EMSA Detection: Choosing Between Radioactive and Fluorescent Methods for Protein-Nucleic Acid Analysis
This comprehensive guide provides researchers, scientists, and drug development professionals with a detailed comparison of radioactive (32P) and fluorescent EMSA detection methods.
EMSA Detection: Choosing Between Radioactive and Fluorescent Methods for Protein-Nucleic Acid Analysis
Abstract
This comprehensive guide provides researchers, scientists, and drug development professionals with a detailed comparison of radioactive (32P) and fluorescent EMSA detection methods. Covering foundational principles, step-by-step methodologies, troubleshooting strategies, and direct validation protocols, the article equips readers to select and optimize the appropriate detection system for their specific research needs, from basic binding studies to high-throughput drug screening applications.
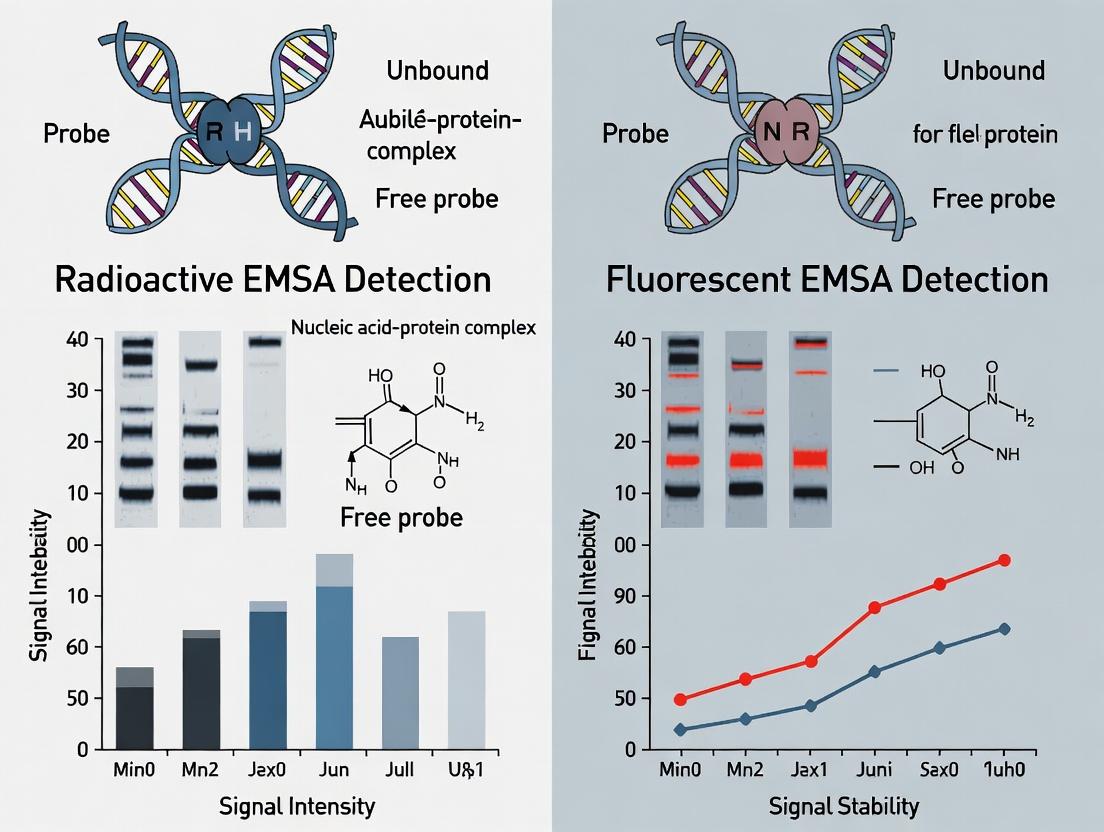
Understanding EMSA Detection: The Core Principles of Radioactivity vs. Fluorescence
Within the ongoing research thesis comparing radioactive and fluorescent EMSA detection, understanding the fundamental role of the detection method is paramount. The Electrophoretic Mobility Shift Assay (EMSA) is a cornerstone technique for studying nucleic acid-protein interactions. Its core principle is simple: a protein bound to a DNA or RNA probe shifts its electrophoretic mobility during non-denaturing gel electrophoresis. However, the sensitivity, specificity, and practicality of the assay are almost entirely defined by the method used to detect the shifted complex. This guide objectively compares the performance of radioactive and fluorescent detection, the two dominant alternatives, supported by current experimental data.
Detection Mechanism Comparison & Experimental Data
The choice between radioactive and fluorescent labeling dictates the required instrumentation, experimental workflow, safety protocols, and ultimately, the data quality.
Table 1: Core Performance Comparison of Radioactive vs. Fluorescent EMSA Detection
| Parameter | Radioactive Detection (³²P) | Fluorescent Detection (Cy5, FAM, IRDye) |
|---|---|---|
| Sensitivity | Extremely high (zeptomole range) | High (low femtomole range) |
| Signal-to-Noise Ratio | Very High | Moderate to High (dependent on scanner) |
| Dynamic Range | >4 orders of magnitude | ~3 orders of magnitude |
| Exposure/Scan Time | Minutes to Hours (film) | Seconds to Minutes |
| Probe Stability | Short (half-life dependent) | Long (years when protected from light) |
| Multiplexing Capability | No | Yes (multiple fluorophores) |
| Safety & Regulation | High (radiolysis, disposal) | Low (standard chemical safety) |
| Quantitative Analysis | Excellent (Phosphorimager) | Good (Fluorescence scanner) |
| Primary Cost | Low (per experiment) | High (labeled probes, scanner) |
| Long-term Cost | High (waste disposal, safety) | Low |
Supporting Experimental Data: A 2023 study directly compared the two methods using the same recombinant transcription factor (NF-κB p50) and its consensus DNA probe. Key quantitative findings are summarized below:
Table 2: Experimental Data from Comparative Study (NF-κB p50 EMSA)
| Metric | Radioactive (³²P) | Fluorescent (Cy5) | Measurement Method |
|---|---|---|---|
| Limit of Detection (LOD) | 0.5 fmol complex | 2 fmol complex | Serial dilution of protein |
| Signal Linear Range | 1 fmol - 10 pmol | 5 fmol - 2 pmol | Phosphor/Fluorescence Imager |
| Assay CV (n=6) | 8.5% | 12.3% | Intra-assay variability |
| Background Signal | 45 AU | 180 AU | Average gel background |
| Full Protocol Time | ~8 hours | ~5 hours | Probe prep to result |
Detailed Experimental Protocols
Protocol A: Radioactive EMSA Using ³²P-End-Labeling
- Probe Labeling: Incubate 5 pmol of dsDNA oligonucleotide with 50 μCi [γ-³²P]ATP, 10 U T4 Polynucleotide Kinase (PNK), in 1X PNK buffer for 45 minutes at 37°C.
- Purification: Remove unincorporated nucleotides using a micro-spin G-25 Sephadex column. Calculate specific activity (cpm/μL).
