect Cells: Harvest ~1x10⁶ cells per condition. Resuspend cell pellet in 100 µL nucleofection solution containing the RNP/dsODN mixture. Perform nucleofection using a 4D-Nucleofector (e.g., program CM-130). Include controls: "No dsODN" and "No nuclease."
- Culture and Harvest: Transfer nucleofected cells to pre-warmed medium. Culture for 72 hours to allow for DSB repair and integration.
- Extract Genomic DNA: Harvest cells and purify genomic DNA according to your kit’s protocol. Quantify DNA by fluorometry.
Part B: GUIDE-seq Library Preparation and Sequencing
Key Reagents:
- Enzymatic shearing kit (e.g., Nextera tagmentation)
- PCR primers containing Illumina adapters
- High-fidelity PCR master mix
- Size selection beads (e.g., SPRIselect beads)
Procedure:
- Fragment DNA: Fragment 500 ng of purified genomic DNA to an average size of 300-500 bp via Covaris sonication or enzymatic tagmentation.
- End Repair & A-Tailing: Perform standard end-repair and dA-tailing reactions to prepare fragments for adapter ligation.
- Adapter Ligation: Ligate Illumina-compatible Y-shaped adapters to the fragments.
- Enrich dsODN-Containing Fragments:
- Perform the first PCR (10-12 cycles) using a primer specific to the GUIDE-seq dsODN (P5-GUIDEseq) and a primer complementary to the ligated adapter (P7-Common).
- Clean up the PCR product with SPRIselect beads (0.8x ratio).
- Final Library Amplification:
- Perform a second, indexing PCR (8-10 cycles) using standard Illumina P5 and indexed P7 primers.
- Clean up the final library with SPRIselect beads (0.8x ratio).
- QC and Sequence: Validate library size distribution (TapeStation/Bioanalyzer) and quantify by qPCR. Pool libraries and sequence on an Illumina platform (e.g., MiSeq, 2x150 bp; or HiSeq, 2x75 bp). Aim for 20-50 million reads per sample.
Part C: Bioinformatic Analysis
A standard pipeline involves:
- Demultiplexing and Quality Control: Use
bcl2fastqorIllumina DRAGEN. - Read Trimming and Alignment: Trim adapter sequences with
cutadapt. Align reads to the reference genome (e.g., hg38) usingbowtie2orBWA, allowing for soft-clipping. - Peak Calling: Use the dedicated GUIDE-seq software or
GUIDE-seqAlignerto identify genomic locations where reads containing the dsODN sequence cluster. This identifies candidate off-target sites. - Annotation and Visualization: Annotate peaks with genomic features. Compare to "no dsODN" and "no nuclease" controls to filter background. Visualize on a genome browser.
Visualizations
GUIDE-seq Experimental Workflow
Method Selection Logic Flowchart
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for GUIDE-seq and Related Methods
| Item | Function & Relevance | Example Vendor/Product |
|---|---|---|
| Phosphorylated dsODN | Double-stranded oligo donor for integration into Cas9-induced DSBs. The cornerstone of GUIDE-seq. | Integrated DNA Technologies (Custom Alt-R GUIDE-seq Oligo Duplex) |
| Recombinant Cas9 Nuclease | High-purity, endotoxin-free Cas9 protein for RNP formation, enabling rapid delivery and cleavage. | Thermo Fisher Scientific (TrueCut Cas9 Protein v2) |
| Synthetic sgRNA (chemically modified) | Nuclease-resistant, high-activity sgRNA for stable RNP complex formation. | Synthego (sgRNA EZ Kit) |
| Nucleofection Kit | Cell line-specific reagent for high-efficiency delivery of RNP complexes and dsODNs. | Lonza (SF Cell Line 4D-Nucleofector X Kit) |
| Genomic DNA Purification Kit | Robust extraction of high-molecular-weight, PCR-quality genomic DNA from cultured cells. | Qiagen (DNeasy Blood & Tissue Kit) |
| Library Prep Kit with Beads | All-in-one kits for fragmentation, end-prep, adapter ligation, and PCR cleanup via SPRI beads. | Illumina (Nextera DNA Flex Library Prep) |
| High-Fidelity PCR Master Mix | Enzyme mix for accurate, high-yield amplification of GUIDE-seq libraries with minimal bias. | NEB (Q5 Hot Start High-Fidelity 2X Master Mix) |
| Bioinformatic Pipeline | Specialized software for identifying and quantifying dsODN integration sites from sequencing data. | GUIDE-seq (open source, from Tsai Lab) or CRIS.py |
GUIDE-seq remains a leading in vivo method for balanced sensitivity and biological relevance in off-target profiling, directly informing therapeutic safety assessments. Its primary advantage is the capture of nuclease activity within the native chromatin context of living cells. However, the pursuit of a singular "gold standard" may be reductive. The evolving thesis suggests a convergent approach: using hyper-sensitive in vitro methods like CIRCLE-seq for exhaustive, agnostic off-target discovery, followed by orthogonal validation of prioritized sites in therapeutically relevant cells using GUIDE-seq or targeted deep sequencing. This tiered strategy offers a robust framework for drug development professionals to de-risk CRISPR-based therapies.
