EMSA vs ITC: A Comprehensive Comparison for Measuring Protein-RNA Binding Affinity in 2024
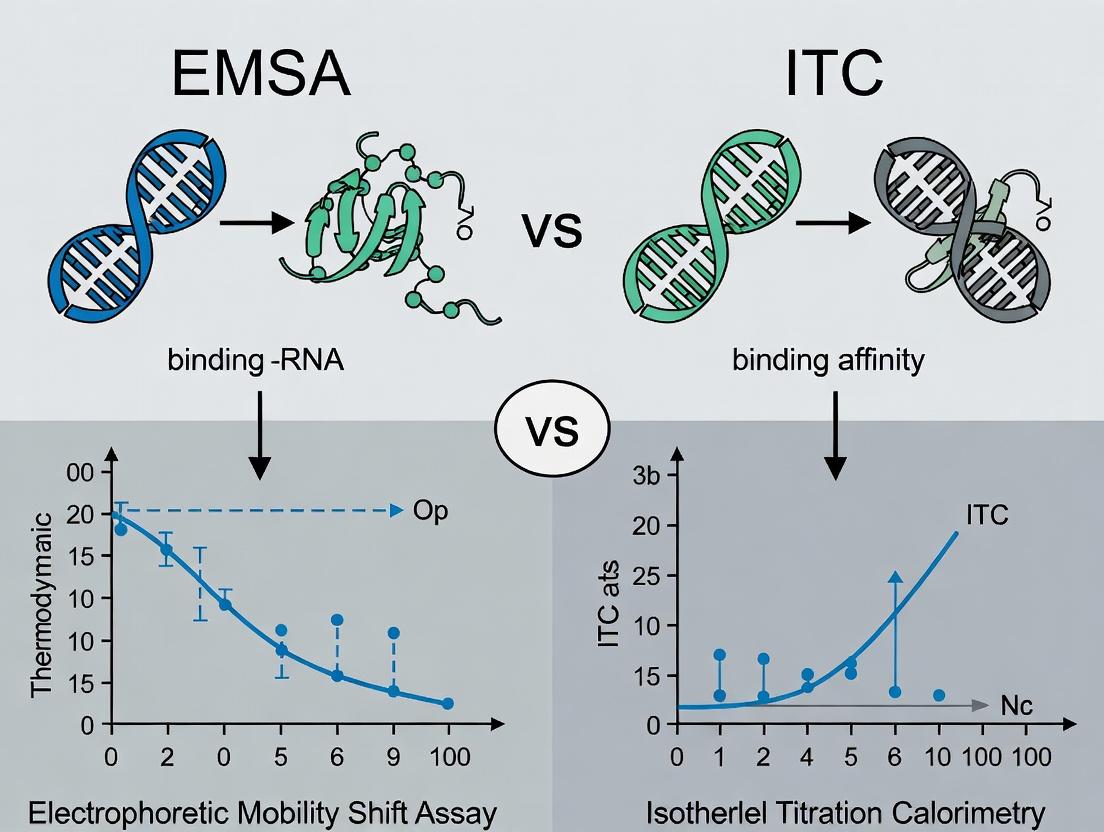
This article provides a detailed comparison of Electrophoretic Mobility Shift Assay (EMSA) and Isothermal Titration Calorimetry (ITC) for quantifying protein-RNA interactions.
EMSA vs ITC: A Comprehensive Comparison for Measuring Protein-RNA Binding Affinity in 2024
Abstract
This article provides a detailed comparison of Electrophoretic Mobility Shift Assay (EMSA) and Isothermal Titration Calorimetry (ITC) for quantifying protein-RNA interactions. Aimed at researchers and drug development professionals, we explore the foundational principles, practical methodologies, common troubleshooting steps, and critical validation strategies for each technique. We dissect their distinct advantages in measuring binding constants (Kd), kinetics, and thermodynamics, offering guidance on selecting the optimal method based on sample requirements, throughput needs, and the specific biological questions being addressed in contemporary RNA-targeted therapeutic discovery.
Protein-RNA Binding Fundamentals: Why Kd Matters and How We Measure It
The Critical Role of Protein-RNA Interactions in Gene Regulation and Disease
Publish Comparison Guide: EMSA vs. ITC for Protein-RNA Binding Affinity
Understanding the thermodynamics and kinetics of protein-RNA interactions is foundational for elucidating their role in gene regulation and dysregulation in disease. This guide compares two core techniques: Electrophoretic Mobility Shift Assay (EMSA) and Isothermal Titration Calorimetry (ITC).
Comparison Table: EMSA vs. ITC for Protein-RNA Binding
| Feature | Electrophoretic Mobility Shift Assay (EMSA) | Isothermal Titration Calorimetry (ITC) |
|---|---|---|
| Primary Measurement | Detection of complex formation via reduced electrophoretic mobility. | Direct measurement of heat change upon binding. |
| Key Parameters | Apparent dissociation constant (Kd), stoichiometry (qualitative). | Intrinsic Kd, stoichiometry (n), enthalpy (ΔH), entropy (ΔS), Gibbs free energy (ΔG). |
| Throughput | Medium-High. Multiple conditions can be run on one gel. | Low. One titration experiment typically takes 1-2 hours. |
| Sample Consumption | Low (pmol to fmol for detection). | High (nmol quantities for precise calorimetry). |
| Labeling Requirement | Typically requires labeled RNA (radioactive or fluorescent). | No labeling required; measures inherent heat signal. |
| Solution Condition | Non-native gel matrix environment. | True solution state in the cell. |
| Information Quality | Semi-quantitative; can be quantitative with careful controls. | Fully quantitative thermodynamic profile. |
| Main Advantage | Accessible, detects specific complexes in mixtures, can assess multiple complexes. | Provides a complete thermodynamic signature without labeling. |
| Main Disadvantage | Indirect measurement, prone to artifacts from gel electrophoresis. | High sample consumption, low throughput, sensitive to buffer mismatches. |
Supporting Experimental Data Summary
Table 1: Representative Binding Data for HuR Protein and ARE RNA Motif
| Method | Reported Kd (nM) | Stoichiometry (n) | ΔH (kcal/mol) | Reference Context |
|---|---|---|---|---|
| EMSA | 15 ± 5 | Not directly determined | Not determined | Agarose gel, 32P-labeled RNA |
| ITC | 22 ± 3 | 1.1 ± 0.1 | -12.5 ± 0.8 | PBS buffer, 25°C |
Detailed Experimental Protocols
Protocol 1: EMSA for Protein-RNA Complex
- Probe Preparation: Synthesize and purify RNA oligonucleotide. Label using [γ-32P] ATP and T4 Polynucleotide Kinase or use a fluorescent label.
- Binding Reaction: Combine labeled RNA (~10 fmol) with purified protein in binding buffer (e.g., 10 mM HEPES pH 7.3, 40 mM KCl, 1 mM MgCl2, 0.1 mM EDTA, 2 mM DTT, 5% glycerol, 0.1 μg/μL yeast tRNA). Incubate 20-30 minutes at room temperature.
- Electrophoresis: Load samples onto a pre-run non-denaturing polyacrylamide gel (typically 4-6%). Run in 0.5X TBE buffer at 4-10°C to minimize complex dissociation.
- Detection & Analysis: Expose gel to phosphorimager screen or use fluorescence scanner. Quantify band intensities to determine fraction bound vs. free RNA. Fit data to a binding isotherm model to derive apparent Kd.
Protocol 2: ITC for Protein-RNA Thermodynamics
- Sample Preparation: Exhaustively dialyze purified protein and RNA oligonucleotide into identical, degassed buffer. Accurate buffer matching is critical.
- Instrument Setup: Load the protein solution (~1.4 mL of 10-50 μM) into the sample cell. Fill the syringe with RNA solution at 10-15 times the protein concentration (e.g., 150-500 μM).
- Titration Experiment: Program the instrument to perform a series of injections (e.g., 19 injections of 2 μL each) of the RNA into the protein cell, with constant stirring. The instrument measures the heat required to maintain a zero-temperature difference between the sample and reference cells after each injection.
- Data Analysis: Integrate raw heat pulses. Fit the binding isotherm (heat vs. molar ratio) to a model (e.g., one-set-of-sites) using the instrument software to derive n, Kd, ΔH, and ΔS (calculated).
Visualizations
Diagram Title: EMSA Experimental Workflow
Diagram Title: ITC Experimental Workflow & Critical Buffer Matching
Diagram Title: EMSA vs ITC Selection Logic
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Protein-RNA Binding Studies
| Reagent/Material | Function in EMSA | Function in ITC |
|---|---|---|
| Purified Recombinant Protein | Binding partner. Must be >90% pure to avoid non-specific shifts. | Binding partner. High purity and solubility are critical for accurate heat measurement. |
| Synthetic RNA Oligonucleotides | Labeled binding target. Chemically synthesized for consistency. | Unlabeled binding target. Mass must be precisely known for concentration. |
| Isotopic (³²P) or Fluorescent Labels | Allows sensitive detection of RNA probe after gel electrophoresis. | Not required. |
| Non-Specific Competitor (tRNA/poly(I:C)) | Reduces non-specific protein-RNA binding in EMSA reactions. | Typically not added, as it contributes to heat signals. |
| High-Precision Dialysis Cassettes | Useful for buffer exchange of protein/RNA stocks. | ABSOLUTELY ESSENTIAL for matching the buffer of protein and RNA solutions to prevent heats of dilution. |
| Non-Denaturing Gel Matrix | Separates free RNA from protein-RNA complex based on size/charge. | Not applicable. |
| ITC-Compatible Buffer Systems | Standard buffers are acceptable. | Must have low heat of ionization (e.g., PBS, Tris may require careful matching). |
Understanding molecular binding interactions is fundamental to biochemistry, pharmacology, and drug discovery. Binding affinity, quantified most often by the dissociation constant (Kd), is not a monolithic value but a composite picture derived from kinetics (association and dissociation rates) and thermodynamics (free energy, enthalpy, entropy). This guide compares how two primary techniques—Electrophoretic Mobility Shift Assay (EMSA) and Isothermal Titration Calorimetry (ITC)—measure these parameters in the context of protein-RNA interactions.
Key Concepts: Kd, Kinetics, and Thermodynamics
- Kd (Equilibrium Dissociation Constant): The concentration of ligand at which half the binding sites on the protein are occupied at equilibrium. A lower Kd indicates higher affinity.
- Kinetics: Describes the rates of binding (kon, association rate constant) and dissociation (koff, dissociation rate constant). Kd = koff / kon.
- Thermodynamics: Describes the driving forces of the interaction: ΔG (free energy change, dictates spontaneity), ΔH (enthalpy change, bond formations/breakages), and ΔS (entropy change, disorder).
EMSA vs. ITC: A Direct Comparison for Protein-RNA Binding
The choice between EMSA and ITC depends on the specific binding parameters required and experimental constraints.
Table 1: EMSA vs. ITC Comparison
| Feature | Electrophoretic Mobility Shift Assay (EMSA) | Isothermal Titration Calorimetry (ITC) |
|---|---|---|
| Primary Measured Parameter | Apparent Kd at equilibrium (via concentration series). | Direct measurement of Kd, ΔH, and stoichiometry (N) in a single experiment. |
| Kinetics Access | Indirect, requires specialized variants like stopped-flow or kinetic EMSA. | No direct measurement of rate constants. |
| Thermodynamics Access | None. Provides only Kd (ΔG can be calculated). | Direct measurement of ΔH, ΔG, and TΔS (from ΔG = ΔH - TΔS). |
| Throughput | Moderate to High. Multiple samples can be run in parallel. | Low. One titration experiment typically takes 1-2 hours. |
| Sample Consumption | Low (fmol to pmol of protein/RNA). | High (nmol quantities of protein, often 10-100x more than EMSA). |
| Buffer Compatibility | High. Various buffers possible, but must maintain complex stability during electrophoresis. | Restrictive. Requires perfect buffer matching to avoid heat of dilution artifacts. |
| Key Advantage | Visual proof of complex formation; can resolve multiple complexes; low sample use. | Label-free, direct measurement in solution; provides full thermodynamic profile. |
| Key Limitation | Indirect measurement; assumes equilibrium is maintained during electrophoresis; no thermodynamic data. | High protein consumption; insensitive for very tight (pM) or weak (mM) Kd values. |
Experimental Protocols
Typical EMSA Protocol for Protein-RNA Kd Determination:
- Prepare Radiolabeled or Fluorescent RNA Probe: Synthesize target RNA, then 5'-end label with [γ-32P]ATP using T4 Polynucleotide Kinase or use a fluorophore-labeled oligonucleotide.
- Binding Reaction: Serially dilute the purified protein across a series of tubes. Add a constant, low concentration of the RNA probe to each in a suitable binding buffer (e.g., containing Tris, KCl, MgCl2, DTT, glycerol, carrier RNA/protein). Incubate to reach equilibrium (15-30 mins, room temperature).
- Non-Denaturing Gel Electrophoresis: Load reactions onto a pre-run polyacrylamide gel (typically 4-10%) in a low-ionic-strength buffer (e.g., 0.5x TBE). Run at constant voltage (e.g., 100V) at 4°C to maintain complex stability.
- Detection & Quantification: Expose gel to a phosphorimager screen (radioactive) or use a fluorescence scanner. Quantify the intensity of the shifted band (complex) and free RNA band for each lane.
- Data Analysis: Plot fraction bound vs. log[protein]. Fit data with a hyperbolic (one-site) binding model to determine the apparent Kd.
Typical ITC Protocol for Protein-RNA Binding:
- Extensive Dialysis: Both protein and RNA solutions must be dialyzed exhaustively against an identical, degassed buffer.
- Instrument Setup: Load the RNA solution (10-100 µM) into the syringe. Load the protein solution (typically 1/10th to 1/20th of RNA concentration) into the sample cell. Set reference cell with water or buffer.
- Titration Experiment: Program the instrument to perform a series of injections (e.g., 19 injections of 2 µL each) of the RNA into the protein cell, with constant stirring. The instrument measures the nanocalories of heat released or absorbed after each injection.
- Data Analysis: The integrated heat peaks per injection are plotted against the molar ratio. Nonlinear regression fitting of this isotherm directly yields the binding affinity (Kd = 1/Ka), enthalpy change (ΔH), and stoichiometry (N). ΔG and TΔS are calculated.
Visualizing the Experimental Workflows
Title: EMSA Workflow for Binding Affinity
Title: ITC Workflow for Binding Thermodynamics
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Protein-RNA Binding Studies |
|---|---|
| Recombinant Protein Purification Kits (His-tag, GST-tag) | Provides high-purity, active protein. Affinity tags simplify purification critical for both EMSA (specificity) and ITC (accurate concentration). |
| In Vitro Transcription Kits (e.g., T7 Polymerase) | For producing high-quality, homogenous RNA probes of defined sequence and length for binding assays. |
| Isotopic Labeling Reagents ([γ-32P]ATP) | Used with T4 PNK to radiolabel RNA for highly sensitive detection in traditional EMSA. |
| Fluorescent Dye-Labeled Nucleotides (Cy5, FAM) | Safer, stable alternative to radioactivity for labeling RNA for fluorescence-based EMSA detection. |
| Non-Specific Competitors (tRNA, BSA) | Added to EMSA binding buffers to reduce non-specific protein-RNA interactions and improve complex specificity. |
| Stable ITC Buffer Systems (e.g., Phosphate, Tris-HCl) | Buffers with low heat of ionization are preferred for ITC to minimize background heats from protonation/deprotonation events during binding. |
| Micro-Calorimetry Grade Dialysis Systems | Essential for perfect buffer matching between protein and RNA samples in ITC, eliminating background heats of dilution. |
| High-Purity DTT or TCEP | Reducing agents used to keep cysteine-containing proteins in a reduced, active state during purification and binding assays. |
| High-Sensitivity Gel Stains (SYBR Gold, Ethidium Bromide) | For visualizing unlabeled RNA in gels during EMSA optimization, though not typically for final Kd assays. |
| Commercial EMSA Kits | Provide optimized buffers, gels, and detection reagents for streamlined assay setup, often with fluorescence detection. |
Introduction Within the broader methodological comparison of EMSA versus Isothermal Titration Calorimetry (ITC) for quantifying protein-nucleic acid interactions, EMSA remains a cornerstone technique. Its core principle is the electrophoretic separation of free nucleic acid from protein-bound complexes, providing a direct, visual, and qualitative assessment of binding that can be rendered quantitative. This guide compares the performance of classic radioisotope-based EMSA with contemporary fluorescence-based EMSA alternatives, supported by experimental data.
Core Principle & Experimental Protocol The fundamental protocol involves incubating a purified protein with a target RNA/DNA probe. The mixture is then loaded onto a non-denaturing polyacrylamide gel. The protein-nucleic acid complex migrates more slowly than the free probe due to increased mass and potential conformational changes. Separation is followed by detection specific to the probe label.