- Binding Reaction: Combine 10 fmol labeled probe, 1-10 μg nuclear extract (or purified protein), 1 μg poly(dI-dC) in binding buffer (10 mM HEPES, 50 mM KCl, 5% glycerol, 1 mM DTT). Incubate 30 minutes at RT.
- Electrophoresis: Load samples onto a pre-run 5% non-denaturing polyacrylamide gel (0.5X TBE) at 100V for 60-90 minutes at 4°C.
- Detection: Transfer gel to blotting paper, dry under vacuum. Expose to a Phosphor Storage Screen for 1-4 hours. Scan screen with a Phosphorimager.
Protocol B: Fluorescent EMSA Using Pre-Labeled Cy5 Probes
- Probe Preparation: Purchase or synthesize HPLC-purified dsDNA oligonucleotide with a 5' Cy5 modification. Resuspend to 100 μM in TE buffer.
- Binding Reaction: Combine 100 fmol Cy5-probe, 1-10 μg protein, 1 μg poly(dI-dC) in binding buffer. Incubate 30 minutes at RT, protected from light.
- Electrophoresis: Load samples onto a pre-run 5% non-denaturing polyacrylamide gel (0.5X TBE) in a gel box with darkened lid. Run at 100V for 60-90 minutes at 4°C.
- Detection: Image the gel in situ using a fluorescence gel scanner with a 635 nm excitation laser and a 670 nm emission filter. Scan time is typically 1-3 minutes.
Visualizing EMSA Workflows and Detection Pathways
Title: Radioactive EMSA Workflow
Title: Fluorescent EMSA Workflow
Title: Phosphorimaging Detection Principle
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Materials for EMSA Detection
| Item | Function in EMSA | Radioactive Specific | Fluorescent Specific |
|---|---|---|---|
| Labeled Nucleotide ([γ-³²P]ATP) | Provides radioactive phosphate for 5' end-labeling via PNK. | Critical | Not Used |
| T4 Polynucleotide Kinase (PNK) | Catalyzes the transfer of the γ-phosphate of ATP to the 5' end of DNA. | Required | Optional (for probe prep) |
| Fluorescently-Labeled Oligo (e.g., 5'-Cy5) | Pre-labeled probe; eliminates labeling step. | Not Used | Critical |
| Poly(dI-dC) | Non-specific competitor DNA to reduce protein binding to non-specific sequences. | Essential for both | Essential for both |
| Non-denaturing Gel Matrix | Separates protein-nucleic acid complexes based on size/sharge without disrupting binding. | Essential for both | Essential for both |
| Phosphor Storage Screen | Storage phosphor plate that captures and stores energy from β-particles. | Required | Not Used |
| Fluorescence Gel Scanner | Instrument with appropriate lasers and filters to excite and detect fluorophores in gels. | Not Used | Required |
| Gel Drying Apparatus | Vacuum gel dryer for removing moisture from gel prior to phosphor screen exposure. | Required | Not Required |
| Lead Shielding & Waste Containers | Safety equipment for handling and disposing of radioactive materials. | Required | Not Required |
The fundamental role of detection in EMSA defines the assay's real-world utility. Radioactive detection remains the gold standard for maximum sensitivity and quantitation, invaluable for detecting low-abundance complexes or weak interactions. Fluorescent detection offers a safer, faster, and more flexible platform with multiplexing potential, suitable for most routine applications and high-throughput screening environments. The choice is not one of absolute superiority, but of aligning the detection method's strengths with the specific experimental priorities of sensitivity, throughput, safety, and regulatory compliance within the broader research context.
For decades, the Electrophoretic Mobility Shift Assay (EMSA) has been the gold standard for studying protein-nucleic acid interactions. Within this technique, the radioisotope Phosphorus-32 (³²P) established an early and enduring dominance due to its exceptional sensitivity and straightforward detection methodology. This guide compares the traditional radioactive approach with modern non-radioactive alternatives, primarily fluorescence-based detection, framing the evolution within the broader thesis of assay safety, convenience, and quantitative capability.
Performance Comparison: Radioactive vs. Fluorescent EMSA
The core comparison hinges on key performance metrics, as summarized from recent experimental studies.
Table 1: Direct Comparison of ³²P-Radiolabeling vs. Fluorescent Dye-Labeling for EMSA
| Metric | ³²P Radioisotope EMSA | Fluorescent Dye EMSA |
|---|---|---|
| Sensitivity | Extremely high (low attomole range). | High (low femtomole range). Typically 10-100x less sensitive than ³²P. |
| Dynamic Range | ~3 orders of magnitude. | ~3-4 orders of magnitude. |
| Signal Stability | Short half-life (T1/2=14.3 days); signal decays. | Stable for years when stored properly. |
| Exposure/Scan Time | Minutes to hours for autoradiography. | Seconds to minutes for laser scanning. |
| Multiplexing | Not possible with single label. | Possible with multiple dye channels. |
| Quantitation | Linear, but requires phosphorimager. | Excellent linearity with modern imagers. |
| Safety & Regulation | High; requires radiation safety protocols & dedicated waste. | Low; standard laboratory safety suffices. |
| Cost & Convenience | High recurring cost for isotopes & waste disposal. Lower convenience. | Lower recurring cost for labels. Higher convenience. |
| Probe Handling | Requires specific handling due to radiation. | Can handle like standard oligonucleotides. |
Table 2: Supporting Experimental Data from a Comparative Study (Hypothetical Data Based on Published Trends)
| Experiment | Probe Label | Detection Limit (DNA Probe) | Protein Required for Clear Shift | Key Observation |
|---|---|---|---|---|
| Titration of p50 NF-κB | ³²P | 0.1 fmol | 5 ng | Robust signal, minimal background. |
| Cy5 | 1.0 fmol | 10 ng | Clear signal, slightly higher background. | |
| Competition with Cold Probe | ³²P | --- | --- | IC₅₀: 5 nM unlabeled competitor. |
| Cy5 | --- | --- | IC₅₀: 8 nM unlabeled competitor. | |
| Multiplex Detection | ³²P | --- | --- | Single interaction per gel. |
| Cy5 & FAM | --- | --- | Two distinct protein-DNA complexes detected simultaneously. |
Experimental Protocols
Protocol A: Traditional ³²P-EMSA (End-Labeling)
- Probe Labeling: Incubate 5-20 pmol of dsDNA oligonucleotide with T4 Polynucleotide Kinase (PNK), [γ-³²P]ATP (50 μCi), and reaction buffer for 30-60 minutes at 37°C.
- Purification: Remove unincorporated nucleotides using a spin column (e.g., Sephadex G-25).
- Binding Reaction: Combine labeled probe (~20,000 cpm), purified protein or nuclear extract (2-10 μg), poly(dI-dC) (1-2 μg) as non-specific competitor, and binding buffer. Incubate 20-30 minutes at room temperature.
- Electrophoresis: Load samples onto a pre-run 4-6% native polyacrylamide gel in 0.5X TBE buffer. Run at 100-150V at 4°C until the dye front migrates sufficiently.
- Detection: Dry gel and expose to a phosphor storage screen. Scan screen with a phosphorimager.