Application Notes Within the broader thesis on genome-wide off-target detection for CRISPR-based therapeutics, the selection of an in vitro sensitivity assay is critical. GUIDE-seq and CIRCLE-seq represent two leading, yet methodologically distinct, approaches. This analysis compares their sensitivity, workflow, and applicability for profiling CRISPR-Cas nuclease off-target effects in a controlled in vitro setting.
Core Principle & Sensitivity Comparison
- GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing): An in cellulo method that detects double-strand breaks (DSBs) in living cells by capturing integrated oligonucleotide tags. Its sensitivity is influenced by cellular delivery efficiency and nuclear uptake of the tag.
- CIRCLE-seq (Circularization for In Vitro Reporting of Cleavage Effects by Sequencing): A purely in vitro, cell-free method that uses circularized genomic DNA as a substrate for Cas nuclease cleavage. It offers ultra-sensitive detection due to reduced sequence complexity and high sequencing depth on target loci.
Quantitative data from key comparative studies are summarized below:
Table 1: Comparative Sensitivity Metrics In Vitro
| Metric | GUIDE-seq | CIRCLE-seq | Notes |
|---|---|---|---|
| Effective Detection Threshold | ~0.1% of INDEL frequency | ~0.01% to 0.001% of reads | CIRCLE-seq detects very rare cleavage events. |
| Background Signal | Moderate (cellular repair noise) | Very Low (controlled enzymatic reaction) | CIRCLE-seq background is primarily from DNA shearing. |
| Genomic Context | Native chromatin (in cells) | Protein-free, fragmented DNA | GUIDE-seq reflects chromatin accessibility; CIRCLE-seq reflects sequence preference. |
| Input DNA Requirement | N/A (requires live cells) | 1 µg – 5 µg of genomic DNA | CIRCLE-seq uses purified genomic DNA. |
| Key Limitation for Sensitivity | Oligonucleotide uptake & integration efficiency. | Adapter circularization & PCR amplification bias. |
Detailed Protocols
Protocol A: GUIDE-seq Workflow for In Vitro Sensitivity Assessment Note: This protocol assesses sensitivity in cultured cells.
- Co-delivery: Co-transfect cultured cells (e.g., HEK293T) with three components: 1) plasmid expressing SpCas9 and sgRNA, 2) GUIDE-seq oligonucleotide duplex (tag), using a high-efficiency method (e.g., nucleofection).
- Incubation: Culture cells for 48-72 hours to allow for DSB formation, tag integration, and repair.
- Genomic DNA Extraction: Harvest cells and extract high-molecular-weight genomic DNA.
- Library Preparation: a. Shear DNA to ~500 bp fragments. b. Perform end-repair, A-tailing, and ligation of sequencing adapters. c. Perform two nested PCRs using an adapter-specific primer and a tag-specific primer to enrich for tag-integrated fragments.
- Sequencing & Analysis: Sequence on an Illumina platform. Map reads to the reference genome, identify tag integration sites, and call off-target sites using the GUIDE-seq computational pipeline.
Protocol B: CIRCLE-seq Workflow for In Vitro Sensitivity
- Genomic DNA Preparation: Extract and purify genomic DNA from target cell type. Fragment by sonication or enzymatic digestion (e.g., ~300 bp).
- DNA Circularization: End-repair, A-tail, and ligate fragmented DNA using a splinter oligo to create single-stranded DNA circles. Ligate the circles using Circligase.
- Cas9 Cleavage In Vitro: Incubate circularized DNA library with recombinant Cas9-sgRNA ribonucleoprotein (RNP) complex in optimized reaction buffer.
- Linearization of Cleaved Circles: Treat reaction with exonuclease to degrade linear DNA. Use a nicking enzyme specific to the adapter sequence to linearize only circles that were cleaved by Cas9.
- Library Preparation: Amplify linearized fragments by PCR with indexed adapters for sequencing.
- Sequencing & Analysis: Perform high-depth sequencing (>50M reads). Map reads, identify breakpoints, and calculate read enrichment at potential off-target sites versus nuclease-free controls.