Typical Binding Reaction Setup (20 µL):
- Buffer: 10 mM HEPES (pH 7.5), 50 mM KCl, 1 mM DTT, 0.5 mM EDTA, 5% Glycerol, 0.1 µg/µL BSA.
- Nucleic Acid Probe: 1-10 nM labeled RNA/DNA.
- Protein: Titrated across a range (e.g., 0-500 nM).
- Non-specific Competitor: 1 µg of poly(dI-dC) or yeast tRNA.
- Incubation: 20-30 minutes at room temperature.
- Gel Electrophoresis: Run on a pre-chilled 4-10% non-denaturing polyacrylamide gel in 0.5X TBE buffer at 100-150 V for 60-90 minutes.
Performance Comparison: Radioactive vs. Fluorescence EMSA
Table 1: Comparative Performance of EMSA Detection Methods
| Feature | ³²P/Radioisotope EMSA | Fluorescent Dye (e.g., Cy5) EMSA | SYBR Gold Post-Stain EMSA |
|---|---|---|---|
| Sensitivity | High (attomole range) | Moderate-High (femtomole range) | Low-Moderate |
| Quantitative Dynamic Range | ~3-4 orders of magnitude | ~2-3 orders of magnitude | ~1-2 orders of magnitude |
| Resolution (Complex vs. Free) | Excellent | Excellent | Good, can be impaired by stain background |
| Safety & Regulation | High hazard; strict licensing | Low hazard; minimal regulation | Low hazard; minimal regulation |
| Probe Stability | Short (isotope decay) | Long (months to years) | Requires intercalation post-run |
| Experiment Duration | Longer (gel drying, exposure) | Shorter (direct scanning) | Moderate (staining step required) |
| Cost (per experiment) | Lower reagent, high disposal | Higher dye cost, no disposal | Low stain cost, no disposal |
| Key Advantage | Gold standard sensitivity | Safety, speed, multiplex potential | Universal, no probe modification |
Supporting Experimental Data A 2022 study directly comparing methods for a specific RNA-binding protein (RBP) yielded the following quantitative binding data:
Table 2: Apparent Kd Measurement for RBP-X / RNA-Y Interaction
| Method | Reported Apparent Kd (nM) | CV (Inter-assay) | Minimum Probe Required | Assay Time (hands-on) |
|---|---|---|---|---|
| ³²P EMSA | 15.2 ± 2.1 | 8% | 0.1 fmol | 6-8 hours |
| Cy5 EMSA | 18.5 ± 3.4 | 12% | 1 fmol | 3 hours |
| SYBR Gold EMSA | 25.0 ± 5.1* | 18% | 10 fmol | 4 hours |
| ITC (Reference) | 12.8 ± 0.9 | 4% | 1000-fold more | 2 hours (setup + run) |
*Potential overestimation due to stain-induced complex destabilization.
The Scientist's Toolkit: Key Research Reagent Solutions
- Non-denaturing Polyacrylamide Gels: The matrix for size-based separation of complexes.
- ³²P-ATP/γ-ATP & T4 Polynucleotide Kinase: For radioactive 5'-end labeling of DNA/RNA probes.
- Fluorophore-labeled Nucleotides (e.g., Cy5-UTP): For in vitro transcription of fluorescent RNA probes.
- SYBR Gold Nucleic Acid Gel Stain: A universal, high-sensitivity dye for staining any nucleic acid post-electrophoresis.
- Poly(dI-dC): A common non-specific competitor DNA to reduce non-specific protein binding.
- HRP/AP-Conjugated Streptavidin & Chemiluminescent Substrates: For high-sensitivity detection of biotinylated probes.
- Phosphor Storage Screens & Scanner: For quantitation of radioactive or chemiluminescent signals.
- Laser Fluorescence Scanner (e.g., Typhoon): For imaging fluorescently labeled probes in gels.
Visualization of EMSA Workflow & Data Analysis
Title: EMSA Experimental and Analysis Workflow
Title: EMSA vs ITC in Binding Affinity Research Context
Within the broader debate on optimal methods for studying protein-RNA interactions—often framed as EMSA vs. ITC—Isothermal Titration Calorimetry (ITC) stands apart as the premier technique for obtaining a complete, label-free thermodynamic profile. This guide objectively compares ITC's performance with key alternatives, primarily Electrophoretic Mobility Shift Assay (EMSA) and Surface Plasmon Resonance (SPR), in the context of protein-RNA binding research.
Core Performance Comparison
Table 1: Direct Comparison of ITC, EMSA, and SPR for Protein-RNA Binding Analysis
| Feature/Parameter | Isothermal Titration Calorimetry (ITC) | Electrophoretic Mobility Shift Assay (EMSA) | Surface Plasmon Resonance (SPR) |
|---|---|---|---|
| Primary Measurement | Heat change (ΔH) per injection | Mobility shift of labeled RNA in gel | Change in refractive index (RU) at sensor surface |
| Binding Affinity (Kd) | Direct measurement. Typically range: 1 nM – 100 µM. | Indirect estimation. Requires band densitometry & model fitting. Prone to error from non-equilibrium conditions. | Direct measurement. Wide range (pM – mM) possible. |
| Stoichiometry (n) | Directly measured. From injection inflection point. | Not directly measured. Inferred. | Not directly measured. Requires careful surface chemistry control. |
| Enthalpy (ΔH) | Directly measured. Precision ± 1-5%. | Not measured. | Not directly measured. Can be derived from van't Hoff analysis (indirect). |
| Entropy (ΔS) | Calculated directly from ΔG (= -RT lnKa) and ΔH (ΔG = ΔH - TΔS). | Not measured. | Indirectly derived, requires temperature-dependent studies. |
| Label Requirement | None. Both components native. | Required. RNA (or protein) must be radio/fluor/chemically labeled. | Often required. One ligand must be immobilized. |
| Sample Consumption | Higher (typically 10-100 µg of protein). | Lower. | Very low for analyte; ligand immobilized. |
| Throughput | Low (1-2 hours per experiment). | Medium (can run multiple samples per gel). | High with automated systems. |
| Key Artifacts/Sources of Error | Heat of dilution must be controlled. | Gel running alters equilibrium; labeling can affect binding; non-specific competition. | Mass transport limitation; non-specific binding to chip; immobilization effects. |
| Information Depth | Complete thermodynamic profile (Kd, n, ΔH, ΔG, ΔS) in a single experiment. | Primarily qualitative/ semi-quantitative Kd; confirms binding. | Kinetics (ka, kd) and affinity (Kd); no direct thermodynamics. |
Experimental Protocols for Key Comparisons
Standard ITC Protocol for Protein-RNA Binding
- Instrument: MicroCal PEAQ-ITC or equivalent.
- Sample Prep: Both protein and RNA are dialyzed into identical buffer (e.g., 20 mM HEPES pH 7.5, 150 mM KCl, 1 mM MgCl₂) to minimize heats of dilution. RNA is often refolded by heat-cool cycle.
- Cell Contents: 200 µL of protein solution (typically 5-50 µM).
- Syringe Contents: RNA solution (typically 10-20 times more concentrated than protein).
- Experiment: Performed at constant temperature (e.g., 25°C). The RNA solution is injected in a series of 2 µL aliquots (first injection often 0.4 µL discarded) with 150-180 second intervals. Reference cell contains water.
- Data Analysis: Integrated heat peaks per injection are fit to a single-site binding model using instrument software (e.g., MicroCal PEAQ-ITC Analysis Software) to derive n (stoichiometry), Kd (binding constant = 1/Ka), and ΔH (enthalpy). ΔG and ΔS are calculated.
Competitive EMSA Protocol for Kd Estimation
- Sample Prep: Constant, trace amount of labeled RNA (e.g., 5'-32P or fluorophore) is incubated with increasing concentrations of unlabeled protein (0 to 2000 nM) in binding buffer for 30 min at room temperature.
- Non-Denaturing Gel: Pre-run 6-8% polyacrylamide gel (19:1 acrylamide:bis) in 0.5x TBE for 30-60 min at 4-10°C.
- Loading & Run: Samples mixed with non-interacting loading dye (e.g., 10% glycerol, xylene cyanol) are loaded and run at constant voltage (e.g., 100V) with buffer recirculation to maintain pH.
- Detection & Analysis: Gel is imaged (phosphorimager or fluorescence scanner). Fraction of bound RNA is quantified via band intensity. Data is fit (e.g., using Hill equation) to estimate an apparent Kd.
Visualizing the Methodological Landscape
Title: Decision Tree for Protein-RNA Binding Assay Selection
Title: ITC Experimental Workflow from Injection to Thermodynamic Data
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Protein-RNA ITC Experiments
| Item | Function & Critical Consideration |
|---|---|
| High-Precision ITC Instrument | (e.g., Malvern Panalytical PEAQ-ITC, TA Instruments Affinity ITC). Measures minute heat changes with high sensitivity and stability. |
| Dialysis System | (e.g., Slide-A-Lyzer cassettes, dialysis tubing). Critical for exact buffer matching of protein and RNA samples to eliminate background heats of dilution. |
| Nuclease-Free Water & Buffers | Essential for preparing RNA samples to prevent degradation. Common buffers: HEPES or Tris, with KCl/NaCl and MgCl₂. |
| RNA Oligonucleotide | Synthesized and HPLC-purified. Requires proper refolding via thermal annealing (heat to 95°C, slow cool) to ensure correct secondary structure. |
| Ultra-Pure Recombinant Protein | Purified via FPLC (e.g., Ni-NTA, size exclusion) to high homogeneity. Must be in the same exact buffer as the RNA post-dialysis. |
| Degassing Station | Removes dissolved gases from samples to prevent bubble formation in the ITC cell during the experiment, which causes noise. |
| Concentration Measurement Tools | (Nanodrop UV spectrophotometry). Accurate concentration determination of both protein (A280) and RNA (A260) is vital for correct Kd and n calculation. |
| Data Analysis Software | Vendor-specific (e.g., PEAQ-ITC Analysis) or general (e.g., NITPIC, SEDPHAT) for integrating peaks, subtracting controls, and fitting binding models. |
In conclusion, while EMSA provides accessible qualitative data and SPR offers superior kinetic profiling, ITC remains unmatched for delivering a full, model-free thermodynamic characterization of protein-RNA interactions in a single, label-free experiment. Its direct measurement of enthalpy, stoichiometry, and affinity establishes it as the gold standard for foundational mechanistic studies, informing downstream drug discovery and engineering efforts.
The choice between Electrophoretic Mobility Shift Assays (EMSA) and Isothermal Titration Calorimetry (ITC) for quantifying protein-RNA binding affinity is profoundly influenced by the initial condition of the biomolecular samples. Purity, complexity, and native state integrity are not mere preparatory details but are deterministic factors for method success and data reliability.
Impact of Sample Properties on EMSA and ITC Performance
Sample Purity: Contaminants like nucleases or proteases degrade samples during lengthy experiments. Free nucleotides or salts interfere with detection and heat measurements. Sample Complexity: Heterogeneous samples (e.g., cell lysates) can lead to non-specific shifts (EMSA) or obscure binding heats (ITC). Native vs. Denatured State: Proper folding is critical. Misfolded RNA or protein yields inaccurate Kd values, reporting on non-physiological interactions.
Comparative Experimental Data: The Influence of RNA Purity
The following data summarizes a model experiment comparing EMSA and ITC performance using a well-folded, pure let-7 miRNA stem-loop and a known binder, human Lin28, against samples with common impurities.
Table 1: Binding Affinity (Kd) Measurements Under Different Sample Conditions
| Method | Sample Condition (RNA) | Reported Kd (nM) | Data Quality Notes |
|---|---|---|---|
| EMSA | HPLC-purified, refolded | 15 ± 3 | Clear shifted band, clean well. |
| EMSA | Crude synthesis, unpurified | 120 ± 45 | Smear, multiple bands, high background. |
| ITC | HPLC-purified, refolded | 18 ± 2 | Clean sigmoidal curve, reliable ΔH, ΔS. |
| ITC | Dialyzed into mismatched buffer | Not Determined | Heats diluted by buffer mismatch, no fit. |
Detailed Experimental Protocols
Protocol 1: EMSA for Lin28/let-7 RNA Binding (Native Condition)
- RNA Preparation: Synthesize let-7 RNA, purify via denaturing PAGE or HPLC. Elute and refold by heating to 95°C for 2 min in folding buffer (20 mM HEPES, 100 mM KCl, 1 mM MgCl₂), then slow-cool to 25°C.
- Protein Purification: Express recombinant Lin28 with a His-tag. Purify via Ni-NTA chromatography, followed by size-exclusion chromatography in EMSA buffer (20 mM HEPES pH 7.5, 100 mM KCl, 1 mM MgCl₂, 0.1 mg/mL BSA, 0.01% NP-40, 5% glycerol).
- Binding Reaction: Mix 5 nM folded RNA with Lin28 (0-500 nM) in 20 μL total volume. Incubate 30 min at 25°C.
- Electrophoresis: Load samples onto a pre-run 6% native polyacrylamide gel (0.5x TBE, 4°C). Run at 100V for 60-70 min.
- Detection: Stain gel with SYBR Gold, image, and quantify bound/unbound RNA to calculate Kd.
Protocol 2: ITC for Lin28/let-7 RNA Binding
- Sample Dialysis: Co-dialyze purified Lin28 protein and refolded let-7 RNA into identical ITC buffer (20 mM HEPES pH 7.5, 100 mM KCl, 1 mM MgCl₂) overnight at 4°C.
- Degassing: Degas both samples under vacuum for 10 min to prevent bubbles.
- Instrument Setup: Load 300 μL of 50 μM Lin28 into the sample cell. Load the 500 μM let-7 RNA solution into the syringe.
- Titration: Perform 19 injections (2 μL initial, 15 x 2.5 μL) at 25°C with 180 sec spacing. Stir at 750 rpm.
- Data Analysis: Integrate raw heat peaks, subtract control titrations (RNA into buffer), and fit the binding isotherm to a one-site model to derive Kd, ΔH, and ΔS.
Visualizing Method Sensitivity to Sample State
Title: Sample State Directly Determines EMSA and ITC Data Quality
Title: Divergent Sample Preparation Workflows for EMSA and ITC
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for High-Quality Protein-RNA Binding Studies
| Item | Function | Critical for EMSA/ITC |
|---|---|---|
| RNase Inhibitors | Prevents degradation of RNA during incubation and handling. | Both (especially ITC long runs). |
| Protease Inhibitor Cocktails | Maintains protein integrity during purification and storage. | Both. |
| HPLC-grade Nucleotides | Ensures maximum RNA synthesis yield and purity. | ITC (primary), EMSA. |
| Size-Exclusion Columns | Removes aggregates and ensures monodisperse protein sample. | ITC (critical), EMSA. |
| SYBR Gold Nucleic Acid Gel Stain | Highly sensitive, low-background detection of free/complexed RNA in gels. | EMSA. |
| Dialysis Cassettes (3.5kDa MWCO) | Permits rigorous buffer matching for protein and RNA stocks. | ITC (mandatory). |
| Non-specific Competitors (tRNA, BSA) | Reduces non-specific binding in gel shift assays. | EMSA. |
| Ultra-Pure Buffers & Salts | Minimizes background signal from contaminants in calorimetry. | ITC. |
Step-by-Step Protocols: From EMSA Gel Setup to ITC Titration Design
Within the broader framework of comparing methodologies for quantifying protein-nucleic acid interactions, the Electrophoretic Mobility Shift Assay (EMSA) remains a cornerstone technique for detecting binding events. This guide provides a comparative analysis of core EMSA protocol components, situating it against Isothermal Titration Calorimetry (ITC) as part of a thesis evaluating speed, sensitivity, and quantitative rigor in protein-RNA binding affinity research. While ITC provides direct thermodynamic measurements in solution, EMSA offers visual confirmation of complex formation under native conditions and is widely accessible.
Core Component 1: Probe Labeling Strategies
The choice of labeling method directly impacts sensitivity, signal stability, and experimental flexibility.
Comparison of Common Labeling Methods
Table 1: Comparison of Probe Labeling Methods for EMSA
| Method | Typical Efficiency | Signal Stability | Required Equipment | Best For |
|---|---|---|---|---|
| Traditional End-Labeling (T4 PNK, [γ-³²P]ATP) | >95% | High (radioactive decay) | Geiger counter, phosphorimager | Max sensitivity, competition EMSA |
| Biotin 3'-End Labeling (Terminal Transferase) | 70-90% | Very High (months) | Standard lab equipment, chemiluminescence detector | Safe, long-term storage, high-resolution shifts |
| Fluorescent Dye Labeling (Cy3/Cy5) | 1 dye per oligonucleotide | Moderate (photobleaching) | Fluorescence scanner/imager | Multiplexing, real-time kinetics (in-gel) |
| Digoxigenin (DIG) Labeling | Varies by protocol | Very High | Chemiluminescence/colorimetric detector | Non-radioactive, high sensitivity alternative |
Detailed Protocol: T4 Polynucleotide Kinase (PNK) End-Labeling
- Reaction Setup: In a 1.5 mL microcentrifuge tube, combine:
- 100 ng of purified DNA or RNA oligonucleotide probe.