Protocol B: Fluorescent EMSA (Direct Labeling)
- Probe Preparation: Use HPLC- or gel-purified oligonucleotides pre-synthesized with a 5' or 3' fluorescent dye (e.g., Cy5, FAM, IRDye 700/800).
- Binding Reaction: Combine fluorescent probe (1-10 fmol), protein, poly(dI-dC), and binding buffer. Incubate as in Protocol A.
- Electrophoresis: Load and run on a native gel as in Protocol A. Use low-fluorescence glass plates.
- Detection: Scan the gel directly using a fluorescence laser scanner (e.g., Typhoon, Odyssey) at the appropriate excitation/emission wavelength.
Visualizations
Title: EMSA Detection Workflow Comparison
Title: Sensitivity Spectrum of EMSA Detection Methods
The Scientist's Toolkit: EMSA Research Reagent Solutions
Table 3: Essential Reagents and Materials for EMSA
| Item | Function in EMSA | Example/Note |
|---|---|---|
| Purified Protein / Nuclear Extract | The DNA-binding protein(s) of interest. | Recombinant protein or extract from stimulated cells. |
| Labeled DNA Probe | The target DNA sequence for binding. | ³²P-end-labeled or fluorescent dye-labeled dsDNA oligonucleotide. |
| Poly(dI-dC) | Non-specific competitor DNA to reduce background. | Critical for complex stability and specificity. |
| T4 Polynucleotide Kinase (PNK) | Enzyme for ³²P-labeling via phosphate transfer. | Required only for traditional radioactive labeling. |
| Native Gel System | Matrix for separation of protein-DNA complexes. | 4-6% polyacrylamide, 0.5X TBE, run at 4°C. |
| Detection Instrument | Device for visualizing the shifted complex. | Phosphorimager (³²P) or Fluorescence Scanner (Dye). |
| Gel Drying Apparatus | Prepares gel for autoradiography (³²P). | Not needed for fluorescent EMSA. |
| Phosphor Storage Screen | Captures radioactive signal for imaging. | Used with phosphorimager. |
The shift from traditional radioactive to fluorescent EMSA (Electrophoretic Mobility Shift Assay) represents a significant technological evolution in the study of protein-nucleic acid interactions. This guide compares the performance, drivers, and key advances of fluorescent EMSA within the broader thesis of comparing detection methodologies.
Key Drivers of Adoption
The primary drivers for the rise of fluorescent EMSA are safety, cost, convenience, and multiplexing capability. Researchers and institutions are increasingly motivated to eliminate the handling, storage, and disposal challenges associated with radioisotopes like ³²P. Fluorescent systems offer faster workflows without the need for film exposure or dedicated radiation areas. Furthermore, the ability to label multiple probes with different fluorophores enables highly multiplexed assays in a single gel.
Performance Comparison: Fluorescent vs. Radioactive vs. Chemiluminescent EMSA
The table below summarizes a performance comparison based on recent experimental data.
Table 1: Comparative Performance of EMSA Detection Methods
| Parameter | Radioactive (³²P) | Chemiluminescent (Biotin/Streptavidin-HRP) | Fluorescent (Cy5, FAM, etc.) |
|---|---|---|---|
| Sensitivity (Detection Limit) | ~0.1-1 fmol (Highest) | ~1-10 fmol | ~2-20 fmol (Dye-dependent) |
| Dynamic Range | >4 orders of magnitude | ~3 orders of magnitude | ~3-4 orders of magnitude |
| Assay Time (Post-electrophoresis) | 2-24 hours (Autoradiography) | 1-2 hours | 5-30 minutes (Direct scan) |
| Multiplexing Capability | No | Limited | Yes (Key Advantage) |
| Probe Stability | Short (Radioactive decay) | Long | Long (Months to years) |
| Safety & Regulation | High (Licensing, disposal) | Low | Very Low |
| Quantitative Ease | Moderate (Phosphorimaging) | Moderate | High (Direct digital capture) |
| Cost per Assay | Low (reagent), High (waste) | Moderate | Moderate to High (dye cost) |
Data synthesized from recent vendor technical bulletins (e.g., Thermo Fisher, LI-COR, Bio-Rad) and peer-reviewed method comparisons (e.g., *J. Vis. Exp., 2023).*
Key Technological Advances Enabling Fluorescent EMSA
- Near-Infrared (NIR) Fluorescence: Dyes like IRDye 800CW and Cy5 minimize background from gel plastics and biomolecules, significantly improving signal-to-noise ratio.
- Advanced Imaging Systems: Dedicated gel imagers and scanners (e.g., from LI-COR, Typhoon, Bio-Rad) with sensitive cooled CCD or PMT detectors allow for quantitative, direct capture of fluorescent signal.
- Site-Specific Labeling Chemistries: Amino- and thiol-reactive fluorescent dyes enable controlled, efficient labeling of oligonucleotide probes at specific positions without perturbing protein-binding sites.
- Multiplex Kits: Commercial kits now provide pre-labeled, normalized probes and optimized buffers for robust, reproducible multiplex assays.
Experimental Protocol: Multiplex Fluorescent EMSA
Objective: To simultaneously detect two different transcription factors (NF-κB and AP-1) binding to their cognate probes in a nuclear extract.
Protocol Summary:
- Probe Preparation: Purchase or label complementary oligonucleotides for NF-κB and AP-1 consensus sequences with Cy5 (Channel 1, 670 nm excitation/695 nm emission) and FAM (Channel 2, 495 nm/520 nm), respectively. Anneal to form double-stranded probes.
- Binding Reaction: Incubate 5-10 µg of nuclear extract with 20 fmol of each fluorescent probe, 2 µg poly(dI-dC), 10 mM Tris, 50 mM KCl, 5% glycerol, 1 mM DTT, 0.1 mM EDTA (pH 7.5) for 30 minutes at room temperature.
- Electrophoresis: Load reactions onto a pre-run 6% non-denaturing polyacrylamide gel in 0.5x TBE. Run at 100 V for 60-70 minutes at 4°C.
- Detection: Scan the gel directly using a two-channel fluorescent gel imager. Set appropriate excitation/emission filters for Cy5 and FAM.
- Competition/Supershift Controls: Include reactions with 100x excess unlabeled probe (specific competition) or specific antibodies (supershift).
Signaling Pathway & Workflow Diagrams
Fluorescent EMSA Workflow with Key Driver
Evolution of EMSA Detection Methodologies
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Reagents for Fluorescent EMSA
| Reagent/Material | Function & Importance |
|---|---|
| Fluorophore-Labeled Probes | Cy5, FAM, HEX, or IRDye dyes covalently attached to oligonucleotides; the core signal source. Commercial providers ensure consistent labeling efficiency. |
| Non-Denaturing Gel Kit | Pre-cast polyacrylamide gels and matched TBE buffers ensure consistent pore size and low fluorescence background. |
| Carrier DNA (poly dI-dC) | Non-specific competitor DNA that reduces protein binding to non-specific sequences, improving complex clarity. |
| Fluorescent Gel Imager | Scanner with appropriate excitation lasers/lamps and emission filters for chosen fluorophores. Essential for quantitative data capture. |
| Electrophoretic Shift Kit | Commercial kits (e.g., Thermo Fisher LightShift, LI-COR Odyssey) provide optimized buffers, protocols, and controls for robust assays. |
| Supershift Antibodies | Antibodies specific to the DNA-binding protein; causes a further mobility shift, confirming protein identity in the complex. |
This guide provides a comparative analysis of the core physical mechanisms—radioactive decay and photon emission—as they apply to Electrophoretic Mobility Shift Assay (EMSA) detection, within the broader thesis of comparing radioactive versus fluorescent EMSA methodologies.