Visualization
Workflow Comparison: GUIDE-seq vs CIRCLE-seq
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Off-target Detection Assays
| Reagent / Material | Function in Experiment | Primary Assay |
|---|---|---|
| GUIDE-seq Oligoduplex | Double-stranded, blunt-ended, phosphorothioate-modified oligonucleotide that integrates into DSBs, serving as a tag for amplification. | GUIDE-seq |
| High-Efficiency Transfection/Nucleofection Kit | Enables co-delivery of Cas9/sgRNA plasmid and GUIDE-seq oligo into hard-to-transfect cell lines with high viability. | GUIDE-seq |
| Recombinant Cas9 Nuclease (Purified) | High-purity, ready-to-complex protein for forming RNP complexes with sgRNA for in vitro cleavage reactions. | CIRCLE-seq |
| Circligase ssDNA Ligase | Enzymatically ligates the 3' and 5' ends of single-stranded, adapter-flanked DNA fragments to form circles. Critical for CIRCLE-seq. | CIRCLE-seq |
| sgRNA (Synthetic or In Vitro Transcribed) | Target-specific guide RNA for complexing with Cas9. Requires high purity and full-length integrity for optimal activity. | Both |
| Fragmentation Enzyme/Shearing System | For generating consistently sized DNA fragments (200-500 bp) from genomic DNA as input for library construction. | CIRCLE-seq |
| Nicking Enzyme (e.g., Nt.BspQI) | Cuts specifically within the CIRCLE-seq adapter sequence to linearize only circles that were cleaved by Cas9, enabling amplification. | CIRCLE-seq |
| High-Fidelity PCR Master Mix | For accurate, low-bias amplification of tag-integrated (GUIDE-seq) or linearized circle (CIRCLE-seq) fragments during NGS library prep. | Both |
Application Notes
Within a thesis investigating genome-wide off-target profiling for therapeutic CRISPR-Cas9 development, selecting the optimal unbiased detection method is critical. This analysis compares GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) and Digenome-seq (digital genome sequencing) with a focus on sensitivity in cell-free contexts. GUIDE-seq is an in-cell method that relies on the capture of double-strand breaks (DSBs) via integration of a tagged oligonucleotide during repair. Its sensitivity is inherently tied to cellular viability, transfection efficiency, and repair activity. In contrast, Digenome-seq is a cell-free assay where genomic DNA is treated with ribonucleoprotein (RNP) complexes in vitro, followed by whole-genome sequencing to map cleavage-induced linearized fragments. This cell-free nature eliminates cellular context variables, theoretically allowing detection of off-target sites with very low cleavage frequencies (<0.1%), as it directly sequences all fragmented DNA without amplification bias from integration events.
Recent studies highlight that Digenome-seq consistently identifies a larger raw number of potential off-target sites than GUIDE-seq, particularly in GC-rich or repetitive genomic regions. However, this high sensitivity necessitates stringent bioinformatic filtering to reduce false positives from background genomic fragmentation. GUIDE-seq data, while often yielding fewer total sites, is enriched for biologically relevant sites within a cellular context. For therapeutic safety profiling, a convergent approach is recommended: using Digenome-seq for its highly sensitive, unbiased initial screen, followed by GUIDE-seq or targeted sequencing to validate which identified sites are cleaved in relevant cellular or animal models.
Protocols
Protocol 1: Digenome-seq for Cell-Free Off-Target Detection
- Genomic DNA Isolation: Extract high-molecular-weight genomic DNA (>50 kb) from target cell lines using a phenol-chloroform method or a commercial kit. Avoid shearing.
- RNP Complex Formation: For a 50 µL reaction, incubate 4 µg of purified SpCas9 protein with 3.6 pmol of sgRNA (at a 1:2 molar ratio) in 1X Cas9 buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM MgCl2, 10% glycerol, 1 mM DTT) for 10 min at 25°C.
- In Vitro Cleavage: Add 1 µg of genomic DNA to the RNP complex. Adjust volume to 50 µL with nuclease-free water and 1X NEBuffer 3.1. Incubate at 37°C for 8 hours.
- DNA Purification: Purify DNA using AMPure XP beads. Elute in 30 µL of low TE buffer.
- Whole-Genome Sequencing Library Prep: Fragment the purified DNA to ~300 bp using a Covaris ultrasonicator. Prepare sequencing libraries using a kit such as Illumina TruSeq DNA Nano, following manufacturer instructions. Include a non-treated genomic DNA control.
- Sequencing & Analysis: Perform paired-end sequencing (≥50x coverage) on an Illumina platform. Align reads to the reference genome. Use Digenome-seq analysis software (e.g., Digenome2.0, Cas-OFFinder) to map cleavage sites identified as genomic positions with significant increases in read start ends (5' ends) in the RNP-treated sample versus control.
Protocol 2: GUIDE-seq for Cellular Off-Target Validation
- GUIDE-seq Oligonucleotide Design: Synthesize a blunt, double-stranded, 5'-phosphorylated GUIDE-seq Oligo (e.g., 5'-/5Phos/NNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGNNNNNN-3', where N are degenerates and * denotes phosphorothioate bonds).
- Cell Transfection: Seed HEK293T or relevant target cells in a 24-well plate. Co-transfect 500 ng of sgRNA expression plasmid (or 250 ng of in vitro transcribed sgRNA), 250 ng of Cas9 expression plasmid (or 250 ng of Cas9 mRNA), and 100 pmol of GUIDE-seq Oligo using a lipid-based transfection reagent (e.g., Lipofectamine 3000). Include controls without the oligo.
- Genomic DNA Harvest: 72 hours post-transfection, harvest cells and extract genomic DNA using a silica-column kit.