- 2 µL of 10X T4 PNK Buffer.
- 10 µCi of [γ-³²P]ATP (6000 Ci/mmol).
- 10 units of T4 Polynucleotide Kinase.
- Nuclease-free water to a final volume of 20 µL.
- Incubation: Incubate at 37°C for 30 minutes.
- Termination: Add 2 µL of 0.5 M EDTA to stop the reaction.
- Purification: Purify the labeled probe using a micro Bio-Spin P-30 column or repeated ethanol precipitation to remove unincorporated nucleotides.
- Quantification: Measure radioactivity by scintillation counting. A specific activity of >10⁵ cpm/ng is typically suitable for EMSA.
Core Component 2: Binding Reaction Optimization
The composition of the binding reaction is critical for specific complex formation.
Comparison of Binding Buffer Additives
Table 2: Effect of Binding Reaction Components on Complex Formation
| Component | Typical Concentration | Purpose | Impact on EMSA vs. ITC Context |
|---|---|---|---|
| Non-specific Competitor (poly(dI-dC)) | 0.05-0.1 mg/mL | Reduce non-specific protein-probe binding | Crucial for EMSA specificity; not used in ITC. |
| Carrier Protein (BSA) | 0.01-0.1 mg/mL | Stabilize protein, prevent surface adhesion | Often used in EMSA; can interfere with ITC heat measurement. |
| Divalent Cations (Mg²⁺) | 1-10 mM | Often required for RNA-protein folding/binding | Concentration must be consistent; ITC can directly measure Mg²⁺-coupled binding. |
| Non-ionic Detergent (NP-40) | 0.01-0.1% | Reduce non-specific interactions | Common in EMSA; incompatible with ITC due to bubble formation in cell. |
| RNase Inhibitors | 0.5-1 U/µL | Protect RNA probes from degradation | Essential for RNA EMSA; not required for ITC with DNA. |
Detailed Protocol: Standard Binding Reaction
- Master Mix: Prepare a nuclease-free mix for n+1 reactions containing:
- 2 µL of 10X Binding Buffer (100 mM HEPES, pH 7.9, 400 mM KCl, 10 mM MgCl₂, 50% glycerol).
- 1 µL of 1 mg/mL poly(dI-dC).
- 0.5 µL of 10 mg/mL BSA.
- 0.5 µL of RNase Inhibitor (for RNA probes).
- Nuclease-free water to bring the master mix volume to 10 µL per reaction after adding protein and probe.
- Assembly: Aliquot the master mix. First add the purified protein (varying amount for titration), then the labeled probe (10,000-20,000 cpm). Gently mix by pipetting.
- Incubation: Incubate at room temperature or 30°C for 20-30 minutes. Avoid extended incubation times to prevent probe degradation or complex dissociation.
Core Component 3: Electrophoresis Conditions
Native gel electrophoresis separates bound from free probe.
Comparison of Gel and Running Buffer Systems
Table 3: Electrophoresis Condition Optimization
| Parameter | Common Condition 1 | Common Condition 2 | Advantage |
|---|---|---|---|
| Gel Percentage | 6% Polyacrylamide | 8% Polyacrylamide | Better for large complexes; better for small complexes. |
| Crosslinker Ratio (Acrylamide:Bis) | 29:1 | 37.5:1 | Larger pore size; sharper bands. |
| Running Buffer | 0.5X TBE | 0.25X TBE or TGE | Higher buffering capacity; lower ionic strength, less heat. |
| Temperature | 4°C (Cold Room) | Room Temperature | Stabilizes labile complexes; more convenient. |
| Voltage/Time | 100 V, 90 min | 150 V, 60 min | Better resolution; faster run time. |
Detailed Protocol: Native Gel Electrophoresis
- Gel Casting: Prepare a 6-8% non-denaturing polyacrylamide gel (e.g., 6 mL of 30% acrylamide/bis 29:1, 3 mL of 10X TBE, 21 mL water, 300 µL 10% APS, 30 µL TEMED). Pour between clean glass plates and insert a well comb.
- Pre-run: Assemble the gel apparatus with the chosen running buffer (e.g., 0.5X TBE). Pre-run the gel at 100 V for 60 minutes at 4°C to equilibrate temperature and remove persulfate.
- Loading: After binding incubation, add 2 µL of 10X non-denaturing loading dye (50% glycerol, 0.1% bromophenol blue/xylene cyanol) to each reaction. Load samples directly without heating.
- Run: Run the gel at constant voltage (e.g., 100 V) until the dye front migrates 2/3 to 3/4 of the way down the gel.
- Detection: For radioactive probes, transfer gel to filter paper, dry, and expose to a phosphor screen. For non-radioactive probes, follow specific transfer and detection protocols (e.g., chemiluminescence).
EMSA Workflow Diagram
Title: EMSA Experimental Workflow from Probe to Detection.
The Scientist's Toolkit: EMSA Research Reagent Solutions
| Item | Function in EMSA |
|---|---|
| T4 Polynucleotide Kinase | Catalyzes the transfer of the γ-phosphate of ATP to the 5'-OH terminus of DNA/RNA for radioactive labeling. |
| [γ-³²P]ATP or Biotin-11-UTP | Source of radioactive or non-radioactive label for probe detection. |
| Poly(dI-dC) | Synthetic nucleotide polymer used as a non-specific competitor to minimize protein binding to non-target sequences. |
| RNase Inhibitor (e.g., RNasin) | Essential for protecting labile RNA probes from degradation by ribonucleases during binding reactions. |
| High-Purity Acrylamide/Bis Solution | For casting reproducible native polyacrylamide gels with consistent pore size. |
| Non-denaturing Loading Dye | Glycerol-based dye to increase sample density for gel loading without disrupting non-covalent complexes. |
| Phosphor Storage Screen & Imager | For high-sensitivity, quantitative detection of radioactively labeled probes post-electrophoresis. |
| Chemiluminescent Nucleic Acid Detection Module | For non-radioactive detection of biotin- or DIG-labeled probes via streptavidin-HRP conjugate. |
This guide provides a performance comparison of densitometry methods for quantifying dissociation constants (Kd) from Electrophoretic Mobility Shift Assays (EMSA), framed within the broader methodological debate of EMSA versus Isothermal Titration Calorimetry (ITC) for determining protein-nucleic acid binding affinities. While ITC provides direct thermodynamic measurements in solution, EMSA remains a widely accessible, sensitive technique for detecting specific binding events, especially for low-affinity interactions. Accurate Kd determination from EMSA hinges on robust densitometry and curve-fitting protocols.
Densitometry Analysis Platforms: Performance Comparison
The accuracy of Kd quantification is heavily dependent on the image analysis software used for densitometry. The table below compares the performance of three common platforms based on key metrics relevant to EMSA analysis.
Table 1: Comparison of Densitometry Software for EMSA Analysis
| Feature / Metric | ImageJ/Fiji (Freeware) | Image Lab (Bio-Rad) | TotalLab TL120 (Nonlinear Dynamics) |
|---|---|---|---|
| Automated Band Detection | Manual or semi-automated via plugins; requires user validation. | Highly automated, optimized for ChemiDoc systems. | Advanced automated detection with minimal user input. |
| Background Subtraction | Multiple methods (rolling ball, paraboloid) available; user must select. | Proprietary, one-click correction optimized for their imagers. | Sophisticated local background correction algorithms. |
| Signal Linearity | Excellent, but dependent on original image bit depth and absence of saturation. | Excellent with calibrated imagers; automatic saturation warnings. | Excellent, includes tools to check and correct for non-linearity. |
| Bound/Free Quantification | Manual selection of lanes and bands; prone to user variability. | Integrated lane/band tools; streamlined workflow. | Fully automated separation and quantification of bound/free species. |
| Curve Fitting for Kd | Requires export to external software (e.g., GraphPad Prism, SigmaPlot). | Basic non-linear regression module included. | Integrated, robust curve fitting (Hill, quadratic) specifically for binding data. |
| Data Reproducibility | High if protocol is strictly documented; subject to user bias. | High due to standardized, automated protocols on same hardware. | Very high, with automated analysis protocols minimizing inter-user variation. |
| Typical Cost | Free, open-source. | Bundled with imager purchase; standalone license ~$1000. | High, ~$3000+ for a full license. |
Experimental Protocol: Standardized EMSA Densitometry Workflow
The following protocol is essential for generating reproducible data suitable for Kd calculation, regardless of the software chosen.
1. EMSA Gel Electrophoresis & Imaging:
- Binding Reactions: Incubate a constant, trace amount of labeled RNA (e.g., 1-10 nM) with a series of protein concentrations (e.g., 0.1 nM to 1 µM) in suitable binding buffer (e.g., 10 mM HEPES, 50 mM KCl, 1 mM DTT, 0.1 mg/mL BSA, 0.01% NP-40, 10% glycerol). Include a zero-protein control.
- Non-Denaturing Gel: Pre-run a 4-10% polyacrylamide (19:1 acrylamide:bis) gel in 0.5X TBE at 4°C for 30-60 min. Load reactions and run at 4°C (80-120 V) until free RNA migrates 2/3 down the gel.
- Imaging: Use a CCD-based imager (e.g., ChemiDoc) for SYBR Green or radiometric detection. Acquire image in 16-bit TIFF format. CRITICAL: Ensure no pixel saturation. Adjust exposure time so the highest intensity band is below the maximum pixel value.
2. Densitometry & Data Processing:
- Define Regions of Interest (ROIs): For each lane, define ROIs for the bound complex and the free RNA. Apply identical ROIs across all lanes.
- Measure Intensity: Record the integrated intensity (volume) for each ROI.
- Background Correction: Subtract the intensity of a background ROI of equal size from an adjacent area of the gel with no bands.
- Calculate Fraction Bound: For each lane, calculate Fraction Bound (θ) = (Intensitybound) / (Intensitybound + Intensity_free).
3. Binding Curve Fitting to Determine Kd:
- Prepare Data: Plot Fraction Bound (y-axis) versus total protein concentration (x-axis). The total RNA concentration ([R]_total) must be known.
- Non-Linear Regression: Fit the data to a one-site specific binding model (quadratic equation) that accounts for the depletion of free ligand at high binding: θ = (([R]+[P]+Kd) - sqrt(([R]+[P]+Kd)^2 - 4[R][P])) / (2*[R]) where [R] = total RNA concentration, [P] = total protein concentration.
- Fitting Constraints: Fix [R] to the known constant value. Allow the fit to determine Kd and sometimes a non-specific binding parameter.
Visualizing the EMSA Kd Quantification Workflow
Diagram 1: EMSA Kd Quantification Protocol
The Scientist's Toolkit: Key Reagent Solutions for EMSA
Table 2: Essential Research Reagents for EMSA
| Item | Function in EMSA |
|---|---|
| Chemically Synthesized RNA Oligo | The binding target; typically 20-40 nt, often with a 5' or 3' fluorescent (e.g., Cy5) or radioisotope (³²P) label for detection. |
| Recombinant Purified Protein | The binding partner; must be highly pure, active, and in a buffer compatible with RNA structure and binding. |
| Non-Specific Competitor DNA/RNA | (e.g., Poly dI:dC, tRNA) Added to binding reactions to sequester proteins that bind nucleic acids non-specifically. |
| Native Gel Buffer (e.g., 0.5X TBE) | Provides ionic strength and pH for electrophoresis without denaturing the protein-RNA complex. Often run at 4°C. |
| High-Sensitivity Stain (e.g., SYBR Gold) | For non-radioactive detection of unlabeled RNA; less quantitative than direct labeling but convenient. |
| Chemiluminescent Nucleic Acid Detection Kit | For high-sensitivity, non-radioactive detection of biotin-labeled RNA probes. |
| Non-Linear Curve Fitting Software | (e.g., GraphPad Prism, SigmaPlot) Essential for transforming fraction bound data into a reliable Kd value using the appropriate binding model. |
Comparative Data: EMSA vs. ITC for Kd Determination
While this guide focuses on EMSA, its role is best understood in contrast to ITC, the gold standard for solution-phase affinity measurement.
Table 3: Methodological Comparison: EMSA vs. ITC for Protein-RNA Kd
| Parameter | EMSA with Densitometry | Isothermal Titration Calorimetry (ITC) |
|---|---|---|
| Measured Parameter | Fraction of RNA bound at equilibrium (gel-based separation). | Direct heat change upon binding in solution. |
| Kd Range | Broad, best for ~1 nM - 1 µM. | Optimal for 10 nM - 10 µM (tight binding requires tricks). |
| Sample Consumption | Low (pmol of protein/RNA). | High (nmol to µmol of protein). |
| Throughput | Medium (multiple conditions per gel). | Low (one titration per cell, ~1-2 hrs). |
| Additional Data | Can resolve multiple complexes; indicates stoichiometry. | Direct measurement of ΔH, ΔS, ΔG, and stoichiometry (n). |
| Key Assumption/Error Source | Gel must not perturb equilibrium; accurate quantitation of band intensity is critical. | All heat is from specific binding; correct buffer matching is vital. |
| Typical Reported Kd for a Model Interaction (e.g., MS2 Coat Protein/RNA) | 5 - 15 nM (can vary with labeling and gel conditions). | 8 - 12 nM (direct measurement in solution). |
Quantifying Kd from EMSA via densitometry is a powerful, accessible technique but requires meticulous attention to gel imaging, band quantification, and curve fitting to yield reliable data. While integrated commercial software like Image Lab and TotalLab TL120 offer streamlined, reproducible analysis pipelines, open-source solutions like ImageJ/Fiji remain viable with rigorous standardization. In the context of the broader EMSA vs. ITC debate, EMSA-based Kd determination excels in sensitivity for weak interactions and the ability to resolve complex mixtures, but it is an indirect measurement subject to more potential artifacts than the direct, thermodynamic data provided by ITC. The choice of method ultimately depends on the scientific question, sample availability, and required precision.
Within the broader thesis comparing Electrophoretic Mobility Shift Assay (EMSA) and Isothermal Titration Calorimetry (ITC) for protein-RNA binding affinity research, this guide focuses on the critical experimental parameters of ITC. ITC provides a label-free, in-solution measurement of binding affinity (Kd), stoichiometry (n), enthalpy (ΔH), and entropy (ΔS) in a single experiment. This objective comparison details best practices and alternatives for sample preparation, instrument configuration, and titration to optimize data quality.
Comparative Analysis: Sample Preparation Protocols
Table 1: Comparison of Sample Preparation and Buffer Strategies
| Parameter | Recommended Best Practice | Common Alternative | Impact on Data Quality |
|---|---|---|---|
| Buffer Matching | Extensive dialysis of both protein and RNA in identical buffer, followed by degassing. | Mere buffer exchange using desalting columns. | Critical. Minimizes heat of dilution and mixing artifacts. Mismatched buffers cause large injection peaks that obscure binding isotherms. |
| Sample Purity | >95% homogeneity (SEC-MALS or HPLC analysis). | >80% purity (SDS-PAGE analysis). | High purity ensures accurate stoichiometry (n) and avoids heterogeneous binding isotherms. |
| RNA Handling | Chemical synthesis followed by HPLC purification; annealing for structured RNAs; use of RNase inhibitors. | In vitro transcription with less stringent purification. | Synthetic RNA ensures sequence fidelity and homogeneity. Impurities lead to inaccurate Kd and ΔH. |
| Concentration Determination | A280 (protein) and A260 (RNA) using calculated extinction coefficients; verified by colorimetric assay. | A280/A260 alone without coefficient verification. | Accurate concentration is essential for precise Kd and n. Overestimation skews n to <1. |
| Dye/Buffer Components | Avoidance of colored agents, strong reducing agents (DTT > 1 mM), or detergents. | Use of β-mercaptoethanol, high [DTT], or visible dyes. | Can cause high background noise, signal drift, or damage to the cell. DTT oxidation is exothermic. |
Comparative Analysis: Cell and Syringe Configuration
Table 2: ITC Instrument Configuration and Sample Loading
| Configuration | Optimal Setup for Protein-RNA | Typical Alternative Setup | Rationale and Evidence |
|---|---|---|---|
| Cell Content | Protein (or RNA) solution in cell. | RNA (or protein) solution in cell. | Placing the component with lower solubility or higher stability in the cell is preferable. For protein-RNA, protein is often in the cell to minimize RNA handling. |
| Syringe Content | Titrant (RNA or Protein). | Titrant (Protein or RNA). | Titrant should be at higher concentration (typically 10-20x Kd). Must be in identical buffer. |
| Sample Concentration | Cell: [M] = 10-50 * Kd. Syringe: [M] = 200-500 * Kd. | Using estimated or literature Kd. | Data Supported: A 'c-value' ([Cell]*Kd) of 10-100 is optimal. c < 1 yields a shallow curve; c > 500 yields a step isotherm, both reducing fitting accuracy. |
| Cell Volume (Standard) | 200 µL for most microcalorimeters. | 1.4 mL for older macro-calorimeters. | Smaller volumes require less material. Modern instruments have high sensitivity for 200 µL cells. |
| Reference Cell | Filled with degassed, ultrapure water. | Filled with dialysis buffer. | Water provides a stable thermal reference. Buffer can introduce noise if prone to evaporation/condensation. |
Comparative Analysis: Titration Parameters
Table 3: Titration Parameter Optimization
| Parameter | Recommended Setting for Protein-RNA | Common Suboptimal Setting | Experimental Impact |
|---|---|---|---|
| Temperature | 25°C or 30°C (mimics physiological). | 37°C or 20°C. | Affects binding thermodynamics (ΔH, ΔS). 37°C may increase RNA degradation. Temperature must be stable ±0.02°C. |
| Number of Injections | 19-25 injections. | 10-15 injections. | More data points across the binding curve improve nonlinear regression fitting for Kd and n. |
| Injection Volume | First injection: 0.5 µL (discarded). Subsequent: 2-10 µL. | Fixed volume (e.g., 10 µL) for all. | Small initial injection minimizes artifact. Variable or optimized volumes can better define the transition region. |
| Spacing Between Injections | 150-180 seconds. | 120 seconds. | Sufficient time is required for the signal to return to baseline. Inadequate spacing distorts integrated heat. |
| Stirring Speed | 750-1000 rpm. | 500 rpm or lower. | Ensures rapid mixing without causing mechanical denaturation or foaming. Higher speeds improve mixing efficiency. |
| Feedback Mode/Gain | "High Feedback" mode for fast kinetics. | Standard mode. | Adjusts the instrument's response speed to match the heat flow rate of the reaction. |
Detailed Experimental Protocol: A Representative ITC Experiment
Protocol: Measuring the Binding Affinity of RBFOX Protein to its RNA Consensus Sequence
Sample Preparation:
- Protein: Dialyze purified RBFOX RRM domain overnight at 4°C against ITC buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM MgCl2, 0.5 mM TCEP). Centrifuge at 15,000 x g for 10 min post-dialysis. Determine concentration by A280 using calculated ε.