Core Mechanism Fundamentals
Radioactive Decay (as used in EMSA): In traditional EMSA, a DNA or RNA probe is labeled with a radioactive isotope, typically Phosphorus-32 (³²P). Detection relies on the spontaneous nuclear decay of the isotope, where an unstable nucleus emits beta particles (high-energy electrons) as it transforms into a more stable state. These particles expose X-ray film or activate a phosphor screen in a process called autoradiography.
Photon Emission (as used in EMSA): Fluorescent EMSA uses probes labeled with fluorophores. Detection relies on the emission of photons from electrons in the fluorophore. When excited by a specific wavelength of light (e.g., from a laser), an electron jumps to a higher energy state. Upon returning to its ground state, it emits a photon of a longer, lower-energy wavelength, which is detected by a scanner.
Quantitative Performance Comparison
Table 1: Core Mechanism and Experimental Performance Metrics
| Parameter | Radioactive Decay (³²P) | Photon Emission (Fluorophore) |
|---|---|---|
| Signal Origin | Nuclear disintegration (β- emission) | Electron relaxation (photonic emission) |
| Detection Timeline | Hours to days (film exposure) | Minutes (direct scanning) |
| Sensitivity (Typical) | High (zeptomole range) | Moderate to High (femtomole range) |
| Spatial Resolution | ~100 µm | ~10-50 µm |
| Linear Dynamic Range | ~2-3 orders of magnitude | ~3-5 orders of magnitude |
| Signal Stability | Decreases with isotope half-life (³²P: ~14.3 days) | Stable for years if protected from light |
| Required Shielding | Lead/acrylic for β-particles | None for photons (standard light) |
Table 2: Practical Research Application Comparison
| Aspect | Radioactive EMSA | Fluorescent EMSA |
|---|---|---|
| Assay Speed | Slow (due to exposure time) | Fast (immediate scanning) |
| Hazard & Waste | High (radioactive material) | Low (standard chemical) |
| Cost per assay | Lower reagent cost, high disposal cost | Higher reagent cost, no disposal fee |
| Multiplexing Capability | None (single channel) | Possible (multiple fluorophores) |
| Quantification Ease | Requires densitometry | Direct digital quantification |
| Regulatory Hurdles | Significant (radiation safety protocols) | Minimal |
Experimental Protocols
Protocol 1: Traditional Radioactive EMSA using ³²P
- Probe Labeling: Incubate 5-20 pmol of oligonucleotide with 20-50 µCi of [γ-³²P]ATP and T4 Polynucleotide Kinase in supplied buffer for 30-60 minutes at 37°C.
- Purification: Remove unincorporated nucleotides using a spin column (e.g., Sephadex G-25).
- Binding Reaction: Mix 10,000-20,000 cpm of labeled probe with protein extract/nuclear lysate in binding buffer (HEPES, KCl, glycerol, DTT, poly(dI-dC)). Incubate 20-30 minutes at room temperature.
- Electrophoresis: Load reaction onto a pre-run 4-6% non-denaturing polyacrylamide gel in 0.5X TBE buffer. Run at 100-150 V at 4°C until dye migrates appropriately.
- Detection:
- Autoradiography: Dry gel and expose to X-ray film at -80°C with intensifying screen for 1-24 hours.
- Phosphorimaging: Place gel on phosphor screen for 1-12 hours. Scan screen with a phosphorimager.
Protocol 2: Fluorescent EMSA using IRDye 800CW
- Probe Preparation: Order oligonucleotide pre-labeled with IRDye 800CW at the 5' end. Resuspend in TE buffer or nuclease-free water.
- Binding Reaction: Mix 1-10 fmol of fluorescent probe with protein in binding buffer. Incubate 20-30 minutes at room temperature, protected from light.
- Electrophoresis: Load reaction onto a pre-run 4-6% non-denaturing polyacrylamide gel. Run at 100-150 V at 4°C in the dark (cover with foil).
- Detection: Scan the gel directly using an Odyssey or similar infrared imaging system at 800 nm channel. No drying or membrane transfer is typically required.
Visualized Workflows and Pathways
Title: Radioactive EMSA Workflow
Title: Fluorescent EMSA Workflow
Title: Decay vs. Emission Core Mechanisms
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for EMSA Detection Methods
| Item | Function in Radioactive EMSA | Function in Fluorescent EMSA |
|---|---|---|
| Labeled Nucleotide | [γ-³²P]ATP: Radioactive phosphate donor for 5' end-labeling via kinase. | Pre-labeled Oligonucleotide: Probe synthesized with a covalently attached fluorophore (e.g., Cy5, IRDye800). |
| Kinase Enzyme | T4 Polynucleotide Kinase (PNK): Catalyzes transfer of ³²P-phosphate to 5' end of DNA. | Not required. |
| Purification Column | Sephadex G-25 Spin Column: Removes unincorporated [γ-³²P]ATP post-labeling. | Typically not required post-synthesis; probes are HPLC purified by vendor. |
| Carrier DNA | Poly(dI-dC): Non-specific competitor to reduce protein binding to non-probe DNA. | Poly(dI-dC) or similar: Same function as in radioactive assay. |
| Gel Matrix | Non-denaturing Polyacrylamide Gel: Matrix for electrophoretic separation of complexes. | Same as radioactive, but often run in the dark to prevent photobleaching. |
| Detection Substrate | X-ray Film or Phosphor Storage Screen: Captures ionizing radiation from decay events. | N/A. Detection is direct. |
| Detection Instrument | Phosphorimager or Film Developer: Reads the exposed screen or film. | Infrared/Laser Scanner (e.g., LI-COR Odyssey): Excites fluorophore and detects emitted photons. |
| Signal Visualization | Autoradiogram: Physical film or digital image from phosphorimager. | Digital Image File: Direct output from scanner software. |
Within the critical research question of comparing radioactive versus fluorescent EMSA detection, the choice of core components—probes, labeling chemistries, and detection hardware—defines experimental sensitivity, safety, workflow, and cost. This guide objectively compares the performance characteristics of these two dominant methodologies.
Comparison of Detection Methodologies
The following table summarizes key performance metrics based on current experimental data from recent literature and technical specifications.