- Library Preparation for Sequencing:
- Sonication & End-Repair: Shear 1 µg of genomic DNA to ~500 bp. Perform end-repair and A-tailing.
- Adapter Ligation: Ligate annealed, partially double-stranded barcoded adapters with T-overhangs.
- Primary PCR: Amplify using an adapter-specific primer and a primer specific to the GUIDE-seq Oligo.
- Secondary (Nested) PCR: Re-amplify the primary PCR product using indexed Illumina primers.
- Sequencing & Analysis: Sequence on a MiSeq or HiSeq system. Process reads using the GUIDE-seq computational pipeline (e.g.,
guideseqpackage) to identify genomic integration sites of the oligo, which correspond to DSB locations.
Diagrams
Digenome-seq Cell-Free Workflow
GUIDE-seq In-Cell Workflow
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Experiment |
|---|---|
| Recombinant SpCas9 Nuclease | High-purity, endotoxin-free protein for forming RNP complexes in Digenome-seq and in vitro assays. |
| Chemically Modified sgRNA | sgRNA with 2'-O-methyl and phosphorothioate modifications for enhanced stability in cellular GUIDE-seq transfection. |
| GUIDE-seq Oligo (dsODN) | Double-stranded, phosphorothioate-protected oligonucleotide that integrates at DSBs to tag sites for amplification. |
| High-Fidelity DNA Polymerase | For accurate amplification of GUIDE-seq libraries (e.g., Q5, KAPA HiFi) to minimize PCR artifacts. |
| AMPure XP Beads | Solid-phase reversible immobilization (SPRI) beads for size selection and purification of DNA in library prep. |
| Illumina-Compatible Adapters | Dual-indexed adapters with unique molecular identifiers (UMIs) for multiplexed, high-throughput sequencing. |
| Cas9 Buffer System | Optimized reaction buffer (MgCl2, HEPES, KCl) to maintain maximum Cas9 RNP nuclease activity in vitro. |
| Genomic DNA Extraction Kit | Kit for high-molecular-weight DNA isolation (e.g., Qiagen Genomic-tip) critical for Digenome-seq input. |
Quantitative Data Comparison
Table 1: Method Characteristics & Sensitivity
| Parameter | GUIDE-seq | Digenome-seq |
|---|---|---|
| Assay Environment | Intact living cells | Cell-free (purified genomic DNA) |
| Detection Principle | Capture of tagged dsODN during NHEJ | Direct sequencing of in vitro cleaved ends |
| Typical Reported Sensitivity | ~0.1% of alleles (dependent on transfection) | Can detect <0.01% cleavage frequency in vitro |
| Primary Source of False Positives | Random oligo integration, PCR artifacts | Background genomic fragility, sequencing errors |
| Time to Result (wet lab) | 7-10 days (includes cell culture/transfection) | 3-4 days |
| Requires Cellular NHEJ Machinery | Yes | No |
| Compatible with Delivery Methods | Transfection, nucleofection (altered efficiency) | Not applicable (cell-free) |
Table 2: Typical Experimental Output (Based on Recent Studies)
| Output Metric | GUIDE-seq | Digenome-seq | Interpretation |
|---|---|---|---|
| Average # of Off-Target Sites (per sgRNA, standard filters) | 5 - 15 sites | 20 - 60+ sites | Digenome-seq has higher raw sensitivity. |
| Overlap with Validated Sites (by targeted deep sequencing) | High (>80%) | Moderate to High (requires stringent filtering) | GUIDE-seq sites are enriched for biological relevance. |
| Detection in Low-Accessibility Chromatin | Limited (requires active DSB repair) | Possible (chromatin is digested in vitro) | Digenome-seq can identify sites inaccessible in cells. |
| Amenability to High-Throughput Screening | Low to Moderate (scalability limited by cell culture) | High (multiple gRNAs can be tested on pooled DNA) | Digenome-seq更适合初级全基因组筛查. |
This application note serves as a critical comparative analysis within a broader thesis investigating genome-wide, unbiased methods for profiling CRISPR-Cas nuclease off-target effects. The therapeutic promise of CRISPR is contingent upon understanding and minimizing off-target cleavage. While early computational prediction tools were foundational, empirical, genome-wide screening methods are essential for comprehensive risk assessment. This document details and compares three pivotal experimental techniques: GUIDE-seq, SITE-seq, and BLISS, providing protocols and data to guide researchers in selecting the optimal approach for their drug development pipeline.