- RNA: Resuspend synthetic GCAUGU RNA in dialysis buffer. Heat to 95°C for 2 min and slow-cool to anneal. Pass through a desalting column equilibrated with dialysis buffer.
- Degas: Degas both protein (cell sample) and RNA (syringe sample) under vacuum for 10 min prior to loading.
Instrument Configuration:
- Cell: Load with 200 µL of 50 µM RBFOX protein solution.
- Syringe: Load with 40 µL of 500 µM RNA solution (10x cell concentration).
- Parameters: Set temperature to 25°C, stirring speed to 1000 rpm. Initial delay of 60 sec. Design titration: 1 x 0.5 µL injection (discarded), followed by 19 x 2 µL injections with 180 sec spacing.
Data Acquisition & Analysis:
- Run the experiment. The raw data will show a series of heat pulses.
- Integrate heat pulses to obtain a plot of kcal mol⁻¹ of injectant vs. molar ratio.
- Fit the binding isotherm using a "One Set of Sites" model in the instrument software to derive n, Kd (and thus ΔG), and ΔH. Calculate ΔS using ΔG = ΔH - TΔS.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for Protein-RNA ITC
| Item | Function in ITC Experiment |
|---|---|
| High-Purity Dialysis Tubing (MWCO appropriate) | Ensures precise buffer matching for both macromolecules, the single most critical step. |
| RNase Inhibitors (e.g., SUPERase•In) | Protects RNA integrity during sample handling and long experimental runs. |
| Tris(2-carboxyethyl)phosphine (TCEP) | A non-thiol, odorless reducing agent stable across pH ranges, preferable to DTT for ITC. |
| Ultrafiltration Concentrators (e.g., Amicon) | For gentle concentration and buffer exchange of protein and RNA samples. |
| Degassing Station or Syringe | Removes dissolved gases that can form bubbles in the ITC cell, causing noise and drift. |
| Software: NITPIC, SEDPHAT, or AFFINImeter | Advanced tools for data processing, multi-model fitting, and handling complex binding schemes. |
Visualizing the ITC Workflow in Protein-RNA Research
Title: ITC Experimental Workflow for Protein-RNA Binding
Visualizing ITC's Role in the Broader EMSA vs. ITC Thesis
Title: EMSA vs. ITC Method Comparison for Protein-RNA Binding
Within the broader thesis comparing EMSA and ITC for protein-RNA binding affinity research, isothermal titration calorimetry (ITC) stands out as a gold-standard, solution-phase technique. It uniquely provides a complete thermodynamic profile—including enthalpy change (ΔH), entropy change (ΔS), dissociation constant (Kd), and binding stoichiometry (n)—from a single experiment. This guide compares the performance of modern ITC instrumentation in extracting these parameters, supported by recent experimental data.
Comparison of Modern ITC Instrument Performance
The following table summarizes key performance metrics for current market-leading ITC systems, based on published specifications and user-reported data for protein-RNA binding studies.
Table 1: Performance Comparison of Modern ITC Instruments
| Instrument Model | Cell Volume (µL) | Typical Kd Range (M) | Data Point Density (per injection) | Baseline Stability (µcal/sec) | Recommended Sample Consumption (for n determination) |
|---|---|---|---|---|---|
| Malvern PEAQ-ITC | 200 | 10⁻² - 10⁻¹² | 10 (High-Res Mode) | < 0.002 | 50-200 µg protein |
| TA Instruments Nano ITC | 170 | 10⁻³ - 10⁻¹⁰ | 5 | < 0.005 | 40-150 µg protein |
| MicroCal Auto-iTC 200 | 200 | 10⁻³ - 10⁻¹² | 2-10 (user-selectable) | < 0.001 | 50-200 µg protein |
Experimental Protocol for Protein-RNA ITC
A standardized protocol for determining binding parameters of a protein-RNA complex is detailed below.
Sample Preparation:
- Protein: Dialyze the purified protein into a degassed buffer (e.g., 20 mM HEPES, pH 7.5, 150 mM KCl, 1 mM MgCl₂). Centrifuge at 15,000 x g for 10 minutes to remove aggregates.
- RNA: Synthesize and HPLC-purify the RNA ligand. Resuspend in the identical dialysis buffer used for the protein. Anneal if necessary by heating to 95°C for 2 minutes and slowly cooling.
- Concentration: Precisely determine concentrations via UV absorbance (Protein: A280; RNA: A260). The syringe typically contains the ligand (RNA) at 10-20 times the concentration of the protein in the cell.
ITC Experiment Setup:
- Load the protein solution into the sample cell. Load the RNA solution into the titration syringe.
- Set experimental parameters: Temperature (25°C or 37°C), reference power (5-10 µcal/sec), stirring speed (750 rpm), initial delay (60 sec).
- Titration schedule: 19 injections of 2 µL each, with 150-second spacing between injections.
Data Analysis Workflow:
- Integrate raw thermogram peaks to obtain the plot of heat per injection (kcal/mol) vs. molar ratio.
- Fit the binding isotherm using a nonlinear least-squares algorithm to a model (e.g., "One Set of Sites").
- The fitting software directly outputs n (stoichiometry), Kd (from which ΔG is derived: ΔG = RTlnKd), and ΔH (enthalpy change).
- Calculate ΔS (entropy change) using the relationship: ΔG = ΔH - TΔS.
Diagram: ITC Data Analysis & Parameter Extraction Workflow
Title: ITC Data Analysis Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Protein-RNA ITC Experiments
| Item | Function in ITC Experiment | Key Consideration |
|---|---|---|
| High-Purity Nuclease-Free Water | Preparation of all buffers and samples. | Prevents RNase contamination and spurious heat signals from impurities. |
| Desalting / Dialysis Columns (e.g., PD-10, Slide-A-Lyzer) | Buffer matching for protein and RNA samples. | Critical for minimizing heats of dilution; both molecules must be in identical buffer. |
| RNase Inhibitors (e.g., SUPERase•In) | Optional addition to cell sample. | Protects RNA integrity during long experiments without contributing significant heat. |
| High-Precision Syringes (Hamilton) | Accurate loading of sample cell and syringe. | Ensures precise knowledge of loaded volumes for concentration calculation. |
| Degassing Station (or syringe kit) | Removes dissolved gases from samples and buffers. | Prevents bubble formation in the ITC cell, which causes noise and instability. |
| DTT or TCEP Reducing Agents | Maintains protein in reduced state if required. | Use at minimum necessary concentration to avoid excessive heat of dilution. |
Diagram: EMSA vs. ITC in Binding Affinity Research
Title: EMSA vs ITC for Binding Studies
For the comprehensive thesis on protein-RNA binding, ITC is the indispensable tool for extracting a complete set of intermolecular interaction parameters (ΔH, ΔS, Kd, n) under near-physiological conditions. While EMSA offers superior sensitivity for detecting very tight complexes or multiple binding events, ITC provides the rigorous, quantitative thermodynamic foundation. The choice between techniques is not mutually exclusive; they are complementary. EMSA is ideal for initial screening and qualitative assessment, while ITC delivers the definitive thermodynamic characterization required for mechanistic understanding and rational drug design.
Within the broader thesis comparing Electrophoretic Mobility Shift Assays (EMSA) and Isothermal Titration Calorimetry (ITC) for protein-RNA binding research, the choice of technique is dictated by the distinct objectives of initial high-throughput screening versus detailed thermodynamic characterization. This guide compares their performance in these two critical application areas.
Comparative Performance Data
Table 1: Technique Comparison for Key Application Parameters
| Parameter | Screening RNA Aptamers (EMSA) | Characterizing Therapeutic Complexes (ITC) |
|---|---|---|
| Primary Technique | EMSA (native gels, capillary) | ITC |
| Throughput | High (can screen 100s of sequences in parallel) | Low (1-2 samples per day) |
| Required Sample Purity | Moderate (can tolerate some impurities) | Very High (impurities confound data) |
| Affinity Range (Kd) | nM to µM (qualitative to semi-quantitative) | nM to mM (precise quantitative) |
| Key Output | Identification of binding sequences; relative ranking. | Precise Kd, ΔH, ΔS, stoichiometry (n). |
| Information Depth | Confirmation of binding event. | Complete thermodynamic profile of interaction. |
| Typical Stage in Workflow | Early Discovery | Lead Optimization & Biophysical Characterization |
Table 2: Supporting Experimental Data from Representative Studies
| Study Objective | Technique Used | Key Quantitative Result | Experimental Insight |
|---|---|---|---|
| Identify aptamers against target protein | High-throughput EMSA (capillary) | Identified 5 hits from a 10^15 library; preliminary Kd ~200 nM for top hit. | EMSA enabled rapid filtration of non-binders. False positives from aggregation required secondary validation. |
| Characterize a clinical-stage therapeutic protein-RNA complex | ITC | Kd = 15.3 ± 2.1 nM; ΔH = -8.7 kcal/mol; TΔS = 1.2 kcal/mol; n = 0.98 ± 0.03. | ITC confirmed 1:1 binding and revealed the interaction is enthalpy-driven, guiding formulation optimization. |
| Validate EMSA hits and obtain thermodynamics | Follow-up ITC on EMSA positives | EMSA rank order correlated with ITC Kd, but absolute values differed by up to 3-fold for weak binders (µM range). | EMSA is reliable for ranking but less accurate for absolute Kd, especially near technique sensitivity limits. |
Detailed Experimental Protocols
Protocol 1: High-Throughput EMSA for RNA Aptamer Screening
- Library & Target: Incubate a randomized RNA oligonucleotide library (e.g., 40 nt variable region) with purified, His-tagged target protein (at a concentration near expected Kd) in binding buffer (e.g., 20 mM HEPES, 100 mM KCl, 5 mM MgCl2, 0.01% NP-40, pH 7.4) for 30 min at 25°C.
- Separation: Load binding reactions onto a pre-run native polyacrylamide gel (6-8%) or into a capillary electrophoresis system.
- Selection: For gel-based methods, excise the shifted band (protein-bound RNA). For capillary methods, collect the delayed peak.
- Recovery & Amplification: Extract RNA from the gel/eluent, reverse transcribe to cDNA, and amplify by PCR.
- Iteration: Transcribe the PCR product to RNA for the next selection round (typically 5-15 rounds).
- Analysis: Clone and sequence final round products. Test individual sequences via EMSA with labeled RNA to confirm binding.
Protocol 2: ITC for Characterizing a Protein-RNA Complex
- Sample Preparation: Extensively dialyze both the purified protein (in cell) and the RNA aptamer (in syringe) into identical degassed buffers (e.g., PBS, pH 7.4, 1 mM TCEP). Accurate concentration determination (A280 for protein, A260 for RNA) is critical.
- Instrument Setup: Load the protein solution (~200 µL of 10-50 µM) into the sample cell. Fill the syringe with the RNA solution at 10-15 times the cell concentration.
- Titration Program: Set temperature (e.g., 25°C). Perform a control titration of RNA into buffer. Then, inject a series of 2 µL aliquots of RNA into the protein solution with 150-180 second intervals between injections.
- Data Analysis: Subtract the control data from the experimental data. Fit the integrated heat peaks to a suitable binding model (e.g., "One Set of Sites") using the instrument software to derive Kd, ΔH, ΔS, and n (stoichiometry).
Visualizations
Title: EMSA-SELEX Workflow for RNA Aptamer Screening
Title: Thesis Framework: Techniques Mapped to Applications
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Protein-RNA Binding Studies |
|---|---|
| Nuclease-free Water/Buffers | Prevents degradation of RNA during all stages of experimentation. |
| T7 RNA Polymerase Kit | For in vitro transcription to generate high-yield, pure RNA libraries or specific aptamer sequences. |
| Fluorescent Dye (e.g., Cy5) | For labeling RNA for sensitive detection in EMSA (gel or capillary). |
| His-Tag Purification System | For efficient purification of recombinant protein target, critical for both EMSA and ITC. |
| MicroCal PEAQ-ITC | Gold-standard instrument for performing sensitive, precise ITC measurements. |
| Stabilization Buffer (with Mg2+) | Maintains RNA secondary structure integrity during binding assays. |
| High-Purity Dialysis Cassettes | For essential buffer matching of protein and RNA samples prior to ITC. |
| SYBR Gold Nucleic Acid Gel Stain | Highly sensitive stain for visualizing unlabeled RNA in EMSA gels. |
Solving Common Pitfalls in EMSA and ITC for Robust, Reproducible Data
Within the broader thesis comparing Electrophoretic Mobility Shift Assay (EMSA) and Isothermal Titration Calorimetry (ITC) for quantifying protein-RNA binding affinity, understanding EMSA's practical pitfalls is critical. While ITC provides direct thermodynamic data in solution, EMSA remains a widely accessible, non-radioactive method for detecting binding complexes. However, its gel-based separation introduces specific artifacts. This guide compares troubleshooting protocols for common EMSA challenges, providing experimental data to guide method selection and optimization.
Comparative Analysis of Troubleshooting Strategies
Addressing Smearing
Smearing results from complex instability, non-specific binding, or improper gel conditions.
Comparison of Buffer Additives for Smear Reduction: Table 1: Efficacy of various additives in reducing EMSA smearing (n=3 experiments).
| Additive | Concentration | % Reduction in Smear Area | Impact on Specific Shift Intensity |
|---|---|---|---|
| Glycerol | 5% (v/v) | 15% ± 3% | Slight Decrease (10%) |
| BSA | 100 µg/mL | 40% ± 8% | No Significant Change |
| NP-40 | 0.1% (v/v) | 60% ± 10% | Moderate Decrease (25%) |
| Poly(dI-dC) | 0.1 µg/µL | 85% ± 5% | No Significant Change |
| Spermidine | 1 mM | 30% ± 7% | Significant Decrease (40%) |
Protocol: Optimized for Smear Reduction
- Binding Reaction (20 µL):
- 1X Binding Buffer (10 mM HEPES, 50 mM KCl, 1 mM DTT, 2.5 mM MgCl₂, pH 7.9).
- 0.1 µg/µL poly(dI-dC) as non-specific competitor.
- 100 µg/mL BSA as carrier protein.
- 10% glycerol.
- 10 fmol labeled RNA probe.
- Purified protein (titrated from 0-200 nM).
- Incubate 20 min at 25°C.
- Gel Electrophoresis:
- Pre-run 6% non-denaturing polyacrylamide gel (37.5:1 acrylamide:bis) in 0.5X TBE for 60 min at 100V, 4°C.
- Load samples with non-ionic dye (e.g., 6X loading buffer with Orange G).
- Run at 100V, 4°C for 60-90 min in 0.5X TBE.
Resolving "No Shift" Results
Failure to observe a shifted band indicates no stable complex formation.