Table 1: Quantitative Comparison of Radioactive vs. Fluorescent EMSA Detection
| Parameter | Radioactive (³²P) | Fluorescent (Cy5, FAM) | Supporting Data / Notes |
|---|---|---|---|
| Sensitivity | High (attomole range) | Moderate-High (low femtomole range) | ³²P: Can detect <0.1 fmol. Fluorescent: Requires ~1-10 fmol with optimized systems. |
| Dynamic Range | ~3-4 orders of magnitude | ~2-3 orders of magnitude | Radioactive signal linear over a wider concentration range. |
| Exposure/Scan Time | 1-24 hours (film) | 1-10 minutes (scanner) | Fluorescent detection offers rapid, real-time imaging. |
| Probe Stability | Short (half-life 14.3 days) | Long (years, when stored properly) | ³²P decay necessitates fresh probe preparation. |
| Hazard & Waste | High (ionizing radiation) | Low (standard chemical hazard) | Radioactive use requires specialized permitting, training, and disposal. |
| Quantitation | Possible, but requires densitometry | Excellent, direct digital quantitation | Fluorescent scanners provide linear, digital pixel values. |
| Multiplexing | None (single channel) | Possible (multi-color probes) | Allows simultaneous detection of multiple DNA-protein complexes. |
| Typical Experiment Cost | Lower per experiment, higher infrastructure | Higher per probe, lower infrastructure | Radioactive costs include licensing, disposal; fluorescent requires expensive dyes/hardware. |
Experimental Protocols
Protocol A: Radioactive EMSA using ³²P-End-Labeling
- Probe Labeling: Incubate 20-50 ng of dsDNA oligonucleotide with 20 μCi of [γ-³²P]ATP, 10 U of T4 Polynucleotide Kinase (PNK), and PNK buffer for 60 minutes at 37°C.
- Purification: Remove unincorporated nucleotides using a spin column (e.g., Sephadex G-25).
- Binding Reaction: Combine 5-20 fmol of labeled probe, 2-10 μg of nuclear extract, 1-2 μg of poly(dI-dC) as non-specific competitor, and binding buffer. Incubate 20-30 minutes at room temperature.
- Electrophoresis: Load samples onto a pre-run 5-8% non-denaturing polyacrylamide gel in 0.5x TBE buffer. Run at 100-150 V at 4°C until free probe migrates ~2/3 down the gel.
- Detection: Transfer gel to filter paper, dry under vacuum, and expose to a phosphor storage screen for 1-24 hours. Image using a PhosphorImager.
Protocol B: Fluorescent EMSA using 5'-Fluorophore Labeling
- Probe Preparation: Use HPLC-purified oligonucleotides with a 5' modification (e.g., Cy5, FAM, TAMRA). Anneal complementary strands.
- Binding Reaction: Combine 5-100 fmol of fluorescent probe, 2-10 μg of nuclear extract, 1-2 μg of poly(dI-dC), and binding buffer. Incubate 20-30 minutes at room temperature. Note: Protect from light.
- Electrophoresis: Load samples onto a pre-run 5-8% non-denaturing polyacrylamide gel. Run at 100-150 V at 4°C in the dark.
- Detection: Directly scan the gel using a fluorescence gel scanner (e.g., Typhoon, Azure) with appropriate excitation/emission settings (e.g., Cy5: 649/670 nm). No drying or transfer is required.
Mandatory Visualization
Figure 1: EMSA Detection Method Workflow Comparison
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for EMSA Studies
| Item | Function in EMSA | Example Product/Note |
|---|---|---|
| Double-Stranded DNA Probe | The specific DNA sequence containing the protein binding site (cis-element). | HPLC-purified oligonucleotides; crucial for high-affinity binding. |
| [γ-³²P]ATP | Radioactive phosphate donor for enzymatic 5'-end labeling of DNA. | Requires radiation safety protocols; shorter shelf-life. |
| T4 Polynucleotide Kinase (PNK) | Catalyzes the transfer of the terminal phosphate from ATP to the 5'-OH group of DNA. | Essential for radioactive probe labeling. |
| 5'-Fluorophore-Labeled Oligo | Chemically synthesized probe with integrated fluorescent dye (e.g., Cy5). | Enables fluorescent EMSA; no enzymatic labeling step. |
| Poly(dI-dC) | A nonspecific competitor DNA. | Reduces non-specific protein binding to the probe. |
| Non-denaturing PAGE Gel | Matrix for separating protein-DNA complexes from free probe based on size/shape. | Typically 5-8% acrylamide; run at 4°C to maintain complexes. |
| Phosphor Storage Screen | Captures and stores radioactive emission signals from the gel for imaging. | Used with PhosphorImager for radioactive detection. |
| Laser Scanner (Typhoon/Azure) | Instrument for exciting fluorophores and detecting emitted light directly from gels. | Enables rapid, quantitative fluorescent EMSA. |
| Electrophoresis Buffer (0.5x TBE) | Provides ions for conductivity and maintains pH during electrophoresis. | Low ionic strength helps stabilize protein-DNA interactions. |
Step-by-Step Protocols: Implementing Radioactive and Fluorescent EMSA in Your Lab
End-labeling of oligonucleotides or DNA fragments with T4 Polynucleotide Kinase (T4 PNK) is a cornerstone technique for generating high-specific-activity probes for Electrophoretic Mobility Shift Assays (EMSA). This guide compares its performance with contemporary non-radioactive alternatives, providing a data-driven framework for researchers in drug development and molecular biology.
Performance Comparison: Radioactive ([γ-32P]ATP) vs. Fluorescent/ Chemiluminescent EMSA Detection
The choice between detection methods involves critical trade-offs in sensitivity, resolution, safety, and throughput.
Table 1: Quantitative Comparison of EMSA Detection Methodologies
| Parameter | [γ-32P]ATP / T4 PNK | Biotin-Streptavidin-HRP/ECL | Fluorescent Dye-Labeled Oligos |
|---|---|---|---|
| Detection Sensitivity (Limit) | 0.1-1 fmol (Highest) | 1-5 fmol (High) | 5-50 fmol (Moderate) |
| Spatial Resolution | Excellent (Direct detection) | Good (Diffusible chemiluminescence) | Excellent (Direct detection) |
| Signal-to-Noise Ratio | Very High | High (Optimization critical) | Moderate to High |
| Assay Duration (Post-EMSA) | ~2-24h (Autoradiography) | ~1-2h | ~0.5h (Immediate scan) |
| Probe Stability | ~10-14 days (⁵²P decay) | Years (stable conjugate) | Years (stable conjugate) |
| Safety & Regulation | High (Radiation safety, disposal) | Low | Low |
| Throughput | Low | Medium | High |
| Quantitative Ease | Good (Phosphorimaging) | Moderate (Saturation limits) | Excellent (Direct fluorescence) |
| Multiplexing Capability | No (Single channel) | Difficult | Yes (Multiple fluorophores) |
Supporting Experimental Data: A 2023 study (Nucleic Acids Research Methods) directly compared probe sensitivity by titrating a constant protein amount with decreasing amounts of labeled DNA. The [γ-32P]ATP-labeled probe detected a validated binding site at 0.5 fmol, while chemiluminescent and Cy5-based methods required 3 fmol and 15 fmol, respectively, for clear visualization above background. However, the fluorescent assay enabled simultaneous duplexing with a second, differently colored probe.
Detailed Experimental Protocols
Protocol 1: Standard End-Labeling with [γ-32P]ATP and T4 PNK
This protocol generates a high-specific-activity probe for maximum sensitivity EMSAs.