Table 1: Core Principle and Detection Mechanism
| Method | Acronym Expansion | Core Principle | Detection Molecule |
|---|---|---|---|
| GUIDE-seq | Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing | Captures double-strand breaks (DSBs) via integration of a blunt, double-stranded oligodeoxynucleotide (dsODN) tag. | Exogenous dsODN |
| SITE-seq | Selective Enrichment and Identification of Tagged Genomic DNA Ends by Sequencing | Uses a biotinylated adaptor ligated to in situ nick-translated DSB ends, followed by streptavidin enrichment. | Biotinylated adaptor |
| BLISS | Breaks Labeling In Situ and Sequencing | Directly labels DSBs in situ with barcoded sequencing adaptors within fixed cells or nuclei. | Barcoded sequencing adaptors |
Table 2: Key Performance Metrics and Practical Considerations
| Parameter | GUIDE-seq | SITE-seq | BLISS |
|---|---|---|---|
| Sensitivity | High (Can detect low-frequency events) | Very High (Enhanced by selective enrichment) | Moderate to High (Depends on labeling efficiency) |
| Input Requirement | 1-5 x 10⁵ transfected cells | ~2 x 10⁶ cells (or nuclei) | 5 x 10³ - 10⁵ cells/nuclei |
| Working Environment | Live cells (requires ODN delivery) | Cell lysates/Nuclear extracts | In situ (Fixed cells/tissues) |
| Primary Application | Off-target profiling in cell lines | Sensitive off-target discovery and quantification | Off-target detection in primary cells, tissues, & in vivo models |
| Key Advantage | Robust, relatively simple workflow | High sensitivity; no ODN transfection bias | Preservation of spatial/nuclear context; low input |
| Key Limitation | Requires efficient ODN delivery/transfection | Complex protocol; high input requirement | Potential for background from sample handling. |
Detailed Experimental Protocols
Protocol 1: GUIDE-seq (Adapted from Tsai et al., 2015)
Key Reagent: GUIDE-seq dsODN (annealed, HPLC-purified).
Procedure:
- Co-transfection: Co-deliver CRISPR-Cas9 RNP (or plasmid) and the GUIDE-seq dsODN (e.g., 100 pmol) into 1-5x10⁵ cells using an appropriate transfection reagent.
- Genomic DNA Extraction: Harvest cells 72 hours post-transfection. Extract genomic DNA using a silica column-based kit.
- DSB End Capture & Amplification:
- Fragment DNA by sonication or enzymatic shearing to ~500 bp.
- End-repair, A-tailing, and ligate Illumina-compatible adaptor (Y-shaped or blunt) using a DNA library prep kit.
- PCR Enrichment: Perform nested PCR using a primer specific to the integrated dsODN and a primer for the Illumina adaptor. Use a high-fidelity polymerase.
- Sequencing & Analysis: Purify PCR amplicons, sequence on an Illumina platform (2x150 bp recommended). Align reads to the reference genome and identify dsODN integration sites using the GUIDE-seq analysis software (available on GitHub).
Protocol 2: SITE-seq (Adapted from Cameron et al., 2017)
Key Reagent: Biotinylated dsDNA Adaptor with 5' dA-overhang.
Procedure:
- Nuclei Isolation & In Situ Nicking: Harvest ~2x10⁶ CRISPR-treated cells. Isolate nuclei. Treat nuclei with a nicking enzyme (e.g., DNase I) in a controlled reaction to create 5' phosphorylated nicks near DSB sites.
- Nick Translation & Biotinylation: Use E. coli DNA Polymerase I to initiate nick translation from nicked sites, incorporating biotinylated dUTP, thereby labeling DSB ends.
- Genomic DNA Extraction & Shearing: Purify and shear genomic DNA to ~300 bp.
- Streptavidin Enrichment: Bind biotinylated fragments to streptavidin magnetic beads. Wash stringently.
- Library Construction & Sequencing: On-bead, perform end-repair, A-tailing, and ligation of Illumina adaptors. Elute and amplify via PCR. Sequence and analyze using the SITE-seq pipeline to map enriched break ends.
Protocol 3: BLISS (Adapted from Yan et al., 2017)
Key Reagent: BLISS Adapters (barcoded, duplex oligos with a 5' dT-overhang).
Procedure:
- Fixation & Permeabilization: Fix cells or tissue sections (5x10³ - 10⁵ cells) with formaldehyde. Permeabilize with Triton X-100.
- In Situ Ligation: In a reaction chamber, ligate BLISS adapters directly to DSB ends on the chromatin using T4 DNA Ligase. This step labels breaks in situ.
- DNA Extraction & Library Prep: Reverse crosslinks and purify DNA. Perform tagmented library preparation (e.g., using Th5 transposase) specifically on adapter-ligated DNA.
- Indexing PCR & Sequencing: Amplify libraries with indexed primers. Sequence. Analyze data with the BLISS analysis code to decode barcodes and map DSB locations.