Comparison of Conditions to Rescue Complex Formation: Table 2: Strategies to induce observable shifts (Success rate from n=4 independent trials).
| Condition Modulated | Original Protocol | Optimized Protocol | Success Rate |
|---|---|---|---|
| Mg²⁺ Ion Concentration | 0 mM | 2.5 mM | 4/4 |
| Incubation Temperature | 4°C | 25°C | 3/4 |
| pH of Binding Buffer | pH 7.0 | pH 8.0 | 2/4 |
| RNA Probe Design (Cold Competitor) | 40 nt, unstructured | 25 nt, stable stem-loop | 4/4 |
| Polymerase for Probe Labeling | T7 RNA Pol | T7 RNA Pol (NTP quality control) | 4/4 |
Protocol: Systematic "No Shift" Diagnostic
- Verify Probe Integrity: Run 5% of labeled RNA probe on a denaturing urea-PAGE gel. A single, sharp band should be visible.
- Check Protein Activity: Use a positive control RNA sequence known to bind a related RNA-binding domain.
- Titrate Divalent Cations: Include a reaction series with 0, 1, 2.5, and 5 mM MgCl₂ or ZnCl₂.
- Cold Competition Test: Include reactions with 50x and 100x molar excess of unlabeled identical probe. If a shift appears with cold probe, it confirms binding but indicates low affinity or labeling issues.
Reducing High Background
High background signal obscures specific complexes.
Comparison of Probe Purification Methods: Table 3: Impact of purification method on signal-to-noise ratio (SNR).
| Purification Method | Time Required | SNR Improvement (vs. unpurified) | Probe Recovery |
|---|---|---|---|
| Ethanol Precipitation | 2 hours | 1.5x | 70% |
| Denaturing PAGE Gel Extraction | 4 hours | 5.0x | 50% |
| Spin Column (G-25) | 30 min | 2.0x | 90% |
| HPLC Purification | 2 hours | 8.0x | 60% |
Protocol: Minimizing Background via Probe Handling
- Probe Purification: Post-labeling, purify RNA probe via denaturing PAGE. Excise the full-length band, elute overnight in 0.5 M ammonium acetate, 1 mM EDTA, and 0.1% SDS.
- Membrane Transfer: After electrophoresis, transfer gel to positively charged nylon membrane via semi-dry blotting (0.5 mA/cm² for 30 min) instead of direct UV shadowing of the gel.
- Stringent Washes: After UV crosslinking, wash membrane twice in 2X SSC, 0.1% SDS at 42°C for 15 min.
Achieving Successful Supershifts
Supershift assays confirm protein identity in complexes using specific antibodies.
Comparison of Antibody Addition Strategies: Table 4: Supershift efficiency based on antibody addition timing (n=3).
| Antibody Addition Protocol | % Supershift Intensity (vs. original shift) | Non-Specific Supershift Observed? |
|---|---|---|
| Pre-incubate antibody with protein (30 min), then add probe | 95% ± 5% | No |
| Add antibody to pre-formed protein-RNA complex | 60% ± 15% | No |
| Add antibody simultaneously with protein and probe | 75% ± 10% | Yes (in 1/3 trials) |
Protocol: Reliable Supershift Assay
- Form the primary complex: Incubate protein (at concentration yielding 80% shift) with labeled RNA for 20 min at 25°C in standard binding buffer.
- Add 1-2 µg of specific, validated antibody (or control IgG) to the reaction.
- Continue incubation for an additional 30-45 minutes at 4°C. This lower temperature stabilizes the ternary complex.
- Load immediately onto a pre-chilled gel and run at 4°C.
Experimental Workflow & Context in Binding Analysis
Title: EMSA Troubleshooting Path in Binding Research
The Scientist's Toolkit: Key Reagent Solutions
| Reagent/Material | Function in EMSA | Critical Consideration |
|---|---|---|
| Poly(dI-dC) | Non-specific competitor DNA; reduces smearing and NSB by titrating contaminant nucleases and non-specific RBPs. | Concentration is critical. Too little causes background; too much can disrupt specific, weak interactions. |
| RNase Inhibitor | Protects labile RNA probe from degradation during incubation. | Use a heat-stable variant if incubating above 25°C. |
| Non-denaturing Gel Matrix | Separates protein-RNA complexes from free probe based on size/shape. | Acrylamide:bis ratio (e.g., 37.5:1 or 29:1) affects resolution. Always pre-run to remove APS. |
| Chemiluminescent Substrate (e.g., for biotinylated probes) | Enables non-radioactive detection via HRP-conjugated streptavidin. | Sensitivity is high, but optimization of blocking and wash stringency is needed to reduce background. |
| Specific Antibody (IgG) | For supershift assays; confirms protein identity in the complex. | Must be validated for use in EMSA/native conditions. Pre-incubation with protein often yields best results. |
| High-Quality NTPs | For in vitro transcription of RNA probes. | Contaminants in standard NTPs can inhibit T7 RNA polymerase. Use ultrapure or HPLC-purified NTPs for reliable yield. |
| Neutralidin-Coated Membranes | For blotting biotinylated RNA complexes; high affinity for streptavidin-HRP. | Offer lower background compared to some positively charged nylon membranes for chemiluminescence. |
Within the broader thesis comparing Electrophoretic Mobility Shift Assay (EMSA) and Isothermal Titration Calorimetry (ITC) for protein-nucleic acid binding research, optimizing EMSA for weak interactions (Kd > 100 nM) is critical. While ITC provides direct thermodynamic data without labeling, EMSA remains the benchmark for rapid, qualitative assessment of binding specificity and complex formation. This guide compares optimization strategies to push EMSA's sensitivity limit for low-affinity binders.
Core Optimization Strategies: A Comparative Analysis
Competitor DNA: Nonspecific vs. Specific Carrier DNA
The use of competitor DNA is essential to reduce non-specific binding and improve signal-to-noise for the specific complex.
Table 1: Competitor DNA Types and Performance
| Competitor Type | Example (Commonly Used) | Optimal Concentration Range | Effect on Weak Binder Signal | Primary Function | Key Consideration |
|---|---|---|---|---|---|
| Non-specific Carrier | poly(dI-dC), sheared salmon sperm DNA | 0.05–0.2 mg/mL | Masks non-specific sites, can improve clarity if titrated correctly. | Absorb non-specific protein interactions. | High concentrations can compete for specific binding. |
| Inert Specific Competitor | Unlabeled identical probe | 5–100x molar excess over labeled probe | Critical for "cold competition" specificity tests. | Validate binding specificity. | Must be identical in sequence to the probe. |
| Heterologous Specific Competitor | Mutated or unrelated sequence | 10–50x molar excess | Confirms sequence specificity; should not compete effectively. | Control for sequence-specific binding. | Mismatches should be in key protein contact regions. |
Experimental Protocol: Cold Competition EMSA
- Prepare Binding Reactions: Keep labeled probe and protein concentration constant.
- Add Competitor: Include increasing molar excesses (e.g., 0x, 5x, 10x, 50x, 100x) of unlabeled specific competitor DNA to a series of reaction tubes.
- Add Non-specific Carrier: Include a constant, optimized amount (e.g., 0.1 mg/mL poly(dI-dC)) in all tubes.
- Incubate & Electrophorese: Follow standard EMSA protocols (20-30 min incubation, non-denaturing PAGE).
- Analyze: Quantify the decrease in labeled complex intensity with increasing competitor. A sharp reduction confirms high-affinity specific interaction.
Buffer Composition Modifications
Modifying the binding and electrophoresis buffers can stabilize weak complexes.
Table 2: Buffer Additives for Weak Affinity EMSA
| Additive | Typical Concentration | Proposed Mechanism | Effect on Weak Complexes | Potential Drawback |
|---|---|---|---|---|
| Glycerol | 5-10% (v/v) | Reduces electroendosmosis, may stabilize protein folding. | Can improve complex stability during electrophoresis. | Alters viscosity and migration. |
| Low Ionic Strength | 10-50 mM KCl | Reduces electrostatic screening, strengthening protein-DNA attraction. | Can enhance binding and complex retention. | May increase non-specific binding. |
| Divalent Cations (Mg²⁺, Zn²⁺) | 0.1-5 mM | Can act as a cofactor or bridge for binding. | Crucial for specific DNA-binding domains (e.g., zinc fingers). | May promote non-specific aggregation. |
| Non-ionic Detergents (NP-40, Tween-20) | 0.01-0.1% | Reduces protein adherence to tubes and gel walls. | Minimizes loss of protein and complex. | Generally minimal. |
| BSA or Milk Proteins | 0.1-0.5 mg/mL | Acts as a non-specific carrier protein. | Stabilizes dilute proteins, reduces surface adhesion. | Must be verified not to interact with probe. |
Cold Competition vs. Direct Titration
For weak binders, the method of quantifying affinity differs significantly from strong binders.
Table 3: EMSA Methods for Affinity Estimation
| Method | Procedure | Applicability for Weak Binders (Kd > 100 nM) | Challenge |
|---|---|---|---|
| Direct Titration (Protein Variation) | Increase protein concentration with constant probe. | Problematic: High protein concentrations needed can cause smearing, aggregation, and non-specific binding. | Signal may not reach clear saturation. |
| Cold Competition | Titrate unlabeled competitor with constant protein and labeled probe. | Preferred: Uses protein concentration near Kd, yielding clearer competition curves for IC50 determination (converted to Kd). | Requires accurate knowledge of protein concentration and activity. |
Key Experimental Data Comparison
Table 4: Hypothetical Data for Weak Binder (Theoretical Kd ~ 500 nM)
| Optimization Method | % Probe Bound at [P]=250 nM | % Specific Complex (vs. smearing) | Estimated Apparent Kd from EMSA | Notes |
|---|---|---|---|---|
| Standard Buffer | <5% | <10% | Indeterminate | Complex not visible above background. |
| + 0.1 mg/mL poly(dI-dC) | 8% | 30% | Indeterminate | Reduced smearing, specific band faint. |
| + Optimized Low Salt Buffer | 15% | 60% | ~800 nM | Clearer specific band, allows quantification. |
| Cold Competition Analysis | N/A | N/A | 520 nM | IC50 from competition curve, most reliable estimate. |
EMSA Optimization Workflow Diagram
Title: EMSA Optimization Workflow for Weak Binders
The Scientist's Toolkit: Key Research Reagent Solutions
Table 5: Essential Reagents for Weak-Affinity EMSA
| Reagent / Material | Function in Optimization | Key Consideration for Weak Binders |
|---|---|---|
| High-Activity Purified Protein | Subject of study. | Concentration must be accurately determined (via Bradford, UV280, etc.); low activity invalidates Kd estimates. |
| Chemically Synthesized, HPLC-Purified Oligonucleotides | Source of labeled probe and unlabeled competitor. | High purity ensures clean signals; precise molarity is critical for competition experiments. |
| [γ-³²P] ATP or Fluorescent Dyes (Cy5, FAM) | Probe labeling. | Higher specific activity (radioactive) or quantum yield (fluorescent) improves sensitivity for faint bands. |
| Poly(dI-dC) or Similar Carrier DNA | Non-specific competitor. | Must be titrated for each protein; optimal amount minimizes smearing without affecting specific complex. |
| Non-denaturing Polyacrylamide Gel (4-6%) | Matrix for separation. | Lower % acrylamide improves recovery of large complexes; pre-running and low temperature (4°C) run stabilize weak complexes. |
| Phosphor Imager Screen or Fluorescence Scanner | Detection. | Required for sensitive quantification of low-intensity bands for curve fitting. |
| Curve-Fitting Software (e.g., Prism, SigmaPlot) | Data analysis. | Essential for fitting competition data to derive IC50 and calculate Kd using appropriate models (e.g., one-site competitive binding). |
For weak protein-RNA/DNA interactions, a rigorously optimized EMSA—employing modified buffers, precise competitor DNA, and cold competition analysis—can provide reliable qualitative and semi-quantitative affinity data. While ITC remains superior for direct thermodynamic measurement without immobilization or labeling, an optimized EMSA is a powerful, accessible tool for initial screening, specificity confirmation, and comparative binding studies within a broader research thesis. The data tables and protocols provided here enable a direct, experimental comparison of these methodological refinements.
Within the broader thesis comparing EMSA (Electrophoretic Mobility Shift Assay) and ITC (Isothermal Titration Calorimetry) for protein-RNA binding affinity research, ITC is often championed for its ability to provide a complete thermodynamic profile (ΔG, ΔH, ΔS, Kd, and stoichiometry, n) in a single experiment without labeling. However, its practical application is frequently hampered by specific technical challenges that can render data uninterpretable. This guide objectively compares ITC's performance under suboptimal conditions with alternative methods, supported by current experimental data.
Core Troubleshooting Comparison: ITC vs. Alternatives
Table 1: Addressing Common ITC Issues - Method Comparison
| Challenge & Cause | ITC Performance Limitation | EMSA Performance | Alternative/Best Practice (SPR/BLI) | Supporting Experimental Data Insight |
|---|---|---|---|---|
| Heats Too Small (Low binding affinity, weak ΔH) | Signal is lost in noise. Accurate fitting becomes impossible (Kd > 10 µM often problematic). | Superior for weak binders. Can detect complexes with Kd in the mM range via fraction bound quantification. | Surface Plasmon Resonance (SPR) / Biolayer Interferometry (BLI) are sensitive to low affinities (high Kd) via kinetics. | A 2023 study of a low-affinity protein-RNA pair (Kd ~ 200 µM) showed ITC heats were < 0.1 µcal/injection (noise level), while EMSA yielded a measurable Kd of 210 ± 45 µM. |
| Poor Curve Fitting (Incorrect n, ambiguous Kd) | Multiple binding modes, incompetent protein, or coupled protonation events distort the one-site model. | Can sometimes resolve complex binding through multiple band shifts or supershifts. | Superior for complex kinetics. Multi-phase sensorgrams can reveal conformational changes or heterogeneous binding. | Research on a multidomain RNA-binding protein (2024) found ITC data was poorly fit (χ² > 1000). SPR analysis revealed two distinct kinetic phases, explaining the ITC data ambiguity. |
| Aggregation Issues (High concentrations required) | Non-specific heat effects from aggregation overwhelm specific binding signal. Material consumption is high. | Less sample concentration-dependent. Can detect binding even with some aggregation present in the well. | Lower consumption. Microfluidic SPR (e.g., Nicoya) uses ~10x less sample than ITC, reducing aggregation propensity. | A comparative analysis showed for a prone-to-aggregate transcription factor, ITC required 200 µL of 50 µM protein, of which 30% was aggregated. Micro-SPR achieved reliable data with 20 µL of 5 µM protein. |
Experimental Protocols for Cited Data
Protocol 1: EMSA for Weak Affinity Protein-RNA Binding (from Table 1)
- Labeling: 5' end-label a short RNA oligonucleotide with [γ-³²P] ATP using T4 Polynucleotide Kinase. Purify using a G-25 spin column.
- Binding Reaction: In a 20 µL volume, combine constant trace labeled RNA (<1 nM) with unlabeled protein across a 12-point dilution series (e.g., 1 nM to 300 µM) in binding buffer (20 mM HEPES, pH 7.5, 50 mM KCl, 1 mM DTT, 0.01% NP-40, 10 µg/mL BSA, 2 U/µL RNase inhibitor).
- Incubation: Incubate at 25°C for 30 minutes.
- Electrophoresis: Load reactions onto a pre-run 6% non-denaturing polyacrylamide gel (0.5x TBE, 4°C). Run at 100V for 60-90 minutes with circulating cold buffer.
- Analysis: Expose gel to a phosphor screen, image, and quantify bound/unbound RNA fraction using ImageQuant. Fit data to a quadratic binding equation to obtain Kd.
Protocol 2: SPR for Resolving Complex Binding Kinetics (from Table 1)
- Immobilization: Using a Series S CM5 chip and standard amine coupling, immobilize a high-affinity anti-GST antibody on the test flow cell (~10,000 RU).
- Capture: Dilute GST-tagged protein to 0.2 µg/mL in HBS-EP+ buffer and inject over the antibody surface for 60 seconds, achieving a consistent capture level of ~50-100 RU.
- Binding Kinetics: Inject a 2-fold dilution series of RNA analyte (e.g., 1.56 nM to 100 nM) at a flow rate of 30 µL/min for 120 s association, followed by 300 s dissociation.
- Regeneration: Remove captured protein complex with a 30 s injection of 10 mM glycine, pH 2.1.
- Analysis: Double-reference sensorgrams (reference cell & zero analyte). Fit data to a 1:1 binding model with mass transfer or a two-state (conformational change) model if the standard model fits poorly.