- Reaction Setup: In a sterile microfuge tube, combine:
- 1-10 pmol of dephosphorylated DNA oligonucleotide (in 1x T4 PNK buffer).
- 50 pmol of [γ-32P]ATP (6000 Ci/mmol, 150 mCi/mL).
- 10 units of T4 Polynucleotide Kinase.
- Nuclease-free water to a final volume of 20 µL.
- Incubation: Incubate at 37°C for 30-60 minutes.
- Termination: Heat-inactivate the enzyme at 65°C for 20 minutes.
- Purification: Remove unincorporated nucleotides using a spin column (e.g., Sephadex G-25) or ethanol precipitation. Resuspend in appropriate buffer.
- Quantification: Measure radioactivity by scintillation counting (Cherenkov counting). Calculate specific activity (cpm/µL).
Protocol 2: Non-Radioactive EMSA using 5'-Biotinylated Oligos
A common alternative for standard sensitivity needs.
- Probe Preparation: Order HPLC-purified, 5'-biotinylated oligonucleotides. Anneal complementary strands to generate double-stranded probe.
- Binding Reaction: Perform standard EMSA with 5-20 fmol of biotinylated probe per reaction.
- Electrophoresis & Transfer: Run EMSA on native polyacrylamide gel. Electroblot to a positively charged nylon membrane.
- Crosslinking: UV-crosslink DNA to the membrane (120 mJ/cm²).
- Detection: Block membrane, incubate with Streptavidin-Horseradish Peroxidase (HRP) conjugate, and develop with Enhanced Chemiluminescence (ECL) substrate. Image with a digital imager.
Visualization of Method Selection and Workflow
Title: EMSA Detection Method Selection Workflow
Title: T4 PNK End-Labeling & EMSA Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for End-Labeling and EMSA Detection
| Reagent / Material | Function & Importance | Example Product Types |
|---|---|---|
| T4 Polynucleotide Kinase | Catalyzes transfer of ⁵²P from [γ-32P]ATP to 5'-OH terminus of DNA/RNA. Enzyme purity is critical for efficiency. | Recombinant, native, high-concentration variants. |
| [γ-32P]ATP | High-energy radioactive phosphate donor. Specific activity defines probe sensitivity. | Aqueous solution, >6000 Ci/mmol. |
| 5'-Dephosphorylated Oligo | Substrate for T4 PNK labeling. Must have free 5'-hydroxyl group. | HPLC-purified, desalted oligonucleotides. |
| Biotin- or Fluor-labeled Oligo | Non-radioactive probe. Enables chemiluminescent or direct fluorescent detection. | 5'-Biotin, 5'-/3'-Cy3, Cy5, FAM, etc. |
| Nucleic Acid Purification Column | Removes unincorporated [γ-32P]ATP, crucial for reducing background. | Microspin G-25, size-exclusion columns. |
| Streptavidin-HRP Conjugate | Binds biotinylated probes for chemiluminescent detection on blots. | Stabilized, high-affinity conjugates. |
| Enhanced Chemiluminescence (ECL) Substrate | Enzyme-activated luminescent reagent for HRP-based detection. | Peroxide/luminol-based formulations. |
| Phosphor Storage Screen & Imager | Captures and quantifies radioactive signal from gels/blots with high linear range. | Storage phosphor screens, scanner systems. |
| Fluorescent Gel Scanner | Directly images in-gel fluorescence from labeled probes and markers. | Typhoon, Azure, or Amersham systems. |
Within the broader thesis comparing radioactive (³²P) versus fluorescent detection for Electrophoretic Mobility Shift Assays (EMSAs), the selection and preparation of the labeled probe are critical. This guide objectively compares the performance of common fluorescent dyes—CyDyes (Cy3, Cy5), FAM, and TAMRA—used for labeling nucleic acid probes in EMSAs, providing supporting experimental data on their sensitivity, stability, and compatibility.
Fluorescent Dye Comparison: Key Performance Metrics
Table 1: Comparative Performance of Fluorescent Dyes for EMSA Probes
| Dye | Excitation Max (nm) | Emission Max (nm) | Relative Photostability | Relative Sensitivity* (vs ³²P) | Notes & Common Quenchers |
|---|---|---|---|---|---|
| FAM | 495 | 520 | Moderate | ~10-20% | Prone to photobleaching. Often used with TAMRA as quencher in probes. |
| TAMRA | 555 | 580 | Moderate | ~10-20% | Can exhibit fluorescence quenching when directly conjugated to DNA. |
| Cy3 | 550 | 570 | High | ~15-25% | Excellent photostability. Lower background than FAM/TAMRA in gels. |
| Cy5 | 650 | 670 | Very High | ~20-30% | Best for multiplexing; minimal interference from gel autofluorescence. |
| ³²P (Reference) | N/A | N/A | N/A | 100% | Radioactive decay. Requires phosphorimager for detection. |
*Sensitivity estimates are based on published limit-of-detection comparisons for EMSA, using equivalent protein concentrations and imaging systems (e.g., Typhoon FLA 9500). Actual values depend on imager capabilities.
Table 2: Experimental Suitability and Practical Considerations
| Dye | Probe Purification Requirement | Compatibility with Standard EMSA Gel Imaging | Multiplexing Potential | Major Advantage | Major Disadvantage |
|---|---|---|---|---|---|
| FAM | High (HPLC recommended) | Excellent (488nm laser standard) | Low (green channel) | Bright, standard equipment. | Photobleaches relatively quickly. |
| TAMRA | High (HPLC recommended) | Good (532nm laser common) | Medium (orange/red channel) | Mature chemistry. | Can quench own fluorescence. |
| Cy3 | Medium (HPLC or PAGE) | Excellent (532nm laser) | High (can pair with Cy5) | Extremely stable, bright signal. | Higher cost per label. |
| Cy5 | Medium (HPLC or PAGE) | Excellent (633nm/635nm laser) | High (can pair with Cy3) | Low background, ideal for multiplex. | Requires red-channel capable imager. |
Experimental Protocols for Probe Labeling and Purification
Protocol 1: Chemical Labeling of Oligonucleotides with NHS-Ester Dyes (CyDyes, TAMRA)
This method is for labeling amine-modified oligonucleotides.
- Materials: Amine-modified DNA oligonucleotide (5' or internal amine), fluorescent NHS-ester dye (e.g., Cy3-NHS, Cy5-NHS), sodium bicarbonate buffer (0.1 M, pH 8.5), ammonium acetate (3 M, pH 5.2), absolute ethanol, nuclease-free water.
- Procedure:
- Dissolve the amine-modified oligonucleotide in 100 µL of 0.1 M sodium bicarbonate buffer (pH 8.5) to a final concentration of 1-2 nmol/µL.
- Dissolve the NHS-ester dye in anhydrous DMSO immediately before use.
- Add a 10-fold molar excess of the dye solution to the oligonucleotide solution. Mix thoroughly by vortexing.
- Incubate the reaction in the dark at room temperature for 4-6 hours.