Visualization of Workflows
Title: Comparative Workflows of GUIDE-seq, SITE-seq, and BLISS
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Off-Target Detection Assays
| Reagent | Primary Function | Example/Note |
|---|---|---|
| GUIDE-seq dsODN | Blunt, double-stranded oligo that integrates into DSBs as a tag for PCR capture. | HPLC-purified, phosphorothioate modifications recommended for stability. |
| Biotinylated dUTP | Incorporated via nick translation to label DSB ends for streptavidin enrichment in SITE-seq. | Critical for pull-down sensitivity. |
| BLISS Adapters | Barcoded duplex oligos with dT-overhang for direct in situ ligation to DSB ends. | Unique barcodes enable multiplexing and reduce PCR duplicates. |
| High-Fidelity Polymerase | Accurate amplification of library fragments for NGS. | Essential for minimizing PCR errors during library enrichment. |
| Streptavidin Magnetic Beads | Solid-phase capture of biotinylated DNA fragments in SITE-seq. | High binding capacity beads reduce non-specific background. |
| T4 DNA Ligase | Catalyzes the ligation of adapters to DSB ends in BLISS and other workflows. | High-concentration formulation recommended for in situ reactions. |
| Cas9 Nuclease (WT) | The effector protein generating DSBs at on- and off-target sites. | Recombinant protein for RNP delivery is standard for high efficiency. |
| Next-Gen Sequencing Kit | Platform-specific reagents for cluster generation and sequencing. | Illumina platforms are most commonly used. |
Application Notes
Within the broader thesis on GUIDE-seq genome-wide off-target detection, a critical challenge is the validation of predicted off-target sites. GUIDE-seq (Genome-wide, Unbiased Identification of DSBs Enabled by Sequencing) is a powerful, unbiased method for identifying potential off-target cleavage sites for CRISPR-Cas nucleases. However, its findings require orthogonal validation to confirm editing frequencies, assess biological relevance, and meet regulatory standards for therapeutic development. A validation cascade integrating GUIDE-seq with targeted, deep-sequencing methods provides a robust framework for confirming off-target activity.
The core principle is to use GUIDE-seq as a discovery tool to generate a genome-wide list of candidate off-target loci. These candidates are then interrogated with orthogonal, amplification-based Next-Generation Sequencing (NGS) assays, such as amplicon sequencing or Capture-seq. This multi-tiered approach increases confidence, as each method relies on distinct biochemical principles (oligonucleotide tag integration vs. PCR amplification of targeted loci).
Recent literature and product developments emphasize the necessity of this cascade. Studies consistently show that while GUIDE-seq is highly sensitive, orthogonal validation quantifies off-target editing rates with greater depth and precision at specific loci, which is essential for risk assessment. The integration of these data is paramount for creating a comprehensive safety profile of a CRISPR-based therapeutic.
Quantitative Data Summary
Table 1: Comparison of GUIDE-seq and Orthogonal NGS Validation Methods
| Feature | GUIDE-seq (Discovery) | Targeted Amplicon-Seq (Validation) |
|---|---|---|
| Primary Goal | Unbiased, genome-wide identification | Targeted, deep quantification |
| Detection Principle | Integration of dsODN tag into DSBs | PCR amplification of specific loci |
| Typical Sequencing Depth | 20 - 50 million reads (whole genome) | 100,000 - 1,000,000x per amplicon |
| Sensitivity | Can detect edits at ~0.1% frequency | Can detect edits at <0.01% frequency |
| Throughput | High (genome-wide) | Low to Medium (dozens to hundreds of loci) |
| Key Output | List of candidate off-target sites | Precise indel percentage at each validated site |
| Major Advantage | Hypothesis-free, unbiased | Extremely sensitive and quantitative |
| Major Limitation | May miss sites in complex/repetitive regions | Requires a priori knowledge of loci |
Experimental Protocols
Protocol 1: GUIDE-seq for Off-Target Discovery
- Cell Transfection: Co-transfect 1x10^6 HEK293T cells (or target cell line) with 1 µg of plasmid expressing Cas9 nuclease and sgRNA, plus 100 pmol of phosphorylated, HPLC-purified GUIDE-seq dsODN using a recommended transfection reagent. Include controls without dsODN.
- Genomic DNA Extraction: Harvest cells 72 hours post-transfection. Extract genomic DNA using a silica-membrane column kit, ensuring high molecular weight DNA.
- Library Preparation & Sequencing: Follow the published GUIDE-seq wet-lab protocol. Briefly, shear gDNA, perform end-repair and A-tailing, ligate to Illumina adapters, and perform two sequential PCRs (first to enrich for dsODN-tagged fragments, second to add index primers for multiplexing). Purify libraries and sequence on an Illumina platform (2x150 bp PE recommended).
- Data Analysis: Process FASTQ files using the publicly available GUIDE-seq software or related pipelines (e.g., CRISPResso2, pipeGUIDE) to align reads and identify significant off-target sites, requiring a minimum of 2 unique reads per site.
Protocol 2: Orthogonal Validation by Targeted Amplicon Sequencing
- Primer Design: Design PCR primers (amplicon size 250-350 bp) flanking each candidate off-target site from Protocol 1, plus the on-target site. Include Illumina adapter overhangs.
- Primary PCR: Perform first-round PCR on 50-100 ng of the same gDNA used in GUIDE-seq (and control DNA) using high-fidelity polymerase. Use 18-20 cycles.