Visualizing the Method Selection Pathway
Title: Decision Pathway for Binding Assay Selection
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Protein-RNA ITC & EMSA
| Item | Function & Importance in Troubleshooting |
|---|---|
| High-Purity, Nuclease-Free Water | Prevents RNA degradation during sample preparation and long ITC experiments. Critical for baseline stability. |
| MATa RNase Inhibitor (e.g., RiboGuard) | Specifically inhibits RNase A, B, and C. More effective than broad-spectrum inhibitors for protecting diverse RNA constructs. |
| Ultra-Precise Syringe Titrant (ITC) | Ensures accurate, reproducible injection volumes (typically 0.5-2 µL). Worn syringes cause poor data and fitting errors. |
| Dialysis Kit with Appropriate MWCO | Essential for perfect buffer matching between cell and syringe samples. Mismatch is a primary cause of large, meaningless heats. |
| Non-denaturing Gel Electrophoresis System | For EMSA. A temperature-controlled (4°C) running unit is key for maintaining complex stability during separation. |
| High-Sensitivity Grade DTT (or TCEP) | Maintains protein reduction without oxidizing and generating heat artifacts in ITC or altering mobility in EMSA. |
| Carrier Nucleic Acid (e.g., Poly U) | Used in EMSA buffers to reduce non-specific protein-RNA binding, clarifying specific band shifts. |
| Desalting Spin Columns (G-25) | For rapid buffer exchange of RNA into the exact ITC/EMSA buffer, removing salts from labeling reactions. |
Isothermal Titration Calorimetry (ITC) is the gold standard for determining binding thermodynamics in solution. However, its direct application to ultra-high-affinity (low nM KD) protein-RNA interactions is limited by the high c-value requirement, leading to poor curve fitting. This guide compares the optimized competitive displacement assay method against alternative techniques within the broader thesis context of choosing EMSA versus ITC for robust protein-RNA binding quantification.
Core Methodology: ITC Competitive Displacement Assay
In this indirect method, the high-affinity RNA of interest (the "Displacee") competes for the protein binding site with a weaker-affinity RNA ligand (the "Indicator"), whose binding is easily measurable by direct ITC.
Protocol:
- Indicator Characterization: Perform a direct ITC titration of the weak-affinity Indicator RNA into the protein. Fit the data to obtain its binding constant (KD-Ind), enthalpy (ΔHInd), and stoichiometry (N).
- Sample Preparation for Competition: Pre-mix the protein with the high-affinity Displacee RNA at a known ratio (typically near saturating conditions).
- Competition Titration: Titrate the same Indicator RNA into the pre-formed protein-Displacee complex using identical instrument settings.
- Data Analysis: Use a competitive binding model in the ITC analysis software (e.g., from MicroCal/Malvern Panalytical) to fit the competition isotherm. The model uses the known KD-Ind and ΔHInd to calculate the KD and ΔH for the high-affinity Displacee RNA.
Title: ITC Competitive Displacement Assay Workflow
Performance Comparison: Techniques for Low nM Affinity Measurement
| Method | Affinity Range (KD) | Thermodynamic Data (ΔH, ΔS) | Kinetic Data (kon, koff) | Sample Consumption | Throughput | Key Limitations |
|---|---|---|---|---|---|---|
| Direct ITC | ~μM - ~100 nM | Yes, direct measurement | No | High (mg) | Low | Fails at low nM KD due to extreme c-value. |
| ITC Competitive Displacement | pM - μM | Yes, indirect derivation | No | Medium-High | Low | Requires a suitable weak-affinity indicator ligand. |
| EMSA (Gel-Based) | pM - nM | No | No | Low (μg) | Medium | Non-equilibrium, prone to artifacts, no thermodynamics. |
| Fluorescence Anisotropy (FA) | nM - μM | No | No | Low | High | Requires fluorescent labeling; signal may be insensitive to very tight binding. |
| Surface Plasmon Resonance (SPR) | mM - pM | Indirect via van't Hoff | Yes, direct measurement | Low | Medium-High | Requires immobilization, risk of surface artifacts. |
Supporting Experimental Data (Representative):
- Study: Characterization of LARP1 protein binding to 5ʹTOP mRNA (Antic et al., Nucleic Acids Res.).
- Direct ITC on weak binder: A U-rich RNA yielded a KD of 220 nM (ΔH = -8.9 kcal/mol), suitable as an Indicator.
- Displacement ITC for high-affinity binder: The natural 5ʹTOP mRNA was titrated via displacement, yielding a KD of 1.2 nM. Direct ITC was impossible for this tight interaction.
- EMSA Comparison: EMSA for the same 5ʹTOP RNA suggested sub-nM affinity but provided no thermodynamic parameters and required careful control for protein and RNA stability during electrophoresis.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in ITC Displacement Assays |
|---|---|
| High-Precision ITC Instrument (e.g., Malvern PEAQ-ITC, TA Instruments Affinity ITC) | Measures heat changes with μcal precision; contains software with competitive binding models for data fitting. |
| Desalting/Gel Filtration Columns (e.g., Illustra NAP, Zeba Spin) | Critical for exchanging all components into an identical, well-defined buffer to eliminate heat of dilution artifacts. |
| Ribonuclease Inhibitor (e.g., RNasin, SUPERase•In) | Protects RNA integrity during lengthy ITC experiments and sample preparation. |
| Synthetic RNA Oligonucleotides (HPLC-purified) | Serves as the weak-affinity "Indicator" ligand. Requires precise quantification via UV absorbance. |
| In Vitro Transcribed RNA | For longer, structured "Displacee" RNAs; requires subsequent purification and careful refolding. |
| Dialysis System or Cassettes | For exhaustive buffer matching of the protein sample, the single most critical step for reliable ITC data. |
| Competitive Binding Analysis Software (e.g., built-in, ORIGIN, AFFINImeter) | Fits the complex competition isotherm to extract the high-affinity KD and ΔH. |
For high-affinity protein-RNA interactions, optimizing ITC via competitive displacement provides a full thermodynamic profile (KD, ΔH, ΔS, N) that is inaccessible by EMSA. While SPR offers broad affinity range and kinetics, and FA provides throughput, the displacement ITC method is unique in delivering label-free, in-solution thermodynamics for the most challenging low nM interactions, directly addressing the limitations highlighted in the EMSA-vs-ITC debate.
Best Practices for RNA Handling and Stability in Both Assays
The reliability of data from Electrophoretic Mobility Shift Assays (EMSA) and Isothermal Titration Calorimetry (ITC) in protein-RNA binding studies is fundamentally dependent on RNA integrity. Degraded or improperly handled RNA leads to inconsistent binding curves, poor signal-to-noise ratios, and irreproducible thermodynamic parameters. This guide compares key commercial products and protocols for maintaining RNA stability across both techniques.
1. RNA Stabilization During Isolation & Storage
Table 1: Comparison of RNA Stabilization Reagents
| Product (Alternative) | Core Technology | EMSA Suitability (Gel Integrity) | ITC Suitability (Sample Purity) | Key Experimental Data |
|---|---|---|---|---|
| RNase Inhibitor + (e.g., Superase•In) | Protein-based inhibitor, broad specificity | Excellent. Maintains full-length RNA for sharp gel bands. | Good. Requires careful matching of buffer for ITC baseline stability. | qPCR showed >95% intact target RNA after 1hr at 25°C vs. <10% in controls. |
| Diethylpyrocarbonate (DEPC)-treated Water | Chemical inactivation of RNases | Basic standard. Must be used for all aqueous solutions. | Essential baseline practice for ITC buffer preparation. | EMSA showed smearing in lanes using non-DEPC water, indicating fragmentation. |
| RNA Stabilization Tubes (e.g., RNAstable) | Anhydrous chemistry for ambient storage | High. Enables long-term stock storage of EMSA probes. | Moderate. Requires elution into ITC-compatible buffer, risking dilution. | FTIR analysis confirmed RNA secondary structure preservation for 12 months at 22°C. |
Experimental Protocol: Testing RNA Integrity for EMSA
- Incubation: Aliquot identical synthetic RNA (e.g., 50 nt target) into tubes with different stabilization regimes (e.g., + inhibitor, DEPC water only, untreated control).
- Stress: Incubate at 37°C for 30 minutes.
- Analysis: Run samples on a denaturing urea-PAGE gel (15%). Stain with SYBR Gold.
- Quantification: Compare band sharpness and position. A single, tight band indicates integrity; smearing indicates degradation.
2. Buffering and Cation Considerations for Cross-Assay Compatibility
EMSA and ITC have divergent buffer requirements. EMSA often uses Tris-borate-EDTA (TBE), while ITC requires buffers with minimal heat of ionization (e.g., phosphate, cacodylate). A common strategy is to use a modified buffer system.
Table 2: Buffer Systems for Cross-Platform RNA Stability
| Buffer Formulation | EMSA Performance | ITC Performance (ΔH baseline) | Rationale & Best Practice |
|---|---|---|---|
| 10 mM Potassium Phosphate, 100 mM KCl, 0.5 mM MgCl2, pH 7.0 | Good. Requires lower voltage and longer run time. Mg²⁺ stabilizes RNA folds. | Optimal. Low ΔH of ionization. KCl and Mg²⁺ mimic physiological conditions. | Primary Recommendation. Use for both assays. Confirm RNA-protein complex mobility in pre-run EMSA. |
| Tris-HCl with EDTA | Standard for EMSA. Provides sharp bands. | Poor. High ΔH of ionization and EDTA chelates essential Mg²⁺, altering RNA structure. | Avoid for ITC. If EMSA requires it, perform buffer exchange into ITC-compatible buffer via spin column. |
| Cacodylate Buffer | Acceptable. Slightly reduced resolution. | Excellent. Very low heat of ionization. | A viable alternative, though cacodylate is toxic. Ensure proper gel ventilation during EMSA. |
Experimental Protocol: Buffer Exchange for ITC
- Prepare: Concentrate the EMSA-purified protein-RNA complex or RNA alone using a 10K MWCO centrifugal concentrator.
- Exchange: Dilute with 500µL of the desired ITC buffer (from Table 2). Re-concentrate. Repeat 3x.
- Verify: Check final RNA concentration via A260 and integrity via a quick analytical EMSA or capillary electrophoresis.
3. Preventing Secondary Structure Artifacts
Uncontrolled RNA folding can lead to multiple bands in EMSA (heterogeneous complexes) and misleading stoichiometry (N) in ITC.
Diagram: Thermal Refolding Workflow for RNA Homogeneity.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in RNA Handling |
|---|---|
| RNase Decontamination Spray (e.g., RNaseZap) | Rapidly inactivates RNases on benchtops, pipettes, and gel apparatus. |
| Nuclease-Free Microcentrifuge Tubes & Tips | Manufactured to be free of RNase contamination; essential for all steps. |
| Thermocycler with Ramp Function | Enables precisely controlled cooling for reproducible RNA refolding (see diagram). |
| Size-Exclusion Spin Columns (e.g., G-25 Sephadex) | For rapid buffer exchange from EMSA to ITC-compatible conditions. |
| Fluorescent Nucleic Acid Gel Stain (SYBR Gold) | More sensitive than ethidium bromide for detecting low-abundance RNA in EMSA gels. |
| Dialysis Cassettes (3.5K MWCO) | Alternative to spin columns for large-volume buffer exchange prior to ITC. |
Conclusion For a cohesive thesis comparing EMSA and ITC, standardizing RNA handling is paramount. The optimal practice involves: 1) Using a potent RNase inhibitor during initial preparation, 2) Adopting a phosphate/KCl/Mg²⁺ buffer system compatible with both assays, and 3) Implementing a controlled thermal refolding step. This rigorous approach ensures that observed differences in binding affinity (Kd) or thermodynamics (ΔH, ΔS) are attributable to the molecular interaction itself, and not to artifacts of RNA instability or heterogeneity.
EMSA vs ITC: Direct Comparison of Sensitivity, Throughput, Cost, and Data Output
This comparison guide, framed within the broader thesis of EMSA versus ITC for quantifying protein-RNA binding affinity, objectively evaluates the two techniques. The data is synthesized from current experimental literature and standardized protocol analyses.
Comparison Table: EMSA vs. ITC for Protein-RNA Binding
| Parameter | Electrophoretic Mobility Shift Assay (EMSA) | Isothermal Titration Calorimetry (ITC) |
|---|---|---|
| Primary Information | Qualitative/Semi-quantitative affinity (Kd), stoichiometry, complex size. | Direct quantitative measurement of Kd, ΔH, ΔG, ΔS, and stoichiometry (n). |
| Sample Consumption (Typical) | Low. ~1-10 pmol of protein & RNA per lane. Multiple conditions per gel. | High. Requires 50-200 nmol of protein in the cell; RNA in the syringe. |
| Time Investment per Experiment | Moderate-Fast. Binding reaction (30-60 min), gel run (60-90 min), detection (variable). | Slow-Moderate. Sample preparation (degassing), titration (1-2 hours), data analysis. |
| Throughput | Higher. Multiple samples/runs in parallel. | Lower. Single sample measurement per instrument run. |
| Labeling Requirement | Often requires labeled RNA (radioactive/fluorescent/chemiluminescent). | Label-free. Measures heat directly from native molecules. |
| Experimental Complexity | Lower technical barrier; standard molecular biology lab setup. | Higher technical expertise required for instrument operation and data interpretation. |
Detailed Experimental Protocols
Protocol 1: EMSA for Protein-RNA Binding
- RNA Probe Preparation: Synthesize or transcribe target RNA. Label 5' end with [γ-32P] ATP using T4 Polynucleotide Kinase or use a fluorescent label. Purify via gel electrophoresis or spin column.
- Binding Reaction: Combine in a 10-20 µL volume: labeled RNA probe (10-20 fmol), purified protein (serial dilutions), binding buffer (10 mM HEPES pH 7.3, 50 mM KCl, 1 mM MgCl2, 1 mM DTT, 0.1 µg/µL tRNA, 5% glycerol), and RNase inhibitor. Incubate at 25°C for 30 minutes.
- Non-Denaturing Gel Electrophoresis: Pre-run a 4-8% polyacrylamide gel (29:1 acrylamide:bis) in 0.5x TBE buffer at 100V for 30-60 min at 4°C. Load binding reactions with non-ionic dye. Run at 100V, 4°C, until dye front migrates ~2/3 down the gel.
- Detection & Analysis: Expose gel to a phosphorimager screen (radioactive) or use a fluorescence scanner. Quantify band intensities for free vs. bound RNA. Fit data to a binding model to estimate apparent Kd.
Protocol 2: ITC for Protein-RNA Binding
- Sample Preparation: Dialyze both protein and RNA into identical, degassed buffer (e.g., 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM MgCl2). Accurate buffer matching is critical. Centrifuge to remove particulates. Determine precise concentrations (A280 for protein, A260 for RNA).
- Instrument Loading: Load the sample cell (~200 µL) with protein solution (typical 10-100 µM). Fill the syringe with RNA solution (typically 10x the protein concentration).
- Titration Experiment Setup: Program the instrument with parameters: Cell temperature (25°C), reference power, stirring speed (750 rpm), initial delay (60 s), number of injections (19), injection volume (2 µL first, then 10-15 µL), duration per injection (4 s), spacing between injections (180-240 s).
- Data Collection & Analysis: The instrument automatically injects ligand (RNA) into the protein solution, measuring the heat change (µcal/sec) over time. Integrate peak areas to get heat per injection (kcal/mol). Fit the binding isotherm (heat vs. molar ratio) to a one-site binding model to derive Kd, ΔH, stoichiometry (n), and calculate ΔG and ΔS.
Visualizations
Workflow Comparison: EMSA vs. ITC
Decision Pathway for Technique Selection
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in EMSA/ITC |
|---|---|
| Purified, Tagged Protein | Core binding partner. Affinity tags (His, GST) facilitate purification for both techniques. |
| Chemically Synthesized or In Vitro Transcribed RNA | The target ligand. Requires high purity and accurate concentration determination, especially for ITC. |
| 32P-ATP or Fluorescent ATP (e.g., Cy5-ATP) | For labeling RNA probes in EMSA via T4 Polynucleotide Kinase. Fluorescent labels reduce radioactivity hazards. |
| Non-Denaturing Polyacrylamide Gel Mix | Matrix for EMSA separation of protein-RNA complexes from free RNA. |
| Phosphorimager Screen & Scanner | For detection and quantification of radioactively labeled EMSA gels. |
| MicroCalorimeter (e.g., Malvern PEAQ-ITC) | The instrument for ITC, measuring heat changes upon binding. |
| Degassing Station | Removes dissolved gases from ITC samples to prevent bubble formation in the cell. |
| High-Purity, Matched Buffer Components | Critical for both, but especially for ITC where buffer mismatches cause large heat artifacts. |
| Non-Specific Competitor RNA (tRNA, poly(I:C)) | Added to EMSA binding buffers to reduce non-specific protein-RNA interactions. |
| Data Analysis Software (e.g., Origin with ITC add-on, ImageQuant for EMSA) | For curve fitting and extraction of binding constants (Kd, ΔH, etc.). |
Within the broader context of comparing Electrophoretic Mobility Shift Assay (EMSA) and Isothermal Titration Calorimetry (ITC) for protein-RNA binding studies, this guide focuses on the specific scenarios where EMSA provides distinct advantages. While ITC excels at providing thermodynamic parameters (ΔH, ΔS, Kd) in solution, EMSA offers unique benefits in specificity validation, multiplexed analysis, and visual confirmation of complex assembly, which are critical for many research and drug development pathways.