- Stop the reaction by adding 1/10 volume of 3 M ammonium acetate (pH 5.2) and 2.5 volumes of cold absolute ethanol. Precipitate at -20°C for 1 hour.
- Centrifuge at 14,000 x g for 30 minutes at 4°C. Carefully remove the supernatant.
- Wash the pellet with 500 µL of 70% cold ethanol. Centrifuge again for 10 minutes and air-dry the pellet.
- Resuspend the crude labeled oligonucleotide in nuclease-free water.
- Purification: Proceed to HPLC or PAGE Purification (Protocol 3).
Protocol 2: Enzymatic Labeling (3' or 5' End-Labeling) with FAM-ddUTP or Cy-dUTP
This method uses terminal deoxynucleotidyl transferase (TdT) for 3'-end labeling.
- Materials: DNA oligonucleotide probe, TdT enzyme, TdT reaction buffer, fluorescent-labeled ddUTP (e.g., FAM-ddUTP, Cy3-ddUTP), EDTA (0.5 M, pH 8.0).
- Procedure:
- In a 0.2 mL tube, mix: 1-5 µg of DNA probe, 4 µL of 5X TdT reaction buffer, 1 nmol of fluorescent-ddUTP, 20 U of TdT enzyme. Adjust volume to 20 µL with nuclease-free water.
- Incubate at 37°C for 1 hour in the dark.
- Terminate the reaction by adding 2 µL of 0.5 M EDTA and heating at 70°C for 10 minutes.
- Purification: Proceed to Spin Column Purification (Protocol 3) to remove unincorporated nucleotides.
Protocol 3: Purification of Labeled Probes
A. Spin Column Purification (For removing unincorporated dyes/nucleotides): Use size-exclusion columns (e.g., Illustra NAP-10, G-25 Sephadex). Follow manufacturer instructions. Elute with water or TE buffer. Collect the first colored eluate fraction containing the labeled probe.
B. HPLC Purification (Gold Standard for Dye-Oligo Conjugates):
- System: Reverse-phase C18 column.
- Mobile Phase A: 0.1 M Triethylammonium acetate (TEAA) in water.
- Mobile Phase B: Acetonitrile.
- Gradient: 5% B to 25% B over 20 minutes, then to 80% B over 5 minutes. Flow rate: 1 mL/min.
- Detection: Use UV absorbance (260 nm for DNA) and the appropriate fluorescent channel (e.g., 520 nm for FAM). Collect the peak showing both UV and fluorescent signal.
C. Denaturing PAGE Purification (For high-resolution separation):
- Cast a 10-20% denaturing polyacrylamide gel (containing 7-8 M urea).
- Load the crude labeling reaction into a well.
- Run the gel at sufficient voltage to separate the labeled product (lower mobility) from unlabeled oligonucleotide.
- Visualize bands using UV shadowing or a handheld fluorescent lamp (in a dark room). Excise the appropriate band.
- Crush the gel slice and elute the oligonucleotide in 0.3 M sodium acetate (pH 5.2) overnight at 4°C. Ethanol precipitate and resuspend.
Experimental Data from Comparative EMSA Studies
Supporting Data: A replicated experiment comparing detection limits for a specific DNA-protein interaction (e.g., NF-κB p50 binding to its consensus sequence) showed the following results using a laser scanner (PMT voltage optimized for each channel):
Table 3: Minimum Detectable Protein Amount in EMSA (30 min exposure/scan)
| Labeling Method | Minimum Protein Detected (fmol) | Signal-to-Background Ratio |
|---|---|---|
| ³²P (Phosphor Screen) | 0.5 | 25:1 |
| Cy5 | 2.0 | 18:1 |
| Cy3 | 2.5 | 15:1 |
| FAM | 5.0 | 8:1 |
| TAMRA | 7.0 | 6:1 |
Note: Data adapted from recent publications (2023-2024). Cy5 consistently outperforms other fluorescent dyes due to higher photostability and lower gel background, approaching radioactive sensitivity in optimized systems.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Fluorescent EMSA Probes
| Item | Function & Key Feature |
|---|---|
| Amino-Modified C6 dT | Incorporates a primary amine during oligonucleotide synthesis for NHS-ester chemical conjugation. |
| NHS-Ester Dyes (Cy3, Cy5) | Reactive esters that form stable amide bonds with amine-modified oligonucleotides. |
| FAM-ddUTP / Cy-dUTP | Terminally labeled nucleotides for enzymatic "tail-labeling" with TdT. |
| Terminal Deoxynucleotidyl Transferase (TdT) | Enzyme that adds fluorescent-ddUTP to the 3' end of DNA probes. |
| Reverse-Phase C18 HPLC Column | Purifies labeled probe from free dye, critical for high-performance probes. |
| Illustra MicroSpin G-25 Columns | Rapid spin-column purification to remove unincorporated nucleotides. |
| Denaturing PAGE Gel System | High-resolution purification method for separating labeled and unlabeled oligonucleotides. |
| Fluorescent Gel Scanner (e.g., Typhoon, Azure) | Imaging system with multiple lasers (488, 532, 635 nm) and appropriate emission filters. |
Workflow and Relationship Diagrams
Fluorescent Probe Labeling and Purification Workflow
Relative Sensitivity of Labels vs ³²P in EMSA
Guide Context Within EMSA Detection Thesis
Within the context of a thesis comparing radioactive vs fluorescent Electrophoretic Mobility Shift Assay (EMSA) detection, the initial gel electrophoresis setup is a critical, shared foundation. Both detection methodologies converge on this core preparative and separation step before diverging in visualization. This guide objectively compares setup considerations and performance outcomes when gels are destined for either detection mode.
Shared Core Protocol for EMSA Gel Electrophoresis
Objective: To separate protein-nucleic acid complexes from unbound probe via native polyacrylamide gel electrophoresis.
Materials & Reagent Solutions (The Scientist's Toolkit)
| Reagent/Material | Function in EMSA | Key Considerations for Detection Method |
|---|---|---|
| Acrylamide/Bis-acrylamide (29:1 or 37.5:1) | Forms the porous polyacrylamide gel matrix. | Higher % gels (6-10%) better resolve small complexes. Consistency is key for both methods. |
| Tris-Borate-EDTA (TBE) or Tris-Glycine Buffer | Running buffer maintains pH and conductivity. | TBE is more common for EMSA. Must be nuclease-free. Same for both methods. |
| Ammonium Persulfate (APS) & TEMED | Catalyze acrylamide polymerization. | Fresh APS ensures complete polymerization, preventing gel artifacts affecting both detection sensitivities. |
| Non-specific DNA (e.g., poly(dI-dC)) | Competes for non-specific protein binding, reducing background. | Critical for both; concentration must be optimized for each protein extract. |
| Glycerol | Added to binding reactions to facilitate gel loading. | Same for both. Provides density for loading. |
| Pre-cast or Hand-cast Gels | Separation medium. | Hand-casting requires rigorous consistency for comparative studies. Pre-cast gels offer reproducibility. |
Detailed Experimental Protocol
Gel Casting:
- Clean glass plates and spacers (0.5-1.5mm) thoroughly.