- Indexing PCR: Use a second, limited-cycle PCR (8-10 cycles) to attach dual indices and full Illumina sequencing adapters.
- Library Quantification & Pooling: Quantify libraries via fluorometry, normalize, and pool equimolarly.
- Deep Sequencing: Sequence the pool on a MiSeq or HiSeq platform (2x250 bp or 2x300 bp) to achieve a minimum depth of 100,000x per amplicon.
- Analysis: Align reads to reference sequences. Quantify indel frequencies using specialized tools (e.g., CRISPResso2, AmpliconDIVider). Sites with indel frequencies significantly above background (e.g., in untreated or control sgRNA samples) are considered validated.
Visualizations
Diagram Title: Validation Cascade Workflow for CRISPR Off-Target Analysis
Diagram Title: Orthogonal Method Principle: Tag Integration vs. Targeted Amplification
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for the Validation Cascade
| Item | Function/Description |
|---|---|
| Phosphorylated GUIDE-seq dsODN | Double-stranded oligonucleotide tag that integrates into CRISPR-Cas-induced double-strand breaks (DSBs) for genome-wide off-target discovery. |
| High-Fidelity DNA Polymerase | Essential for accurate amplification during both GUIDE-seq library construction and targeted amplicon generation for validation. |
| Illumina-Compatible Adapter & Index Kits | Provides reagents for attaching sequencing adapters and multiplexing indices to libraries for NGS on Illumina platforms. |
| CRISPR-Cas9/sgRNA Expression Vector | Plasmid or RNA for delivering the nuclease and guide RNA components into target cells. |
| Targeted Amplicon Primer Pools | Custom-designed primers for amplifying candidate off-target loci identified by GUIDE-seq for deep sequencing validation. |
| Genomic DNA Extraction Kit (Column-Based) | For high-quality, high-molecular-weight gDNA isolation from transfected cells, critical for both discovery and validation steps. |
| NGS Analysis Software (CRISPResso2, pipeGUIDE) | Specialized bioinformatics tools for analyzing both GUIDE-seq and amplicon sequencing data to identify and quantify indel events. |
| Cell Line-Specific Transfection Reagent | Ensures efficient delivery of GUIDE-seq dsODN and CRISPR machinery into the target cell type under study. |
Within GUIDE-seq genome-wide off-target detection research, multiple experimental and computational methods exist, each yielding potentially divergent results. These discrepancies are not merely noise but arise from inherent methodological biases and strengths. This document provides Application Notes and Protocols for interpreting these differences, enabling researchers to synthesize a more accurate, comprehensive off-target profile for therapeutic CRISPR-Cas systems.
Core Methodologies: Biases & Data Interpretation
The table below summarizes key characteristics of primary genome-wide detection techniques, explaining sources of discrepancy.
Table 1: Method-Specific Biases and Strengths in GUIDE-seq Context
| Method | Principle | Key Strengths | Inherent Biases/Limitations | Primary Discrepancy Sources vs. GUIDE-seq |
|---|---|---|---|---|
| GUIDE-seq | Integration of oligoduplex into DSBs followed by sequencing. | In cells; detects DSBs directly; low false-positive rate. | Requires oligo delivery; may miss low-efficiency or transient breaks. | Gold standard; discrepancies often indicate false positives in other methods. |
| CIRCLE-seq | In vitro circularization & enrichment of cleaved genomic DNA. | Highly sensitive; works on purified genomic DNA. | In vitro conditions may not reflect cellular chromatin state. | Can identify many in vitro sites not detected in cells (chromatin bias). |
| SITE-seq | In vitro cleavage & capture of biotinylated ends. | Sensitive; uses purified Cas9 RNP. | In vitro; requires high concentrations of RNP. | Similar to CIRCLE-seq; discrepancies highlight chromatin/context effects. |
| Digenome-seq | In vitro Cas9 cleavage, whole-genome sequencing of ends. | Genome-wide; no sequence bias from amplification. | In vitro; computationally intensive; high DNA input. | Identifies cleavage potential without cellular repair factors. |
| BLISS | Direct labeling and capture of DSB ends in fixed cells. | Captures endogenous DSBs in situ; single-cell possible. | Lower throughput; complex library prep. | May capture off-targets from staggered nicks or other nucleases. |
| Computational Prediction | In silico scoring based on sequence similarity. | Fast, inexpensive; can scan whole genome. | High false-positive and false-negative rates. | Lacks biochemical/cellular context; discrepancies validate models. |
Framework for Interpreting Discrepant Results
- GUIDE-seq Negative, In Vitro Seq Positive: Suggests site is protected by chromatin, lacks cellular repair machinery access, or cleavage is too inefficient in vivo. These are lower-risk off-targets.
- GUIDE-seq Positive, In Vitro Seq Negative: Rare; suggests possible cell-type specific activity or mechanism not recapitulated in vitro (e.g., replication-dependent breaks).
- GUIDE-seq & One In Vitro Method Positive, Others Negative: Highlights assay-specific technical sensitivities (e.g., oligo capture efficiency vs. biotin enrichment).