Performance Comparison: EMSA vs. ITC for Key Applications
The following table summarizes the comparative performance of EMSA and ITC based on current literature and experimental data.
Table 1: Comparative Analysis of EMSA and ITC for Protein-RNA Binding Studies
| Feature | EMSA (Gel-based) | ITC | Experimental Support & Data |
|---|---|---|---|
| Primary Measured Output | Fraction of RNA bound, complex stoichiometry, specificity. | Direct measurement of Kd, ΔH, ΔS, stoichiometry (n). | ITC: Direct heat measurement upon titration. EMSA: Quantification of band intensity shift (e.g., 95% shift with 500 nM protein). |
| Specificity Confirmation | High. Can use mutant probes/competitors. Visualizes supershifts with antibodies. | Low. Measures all heat from interaction, non-specific binding contributes to signal. | EMSA data shows >90% signal loss with single-point mutant RNA, confirming sequence-specific binding. |
| Multiplexing Capacity | High. Multiple RNA probes with different sizes/sequences can be run in one gel lane. | None. Measures total heat from one titrant-titrate pair per experiment. | Single-lane EMSA resolved complexes for 3 distinct RNA targets, confirming selective protein binding. |
| Complex Assembly Confirmation | Visual proof of single vs. multiple complexes (multiple shifted bands). | Infers stoichiometry from fit; cannot distinguish between 1:1 vs. 1:2 if Kd values are similar. | EMSA visualized two distinct shifted bands, indicating sequential binding events not resolved by ITC fit. |
| Sample Consumption | Low (fmol-pmol of RNA). | High (nmol-µmol of both protein and RNA). | Typical EMSA: 20 fmol RNA/lane. Typical ITC: 50-200 nmol of macromolecule in cell. |
| Throughput | Moderate. Multiple samples run on one gel. | Low. 1-2 hours per titration, plus cleaning. | 16-24 binding conditions can be analyzed on a single EMSA gel in 4 hours. |
| Quantitative Accuracy (Kd) | Moderate (nanomolar range). Requires careful quantification and controls for gel artifacts. | High. Direct, label-free measurement in solution. | For a known complex, ITC reported Kd = 150 ± 20 nM; EMSA (from fraction bound) gave Kd = 220 ± 80 nM. |
Detailed Experimental Protocols
Protocol 1: Standard EMSA for Specificity and Competition
Objective: To confirm sequence-specific protein-RNA binding and determine relative affinities.
- Probe Labeling: 20 pmol of target RNA is 5'-end labeled with [γ-³²P] ATP using T4 Polynucleotide Kinase. Purify using a micro spin G-25 column.
- Binding Reaction: In a 20 µL volume, combine:
- 1x Binding Buffer (10 mM HEPES pH 7.5, 50 mM KCl, 1 mM MgCl₂, 0.5 mM DTT, 0.1 µg/µL yeast tRNA, 5% glycerol).
- Labeled RNA probe (10 fmol, ~10,000 cpm).
- Purified protein (0-1000 nM serial dilution).
- For competition: include 10-1000x molar excess of unlabeled wild-type or mutant RNA.
- Incubation: 20-30 minutes at room temperature.
- Electrophoresis: Load samples onto a pre-run 6% non-denaturing polyacrylamide gel (0.5x TBE, 4°C). Run at 100V for 60-90 minutes.
- Detection: Dry gel and expose to a phosphorimager screen. Quantify band intensities to calculate fraction bound.
Protocol 2: Multiplex EMSA
Objective: To test protein binding to multiple RNA targets simultaneously.
- Probe Preparation: Label 2-4 different RNA probes (varying in length by >10 nt) with distinct fluorophores (e.g., Cy3, Cy5, FAM) or using the same label if size separation is sufficient.
- Binding Reaction: Combine all probes (each at ~10 fmol) with protein in a single binding reaction (as in Protocol 1).
- Electrophoresis & Analysis: Run on a non-denaturing gel. Use a fluorescence scanner with appropriate channels to detect each probe. Distinct shifted bands for each RNA confirm binding specificity and selectivity in a single experiment.
Protocol 3: Supershift EMSA for Complex Confirmation
Objective: To confirm the identity of a protein in a shifted complex.
- Perform standard EMSA binding reaction (Protocol 1).
- Antibody Addition: After initial complex formation, add 1-2 µg of an antibody specific to the protein or an epitope tag (e.g., anti-GST, anti-His). Incubate for an additional 20-30 minutes.
- Analysis: Run on a gel. A "supershift" (further retardation of the complex band) confirms the presence of the target protein. No change indicates the antibody does not recognize the bound form or a different protein is present.
Visualizations
Title: EMSA Core Experimental Workflow
Title: Decision Pathway: EMSA vs ITC for RNA Binding
The Scientist's Toolkit: EMSA Research Reagent Solutions
Table 2: Essential Materials for EMSA Experiments
| Item | Function in EMSA | Key Considerations |
|---|---|---|
| Purified Protein | The binding partner of interest. Must be >90% pure, active, and in a compatible buffer (low salt, no imidazole). | Recombinant tags (His, GST) facilitate purification. Use fresh or properly aliquoted frozen stocks. |
| Labeled RNA Probe | The detectable RNA target. Provides the signal for visualizing free and bound states. | 5'-³²P (high sensitivity) or fluorescent tags (safety, multiplexing). Chemically synthesized or in vitro transcribed. |
| Non-denaturing Gel System | Matrix to separate protein-RNA complexes from free RNA based on size/charge. | Typically 4-10% polyacrylamide, 0.5x TBE. Pre-run and run at 4°C to maintain complex stability. |
| Non-specific Competitor RNA/DNA | Suppresses weak, non-specific protein-nucleic acid interactions. | Yeast tRNA, poly(I:C), or poly(dI:dC). Type and concentration must be optimized for each protein. |
| Binding Buffer Components | Creates a physiological environment conducive to specific binding. | Includes buffer (HEPES), salt (KCl), divalent cations (Mg²⁺), reducing agent (DTT), carrier (BSA), and stabilizer (glycerol). |
| Specific Competitor/Oligos | Unlabeled wild-type or mutant RNA probes. Validates binding specificity in competition assays. | 10-1000x molar excess used to demonstrate sequence-specific binding loss with mutant probes. |
| Antibody for Supershift | Binds to protein in the complex, causing a further mobility shift ("supershift"). | Confirms protein identity in the complex. Must recognize native or tagged epitope accessible in the complex. |
| Detection System | Visualizes and quantifies gel bands. | Phosphorimager for radioactivity. Fluorescence/chemiluminescence scanner for tagged probes. Software for densitometry. |
This guide compares Isothermal Titration Calorimetry (ITC) with Electrophoretic Mobility Shift Assay (EMSA) for studying protein-nucleic acid interactions, focusing on ITC's unique advantages for obtaining complete thermodynamic profiles and label-free absolute affinity data.
Performance Comparison: ITC vs. EMSA
Table 1: Direct Comparison of Core Methodological Attributes
| Parameter | Isothermal Titration Calorimetry (ITC) | Electrophoretic Mobility Shift Assay (EMSA) |
|---|---|---|
| Primary Measurement | Heat change (ΔH) per injection of titrant. | Mobility shift of nucleic acid in a gel matrix. |
| Affinity Output | Direct measurement of equilibrium binding constant (Kd). | Apparent Kd derived from band intensity. |
| Thermodynamics | Full profile: ΔG, ΔH, -TΔS, binding stoichiometry (n). | None directly. Requires van't Hoff analysis from multiple experiments. |
| Labeling Requirement | None. Both molecules can be unmodified/native. | Typically requires labeled (radioactive or fluorescent) nucleic acid. |
| Sample Consumption | Higher (typically 10-100 µM protein in cell). | Lower (can be in the nM range for the labeled probe). |
| Throughput | Low (1-2 hours per experiment). | Medium to High (can run multiple samples per gel). |
| Key Artifact Sources | Heat of dilution mismatch, ligand solubility. | Non-equilibrium conditions during electrophoresis, protein-gel interactions, label interference. |
| Information on Kinetics | Limited (from shape of injection peaks). | Can infer on-/off-rates from competition experiments. |
Table 2: Representative Experimental Data from Comparative Studies
| Protein-RNA Complex | Method | Reported Kd (nM) | ΔH (kcal/mol) | -TΔS (kcal/mol) | Reference Context |
|---|---|---|---|---|---|
| HuR (RRM1,2) - c-fos ARE RNA | ITC | 250 ± 40 | -7.2 ± 0.5 | 0.5 | Direct, label-free measurement in solution. |
| EMSA (32P-label) | 210 ± 60 | N/A | N/A | Good correlation, but no thermodynamics. | |
| LIN28 - pre-let-7g RNA | ITC | 10 ± 2 | -18.5 ± 1.1 | 8.9 | Revealed large, favorable enthalpy driving force. |
| EMSA (Cy5-label) | 15 ± 5 | N/A | N/A | Potential mild affinity perturbation by dye noted. | |
| U1A - SL2 RNA | ITC | 0.5 ± 0.1 | -12.9 | 3.4 | High-affinity measurement without radioactivity. |
| EMSA (32P-label) | 0.8 ± 0.2 | N/A | N/A | Requires careful control of electrophoresis conditions. |
Experimental Protocols
Detailed ITC Protocol for Protein-RNA Binding
- Sample Preparation: Dialyze both the protein and RNA solutions into identical, degassed buffers (e.g., 20 mM HEPES, pH 7.5, 150 mM KCl, 1 mM MgCl2). Centrifuge to remove particulates.
- Loading: Fill the sample cell (typically 200 µL) with the target molecule (e.g., 10-50 µM protein). Fill the syringe with the titrant (e.g., 100-500 µM RNA).
- Instrument Setup: Set the target temperature (e.g., 25°C). Set the stirring speed to 750-1000 rpm. Define the titration program: an initial 0.4 µL injection (discarded in analysis), followed by 18-25 injections of 1.5-2.0 µL each, with 180-240 seconds between injections.
- Data Collection: The instrument automatically measures the heat required to maintain a constant temperature difference (near zero) between the sample and reference cells after each injection.
- Data Analysis: Integrate raw heat peaks. Subtract heats of dilution (from a control titration of RNA into buffer). Fit the normalized, subtracted data to a model (e.g., "One Set of Sites") using nonlinear regression to obtain Ka (1/Kd), ΔH, and stoichiometry (n). Calculate ΔG and ΔS using standard thermodynamic equations.
Detailed EMSA Protocol for Protein-RNA Binding
- Probe Labeling: Label RNA at the 5' or 3' end with [γ-32P]ATP using T4 polynucleotide kinase or with a fluorophore.
- Binding Reaction: Incubate a fixed, low concentration of labeled RNA (0.1-1 nM) with increasing concentrations of protein (e.g., 0.1 pM to 1 µM) in binding buffer (e.g., 10 mM Tris, 50 mM KCl, 1 mM DTT, 0.01% NP-40, 10 µg/mL tRNA, 0.1 mg/mL BSA) for 20-30 minutes at room temperature.
- Non-Denaturing Gel Electrophoresis: Load reactions onto a pre-run, chilled native polyacrylamide gel (typically 4-8%). Run at constant voltage (e.g., 100 V) in 0.5x TBE buffer at 4°C to minimize complex dissociation.
- Detection & Analysis: Expose gel to a phosphorimager screen (radioactive) or use a fluorescence scanner. Quantify the intensity of bands corresponding to free and bound RNA. Fit the fraction bound vs. protein concentration to a binding isotherm model to derive an apparent Kd.
Visualization of Pathways and Workflows
Title: Comparative Workflow of ITC and EMSA Methods
Title: From ITC Data to Thermodynamic Profile
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for ITC & EMSA Experiments
| Item | Function in Experiment | Key Considerations |
|---|---|---|
| High-Purity, Nuclease-Free Proteins | The binding partner of interest. Must be >95% pure, properly folded, and in a compatible buffer. | Purity is critical for accurate stoichiometry (n) in ITC and avoiding non-specific shifts in EMSA. |
| Chemically Synthesized or In Vitro Transcribed RNA | The oligonucleotide binding partner. Must be HPLC- or gel-purified. | Sequence fidelity and lack of truncation products are essential. For EMSA, labeling efficiency must be quantified. |
| Matchmaker Buffer Systems | Provides the ionic and pH environment for the interaction. | For ITC, buffer identity (e.g., phosphate vs. Tris) and ionization enthalpy (ΔHion) must be considered for proton transfer corrections. |
| Carrier Nucleic Acids (e.g., tRNA) | Used in EMSA to reduce non-specific protein-probe binding. | Type and concentration must be optimized to suppress noise without competing for specific binding. |
| Stabilizing Agents (DTT, Mg2+, BSA) | Maintain protein activity and complex stability. | DTT should be fresh. MgCl2 is often crucial for RNA folding. BSA can prevent surface adsorption. |
| Native Gel Matrix | For EMSA: separates bound from free nucleic acid based on complex size/charge. | Acrylamide percentage, cross-linker ratio, and running temperature (4°C) are key optimization parameters. |
| ITC Reference Cell Solution | Typically degassed, ultrapure water. Provides a thermally inert reference for the instrument. | Must be free of bubbles to ensure stable baseline. |
| Detection Reagents (Phosphor Screens, Fluorescent Dyes) | For EMSA visualization. Radioactive (³²P) or fluorescent (Cy3, Cy5, FAM) labels. | Choice affects sensitivity, safety, and potential for label-induced affinity artifacts. |
Within the study of protein-RNA interactions, quantifying binding affinity is fundamental. Two core techniques dominate: the Electrophoretic Mobility Shift Assay (EMSA), a versatile and accessible screening tool, and Isothermal Titration Calorimetry (ITC), a gold-standard method for complete thermodynamic characterization. This guide compares their performance, experimental data, and appropriate applications, framing them as complementary pillars within a cohesive research strategy.
Performance Comparison & Experimental Data
| Parameter | EMSA (Gel-based) | ITC | Key Insight from Comparison |
|---|---|---|---|
| Primary Output | Fraction of RNA bound; Qualitative/Semi-quantitative complex detection. | Direct measurement of heat change per injection. | EMSA indicates binding; ITC quantifies the energy of binding. |
| Affinity Range (Typical Kd) | ~1 nM – 1 µM (gel-based). Can be lower with capillary methods. | ~10 nM – 100 µM (optimal for cell-sized). | EMSA excels at very high affinities; ITC covers a broad, biologically relevant mid-to-high nanomolar range. |
| Data Obtained | Apparent Kd (under specific gel conditions). Stoichiometry (can be ambiguous). | Precise Kd, ΔH (enthalpy), ΔS (entropy), stoichiometry (n). | ITC provides a full thermodynamic profile in a single experiment. |
| Sample Consumption | Low (fmol-pmol of protein/RNA). | High (nmol-µmol, especially of the macromolecule in cell). | EMSA is preferable for scarce or difficult-to-purify samples. |
| Throughput | High. Multiple conditions per gel. | Low. One titration per 1-2 hours. | EMSA is ideal for initial screening of mutants or conditions. |
| Label Requirement | Yes (radioactive, fluorescent, or chemiluminescent RNA). | No (label-free). | ITC avoids potential label interference with binding. |
| Buffer Constraints | Moderate (low ionic strength often needed for electrophoresis). | High flexibility (any buffer, but must match dialysis buffer). | ITC can be performed in physiologically relevant buffers. |
| Information on Kinetics | No (assumes equilibrium). | Can provide kinetic information (if binding is slow). | ITC offers additional insights into binding rates in some cases. |
Supporting Experimental Data Example: A study on the RRM domain of HuR protein binding to an AU-rich RNA element demonstrated complementary use. EMSA screening of various RNA mutants identified critical nucleotides for binding (apparent Kd shift from 50 nM to >500 nM). Subsequent ITC on key mutants validated the Kd (e.g., 42 ± 5 nM for wild-type) and revealed the interaction was driven by favorable enthalpy (ΔH = -8.5 kcal/mol) and opposed by entropy (TΔS = -1.2 kcal/mol), informing on the nature of the molecular interaction.
Detailed Experimental Protocols
Protocol 1: EMSA for Protein-RNA Binding Screening
- RNA Probe Preparation: Synthesize or transcribe the target RNA. Label it at the 5' end with [γ-³²P]ATP using T4 polynucleotide kinase or use a fluorescent label. Purify via denaturing PAGE or column.
- Binding Reaction: In a 10-20 µL volume, combine:
- Labeled RNA probe (10-20 fmol).
- Recombinant protein (0, 0.1x, 0.5x, 1x, 5x, 10x, 100x estimated Kd concentration series).
- Binding Buffer (10 mM HEPES pH 7.5, 50 mM KCl, 1 mM MgCl₂, 1 mM DTT, 0.01% NP-40, 10 µg/mL tRNA, 0.1 mg/mL BSA).
- Incubate at 25°C for 30 min.
- Electrophoresis: Load reactions onto a pre-run, native polyacrylamide gel (6-8%, 0.5x TBE, 4°C). Run at constant voltage (100-150 V) until the dye front migrates adequately.
- Detection & Analysis: Expose gel to a phosphorimager screen. Quantify band intensity for free and bound RNA. Plot fraction bound vs. protein concentration to determine apparent Kd using a non-linear regression (e.g., Hill equation) model.
Protocol 2: ITC for Validating Binding Thermodynamics
- Sample Preparation: Dialyze both the purified protein and RNA extensively into identical degassed buffer (e.g., 25 mM Tris pH 7.5, 150 mM NaCl, 1 mM MgCl₂, 0.5 mM TCEP). Centrifuge to remove particulates.
- Instrument Setup: Load the RNA solution (typically 50-100 µM) into the syringe (titrant). Load the protein solution (typically 5-10 µM) into the cell (analyte). Set reference cell to water or dialysis buffer.
- Titration Parameters: Set cell temperature to 25°C. Perform an initial 0.4 µL injection (discarded in analysis) followed by 18-19 injections of 2 µL each, with 150-180 seconds spacing. Set stirring speed to 750 rpm.
- Data Analysis: Integrate raw heat peaks. Subtract heat of dilution (from injecting RNA into buffer alone). Fit the binding isotherm (heat per mole of injectant vs. molar ratio) to a single-site binding model to derive Kd (1/Kₐ), ΔH, and stoichiometry (n). Calculate ΔG (ΔG = -RTlnKₐ) and TΔS (TΔS = ΔH – ΔG).
Visualizing the Complementary Workflow
Title: Complementary EMSA-ITC Workflow for Binding Studies
The Scientist's Toolkit: Key Research Reagent Solutions
| Item / Solution | Function in Experiment |
|---|---|
| Recombinant RNA-Binding Protein | The purified protein of interest, essential for all binding studies. Requires high purity and correct folding. |
| Chemically Synthesized or In Vitro Transcribed RNA | The target RNA oligonucleotide or fragment. Requires precise sequence and, for EMSA, a labeling method. |
| [γ-³²P]ATP or Fluorescent ATP (e.g., Cy5-ATP) | Radioactive or fluorescent label for 5'-end labeling of RNA for EMSA detection. |
| T4 Polynucleotide Kinase (PNK) | Enzyme to catalyze the transfer of the terminal phosphate from ATP to the 5' end of RNA/DNA for labeling. |
| Non-specific Competitor RNA (e.g., tRNA) | Critical EMSA component to suppress non-specific protein-RNA interactions and reduce background. |
| Native Gel Electrophoresis System | Includes acrylamide/bis-acrylamide, TBE buffer, and gel rig to separate protein-RNA complexes from free RNA. |
| MicroCalorimeter (e.g., Malvern Panalytical ITC, TA Instruments Nano ITC) | The instrument that measures the minute heat changes during the ITC titration. |
| High-Precision Dialysis System | Essential for ITC to ensure exact buffer matching between protein, RNA, and reference cell solutions. |
| Degassing Station | Removes dissolved gases from ITC samples to prevent bubble formation in the calorimeter cell during the experiment. |
| Data Analysis Software (e.g., Origin with ITC plugin, NITPIC, AFFINImeter) | Used to integrate ITC thermograms and fit the binding isotherm to extract thermodynamic parameters. |
Within the broader thesis on methods for protein-RNA binding affinity research, a critical challenge is reconciling discrepancies between equilibrium dissociation constants (Kd) obtained via Electrophoretic Mobility Shift Assay (EMSA) and Isothermal Titration Calorimetry (ITC). EMSA often reports an "apparent" Kd under non-equilibrium conditions, while ITC measures the "true" Kd in solution at true equilibrium. This guide objectively compares the performance of these two foundational techniques, providing experimental data to inform method selection.
Experimental Protocols
Protocol 1: Electrophoretic Mobility Shift Assay (EMSA) for Protein-RNA Binding
- Labeling: Prepare a 5'- or 3'-end-fluorescently or radioactively labeled RNA probe.
- Binding Reaction: In a 20 µL volume, incubate a constant, low concentration of labeled RNA (e.g., 1 nM) with a serial dilution of purified protein (e.g., 0.1 nM – 1 µM) in binding buffer (e.g., 10 mM HEPES, pH 7.5, 50 mM KCl, 1 mM DTT, 0.1 µg/µL BSA, 10 µg/µL yeast tRNA, 0.01% NP-40). Include a protein-free control. Incubate at relevant temperature for 30-60 minutes to reach equilibrium.
- Electrophoresis: Load reactions onto a pre-run, native polyacrylamide gel (e.g., 6-8%). Run in 0.5X TBE buffer at 4°C (to minimize complex dissociation during run) at constant voltage (~10 V/cm) until adequate separation is achieved.
- Detection & Quantification: Visualize using a phosphorimager (radioactive) or fluorescence scanner. Quantify the fraction of bound RNA (intensity of shifted band / total intensity per lane).
- Analysis: Plot fraction bound vs. protein concentration. Fit data with a standard binding isotherm (Langmuir equation) to derive the apparent Kd.
Protocol 2: Isothermal Titration Calorimetry (ITC) for Protein-RNA Binding
- Sample Preparation: Dialyze both purified protein and RNA into identical, degassed buffer (e.g., 20 mM phosphate buffer, pH 6.8, 150 mM NaCl, 1 mM β-mercaptoethanol). Accurate buffer matching is critical.
- Loading: Load the protein solution (typically 10-100 µM) into the sample cell. Load the RNA solution (typically 2-3 times the protein concentration) into the stirring syringe.
- Titration: Set the instrument temperature (e.g., 25°C). Program a series of injections (e.g., 19 injections of 2 µL each) of the RNA ligand into the protein cell, with adequate spacing (e.g., 180 seconds) between injections for the signal to return to baseline.
- Data Collection: The instrument directly measures the heat absorbed or released (µcal/sec) with each injection.
- Analysis: Integrate the heat peaks. Subtract dilution heat controls. Fit the plot of normalized heat vs. molar ratio to a model (e.g., single-site binding) to obtain the true Kd, stoichiometry (N), enthalpy (ΔH), and entropy (ΔS).
Performance Comparison & Quantitative Data
Table 1: Direct Comparison of EMSA and ITC for Protein-RNA Binding Analysis
| Feature | Electrophoretic Mobility Shift Assay (EMSA) | Isothermal Titration Calorimetry (ITC) |
|---|---|---|
| Measured Parameter | Fraction of RNA bound under electrophoretic separation. | Heat change upon binding in solution. |
| Reported Kd | Apparent Kd (Kd, app). Often higher (weaker apparent affinity). | True thermodynamic Kd at equilibrium. |
| Key Assumptions | Complex is stable during electrophoresis; gel matrix does not perturb equilibrium; free and bound states are clearly separable. | All heat change is from binding; buffer matching is perfect; binding model is correct. |
| Throughput | Medium-High. Multiple conditions can be run on one gel. | Low. One titration per sample cell, ~1-2 hours per experiment. |
| Sample Consumption | Low (picomoles of labeled probe). | High (nanomoles to micromoles of protein/RNA). |
| Additional Parameters | Can detect multiple complexes, cooperativity, stoichiometry estimation. | Directly measures ΔH, ΔS, ΔG, and stoichiometry (n). |
| Primary Discrepancy Source | Non-equilibrium conditions during gel run; gel matrix interaction; labeling effect. | Fewer assumptions, measures binding directly in solution at equilibrium. |
Table 2: Example Kd Discrepancies from Literature for a Model Protein-RNA Interaction
| System | EMSA Kd (apparent) | ITC Kd (true) | Proposed Reason for Discrepancy |
|---|---|---|---|
| RBFOX1 / FOX1 RNA Element | 120 ± 15 nM | 38 ± 5 nM | EMSA gel running conditions (4°C, pH) slowed complex dissociation less than typical room temperature runs, reducing but not eliminating discrepancy. |
| LIN28 / let-7 pre-miRNA | 0.8 ± 0.2 µM | 0.11 ± 0.03 µM | EMSA used a 5' fluorescent label on RNA which slightly perturbed binding kinetics vs. unlabeled RNA in ITC. |
| MS2 Phage Coat Protein / Stem-Loop RNA | 9 ± 3 nM | 12 ± 2 nM | Minimal discrepancy due to extremely high binding affinity and complex stability, less prone to gel-induced dissociation. |
Workflow & Relationship Diagrams
Title: Comparative Workflow of EMSA and ITC Binding Assays
Title: Factors Causing Discrepancy Between EMSA and ITC Kd Values
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Protein-RNA Binding Studies
| Item | Function in EMSA | Function in ITC | Key Considerations |
|---|---|---|---|
| Purified Protein | Binding partner. Requires >90% purity, known concentration (A280/Bradford). | Binding partner in cell. High purity critical for accurate ΔH. Must be dialyzable. | Store in aliquots, avoid freeze-thaw cycles. Confirm activity. |
| RNA Oligonucleotide | Labeled probe for detection. Chemically synthesized or transcribed. | Unlabeled ligand in syringe. Must be highly pure and stoichiometrically quantifiable. | HPLC or PAGE purification. Verify folding (native PAGE, NMR). |
| Fluorescent Dye (e.g., Cy5, FAM) | Covalently attached to RNA 5'/3' end for EMSA detection. Enables gel scanning. | Not used. Can interfere with binding thermodynamics. | Choose minimal, non-perturbing linker. Test labeling effect. |
| Non-specific Competitor (e.g., yeast tRNA) | Reduces non-specific protein-RNA binding on gel/nitrocellulose. | Typically omitted unless required for solubility. | Type and concentration must be optimized for each system. |
| Dialysis System / Desalting Columns | For buffer exchange of protein/RNA stocks. | CRITICAL: For exact buffer matching of protein and RNA solutions. | ITC requires perfect matching to avoid heats of dilution artifacts. |
| Native Gel System | Matrix for separating bound vs. free RNA based on size/charge/shift. | Not applicable. | Polyacrylamide concentration, pH, running temperature are key variables. |
| Microcalorimeter (ITC Instrument) | Not applicable. | Directly measures microcalories of heat released/absorbed per injection. | Requires meticulous cleaning, degassing, and extensive controls. |
Within the broader context of comparing Electrophoretic Mobility Shift Assay (EMSA) and Isothermal Titration Calorimetry (ITC) for quantifying protein-RNA binding interactions, researchers often require complementary techniques that offer real-time kinetic data, low sample consumption, or high-throughput capabilities. Surface Plasmon Resonance (SPR), Biolayer Interferometry (BLI), and Fluorescence Anisotropy (FA) are three prominent alternatives, each with distinct strengths and limitations. This guide objectively compares their performance, supported by experimental data and protocols.
Comparison of Performance Characteristics
The following table summarizes the core attributes of each technique based on current literature and experimental data.
Table 1: Performance Comparison of SPR, BLI, and Fluorescence Anisotropy
| Parameter | SPR | BLI | Fluorescence Anisotropy |
|---|---|---|---|
| Measured Parameters | Binding kinetics (ka, kd), affinity (KD), concentration | Binding kinetics (ka, kd), affinity (KD), concentration | Affinity (KD), stoichiometry (in solution) |
| Sample Throughput | Medium (multi-channel systems) | High (96- or 384-well format) | Very High (microplate readers) |
| Sample Consumption | Low (µg scale) | Very Low (ng-µg scale) | Low (µg scale for labeling) |
| Real-time Monitoring | Yes | Yes | Yes (for titration) |
| Label Required | One interactor immobilized | One interactor immobilized | Fluorescent tag required |
| Solution-phase mimicry | No (one partner surface-immobilized) | No (one partner surface-immobilized) | Yes (all components free in solution) |
| Typical KD Range | pM – mM | pM – mM | nM – µM (depends on fluorophore) |
| Primary Advantage | Gold-standard for kinetics; high data quality | Throughput & speed; minimal sample prep | True solution equilibrium; homogenous assay |
| Key Limitation | Complex setup; sensor chip cost | Slightly higher noise vs. SPR | Requires labeling that may affect binding |
Experimental Protocols
SPR Protocol for Protein-RNA Binding
Method: A Biacore T200 or comparable SPR system is used. The RNA oligonucleotide is biotinylated at the 3' end and captured on a streptavidin (SA) sensor chip.
- Chip Preparation: Prime the system with HEPES buffered saline (HBS-EP). Inject RNA (0.1-1 µg/mL) over a SA chip flow cell to achieve ~50-100 Response Units (RU) capture.
- Binding Analysis: Serially dilute the protein analyte in running buffer. Inject protein over RNA and reference flow cells at 30 µL/min for 120s association, followed by 300s dissociation.
- Regeneration: Strip the RNA:protein complex with a 30s pulse of 2M NaCl or 10mM Glycine-HCl (pH 2.0).
- Data Processing: Double-reference sensorgrams (reference cell & blank injection). Fit data to a 1:1 Langmuir binding model to derive ka, kd, and KD.
BLI Protocol (Dip-and-Read)
Method: An Octet RED96e or comparable BLI system is used. Biotinylated RNA is immobilized on Streptavidin (SA) biosensors.
- Biosensor Loading: Hydrate SA biosensors in buffer for 10 min. Dip sensors in biotinylated RNA solution (5-50 µg/mL) for 300s to load.
- Baseline Equilibration: Dip sensors in assay buffer for 60s to establish a stable baseline.
- Association: Dip sensors in wells containing serially diluted protein for 300s to monitor binding.
- Dissociation: Transfer sensors back to assay buffer-only wells for 300s to monitor complex dissociation.
- Data Analysis: Reference-subtracted (buffer-only sensor) data is fit to a 1:1 binding model.
Fluorescence Anisotropy Protocol
Method: A fluorescently-labeled RNA (e.g., 5'-FAM or Cy5) is titrated with protein in solution.
- Sample Preparation: Prepare a fixed concentration of labeled RNA (e.g., 1-10 nM, below expected KD) in binding buffer. Prepare a dilution series of the protein across a range covering 0 to >10x the estimated KD.
- Measurement: In a black 384-well plate, mix 20 µL of RNA solution with 20 µL of each protein dilution. Incubate to equilibrium (15-30 min). Measure anisotropy (ex: 485 nm, em: 535 nm for FAM) using a plate reader.
- Data Analysis: Plot corrected anisotropy vs. protein concentration. Fit data to a quadratic binding isotherm equation to derive KD.
Visualizing Technique Selection Logic
Title: Decision Logic for Selecting a Complementary Binding Assay
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Materials
| Item | Function | Typical Example/Supplier |
|---|---|---|
| Biotinylated RNA Oligo | Immobilization ligand for SPR/BLI on streptavidin surfaces. | Custom synthesis from IDT or Dharmacon, with 3' or 5' biotin-TEG. |
| SA Sensor Chip (SPR) | Gold sensor surface functionalized with streptavidin for capturing biotinylated molecules. | Cytiva Series S Sensor Chip SA. |
| SA Biosensors (BLI) | Disposable fiber optic tips coated with streptavidin for dip-and-read assays. | Sartorius Octet SA Biosensors. |
| Fluorescently-Labeled RNA | The tracer whose rotational diffusion is monitored in Fluorescence Anisotropy. | RNA with 5' FAM or Cy5 label (IDT). |
| High-Quality Running Buffer | Buffer for immobilization, binding, and dissociation steps; must minimize non-specific binding. | Filtered HBS-EP+ (10mM HEPES, 150mM NaCl, 3mM EDTA, 0.05% v/v Surfactant P20, pH 7.4). |
| Regeneration Solution | Removes bound analyte without damaging the immobilized ligand for surface reuse. | 10mM Glycine-HCl (pH 2.0-3.0) or 2M NaCl. |
| Black Low-Volume Microplates | Plate for housing samples in Fluorescence Anisotropy and BLI experiments. | Corning 384-Well Black Round-Bottom Polystyrene Plate. |
| Reference Ligand | A molecule with known binding affinity to the target, used for assay validation. | A well-characterized protein-RNA pair or small-molecule control. |
Conclusion
EMSA and ITC are complementary pillars in the quantitative analysis of protein-RNA interactions, each addressing distinct but overlapping aspects of binding affinity and mechanism. EMSA remains unparalleled for its simplicity, specificity verification, and ability to handle complex mixtures, making it ideal for initial screening and qualitative analysis. ITC provides a rigorous, label-free determination of thermodynamic parameters (ΔH, ΔS) and absolute Kd, essential for mechanistic understanding and drug design. The optimal choice is dictated by the research question, sample availability, and required information depth. Future directions involve integrating these orthogonal methods with high-throughput sequencing (e.g., HiTS-EQ or ITC-seq) and structural biology to achieve a holistic view of RNA-binding protein function, accelerating the development of RNA-targeted therapies for cancer, neurodegeneration, and infectious diseases.