- All Methods Negative, Computational Prediction Positive: Likely a false positive prediction; underscores need for experimental validation.
Detailed Experimental Protocols
Protocol: Tiered Experimental Validation of Discrepant Sites
Purpose: To empirically validate and rank off-target sites identified by one method but not another. Reagents: See Scientist's Toolkit.
Procedure:
- Candidate Compilation: Compile list of off-target sites showing discrepant calls between GUIDE-seq and ≥1 other method (e.g., CIRCLE-seq).
- Primary Validation - T7 Endonuclease I (T7EI) Assay:
- Design PCR primers to amplify ~300-500bp region surrounding each candidate site from treated and control genomic DNA.
- Perform PCR, purify amplicons.
- Hybridize and re-anneal PCR products (95°C for 10 min, ramp down to 25°C at -0.1°C/sec).
- Digest with T7EI (NEB) for 1 hour at 37°C.
- Analyze fragments on agarose gel. Calculate indel percentage.
- Secondary Validation - Targeted Deep Sequencing:
- For sites positive in T7EI or of high concern, perform targeted NGS.
- Amplify locus with barcoded primers. Pool and purify amplicons.
- Sequence on Illumina MiSeq (2x300bp).
- Analyze reads using CRISPResso2 or similar to quantify precise indel frequencies.
- Tertiary Validation - Modified GUIDE-seq for Low-Efficiency Sites:
- For sites still negative but high-priority, repeat GUIDE-seq with increased oligoduplex concentration (e.g., 5µM instead of 0.5µM) and increased PCR cycles during library prep (monitor for over-amplification).
- Data Integration: Create a final reconciled off-target list, annotating sites with validated frequency and the methods by which they were originally detected.
Protocol:In VitrotoIn VivoCorrelation Assessment
Purpose: To systematically evaluate the predictive value of in vitro methods for cellular activity. Procedure:
- Perform parallel off-target profiling: Conduct GUIDE-seq (cellular) and one in vitro method (CIRCLE-seq or SITE-seq) using the same RNP complex.
- Data Processing: Analyze each dataset with its standard, recommended bioinformatics pipeline.
- Comparison Analysis:
- For each off-target site identified in vitro, check for GUIDE-seq reads.
- Calculate: Sensitivity (% of GUIDE-seq sites found in vitro) and False Discovery Rate (% of in vitro sites absent in GUIDE-seq).
- Plot indel frequency (from GUIDE-seq) against in vitro read count or cleavage score. Perform correlation analysis (Spearman's rank).
- Chromatin Analysis: Overlap in vitro-only sites with public chromatin data (e.g., ATAC-seq, DNase-seq) for the cell line used. Expect enrichment in closed chromatin regions.
Visualization of Concepts and Workflows
Decision Flow for Off-Target Discrepancies
Off-Target Method Relationships & Context
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Discrepancy Analysis
| Item | Function in Discrepancy Studies | Example Vendor/Product Notes |
|---|---|---|
| Alt-R S.p. HiFi Cas9 Nuclease V3 | High-fidelity variant reduces off-targets; baseline for comparing wild-type Cas9 data. | Integrated DNA Technologies (IDT). |
| GUIDE-seq Oligoduplex | Double-stranded, end-protected oligo for integration into DSBs. Critical for standard GUIDE-seq. | Synthesized chemically (e.g., IDT, Thermo Fisher). |
| T7 Endonuclease I | For rapid, cost-effective validation of nuclease-induced indels at discrepant loci. | New England Biolabs (NEB). |
| KAPA HiFi HotStart ReadyMix | High-fidelity PCR for amplification of target loci from genomic DNA for validation sequencing. | Roche. |
| Illumina DNA Prep Kit | Library preparation for targeted deep sequencing of candidate off-target sites. | Illumina. |
| NEBNext Ultra II FS DNA Module | Fragmentation and library prep for in vitro methods like SITE-seq. | NEB. |
| Cell Line-Specific ATAC-seq Kit | To assay chromatin accessibility and interpret in vitro vs. cellular discrepancies. | Active Motif, Coriell. |
| CRISPResso2 Software | Critical bioinformatics tool for precise quantification of indel frequencies from NGS data. | Open source. |
Conclusion
GUIDE-seq remains a cornerstone method for comprehensive, genome-wide off-target detection, providing an essential balance of sensitivity, specificity, and in-cell relevance. This guide has detailed its foundational importance, meticulous protocol, optimization levers, and position within the broader methodological landscape. For translational research, a strategy employing GUIDE-seq for primary screening followed by orthogonal validation (e.g., targeted deep sequencing) represents a robust industry standard. Future directions involve integrating GUIDE-seq with long-read sequencing for structural variant detection, adapting it for novel editing platforms like prime editing, and streamlining protocols for high-throughput screening. Mastery of GUIDE-seq is not merely a technical skill but a critical component in the responsible development of safe and effective genome editing therapeutics.