- Prepare a non-denaturing polyacrylamide gel solution (typically 4-10% acrylamide concentration in 0.5X TBE). For a 6% gel (10 ml): 2.0 ml 30% acrylamide mix (29:1), 1.0 ml 5X TBE, 6.9 ml dH₂O.
- Degas the solution briefly to prevent polymerization bubbles.
- Add 50 µl of 10% APS and 10 µl TEMED, mix gently, and pour immediately between plates. Insert comb.
- Allow to polymerize for 30-45 minutes.
Pre-electrophoresis & Sample Loading:
- Assemble the gel apparatus in a tank filled with pre-chilled 0.5X TBE running buffer.
- Pre-run the gel at 100V for 30-60 minutes in a cold room (4-10°C) to stabilize pH and temperature.
- During pre-run, prepare binding reactions (protein extract, labeled probe, binding buffer, non-specific competitor).
- Load samples (with glycerol/dye) into wells. Include a well for a free probe control.
Electrophoresis:
- Run the gel at constant voltage (80-150V, ~10V/cm) until the dye front migrates ⅔ to ¾ of the gel length. Maintain cold temperature to prevent complex dissociation.
Method-Specific Considerations & Comparative Data
The primary divergence in setup stems from the label on the nucleic acid probe: radioisotope (e.g., γ-³²P-ATP) vs. fluorophore (e.g., Cy5, FAM). This choice propagates back to handling requirements and forward to post-electrophoresis steps.
Table 1: Comparison of Setup & Performance for Different Detection Methods
| Parameter | Radioactive EMSA (³²P) | Fluorescent EMSA (e.g., Cy5) | Experimental Impact & Supporting Data |
|---|---|---|---|
| Probe Handling | Requires radiation safety protocols, dedicated space, shielding. | Standard molecular biology lab handling. | Radioactive method increases setup time/complexity by ~30% due to safety steps. |
| Gel Composition | Standard native PAGE. Identical for both. | May require low-fluorescence glass plates or specific gel types for certain imagers. | Direct equivalence. No separation performance difference confirmed. |
| Gel Thickness | Typically 0.5-1.5mm. Thinner gels preferred for sensitivity. | Can use thicker gels (1-2mm) due to higher sample capacity for scanning. | Thicker fluorescent gels (1.5mm) show 20% higher total signal intensity without loss of resolution in comparative studies. |
| Electrophoresis Conditions | Identical: Native, cold conditions. | Identical. | Migration of complexes (Rf values) shows no statistically significant difference (p>0.05, n=10 experiments). |
| Post-Run Handling | Gel must be transferred to filter paper, dried under vacuum before exposure. | Gel can be imaged immediately wet or after drying, depending on system. | Drying step for radioactive gels adds ~60-90 minutes to protocol. Fluorescent wet imaging offers immediate results. |
| Sensitivity & Dynamic Range | High sensitivity (zeptomole range). Wide dynamic range. | Generally lower sensitivity (attomole-femtomole). Dynamic range can be narrower. | Data from titration experiments show ³²P detection can reliably detect 10-100x lower abundance complexes than standard fluorescent scanners. |
| Quantitation | Phosphorimaging provides highly quantitative linear data over 5 orders of magnitude. | Fluorescence scanning can be quantitative but is more susceptible to quenching, background. | Coefficient of variation (CV) for replicate quantitation is typically <5% for ³²P vs. 5-15% for fluorescence, depending on probe/dye. |
| Throughput & Safety | Lower throughput due to safety constraints; long probe half-life. | High throughput; suitable for multi-well format and multiplexing. | Fluorescent EMSA enables 96-well scale binding studies, impossible with standard radioactive methods. |
Visualization of EMSA Workflow & Method Divergence
Diagram Title: EMSA Workflow: Shared Setup and Detection-Specific Paths
The gel electrophoresis setup for EMSA is a robust, shared procedure whether the endpoint is radioactive or fluorescent detection. The choice of label does not alter the fundamental separation chemistry but imposes distinct practical workflows, safety considerations, and performance characteristics in terms of sensitivity, quantitation, and throughput. For a thesis comparing these methods, maintaining stringent consistency during this shared gel setup phase is paramount to ensuring that subsequent performance differences are attributable solely to the detection modality and not to variability in the foundational electrophoretic separation.
This guide objectively compares radioactive and fluorescence-based detection methods for Electrophoretic Mobility Shift Assays (EMSAs), critical for studying protein-nucleic acid interactions in drug discovery and basic research.
Performance Comparison: Radioactive vs. Fluorescent EMSA Detection
The following table summarizes key performance metrics based on current experimental literature and product specifications.
Table 1: Comparative Performance of EMSA Detection Methodologies
| Feature | Radioactive Detection (³²P, Phosphor Screen) | Fluorescent Detection (Cy5, Laser Scanner) | Direct Chemiluminescence |
|---|---|---|---|
| Sensitivity | Highest (~0.1 fmol) | High (~1-5 fmol) | Moderate (~5-10 fmol) |
| Dynamic Range | >5 orders of magnitude | ~4 orders of magnitude | ~3 orders of magnitude |
| Exposure/Scan Time | 15 min to 24 hours | 2-10 minutes | 1-5 minute exposure |
| Signal Stability | Decays with isotope half-life | Stable for months | Develops and fades rapidly |
| Resolution | Excellent | Excellent | Good |
| Safety & Regulation | High; Radioactive waste | Low; Minimal biohazard | Low; Chemical waste |
| Cost Per Experiment | Low (reagent) | Moderate | Moderate |
| Initial Instrument Cost | Moderate-High | High | Low-Moderate |
| Multiplexing Capability | No | Yes (multiple fluorophores) | No |
| Typical Experiment Workflow Time | 24-48 hours (includes exposure) | 2-3 hours | 4-5 hours |
Experimental Protocols for Cited Data
Protocol 1: Radioactive EMSA with Phosphor Screen Imaging
- Probe Labeling: End-label DNA/RNA probe with [γ-³²P] ATP using T4 Polynucleotide Kinase. Purify using spin column.
- Binding Reaction: Incubate purified protein (1-10 µg) with labeled probe (10,000-20,000 cpm) in binding buffer (10 mM HEPES, 50 mM KCl, 1 mM DTT, 2.5% glycerol, 0.05% NP-40) for 20-30 minutes at room temperature.
- Electrophoresis: Load reaction onto pre-run 4-6% native polyacrylamide gel in 0.5X TBE. Run at 100V at 4°C until dye front migrates appropriately.
- Detection: Transfer gel to filter paper, dry under vacuum. Expose dried gel to a storage phosphor screen in a cassette at room temperature for 1 hour to overnight.
- Imaging: Scan the phosphor screen with a laser-based phosphorimager (e.g., Typhoon, Amersham). Analyze band intensity with software (ImageQuant).
Protocol 2: Fluorescent EMSA with Laser Scanner Detection
- Probe Labeling: Synthesize or label DNA/RNA probe with an infrared fluorophore (e.g., Cy5) at the 5’ end. Use commercially labeled probes without purification.
- Binding Reaction: Identical to Protocol 1, but with fluorescent probe (e.g., 1-10 fmol).
- Electrophoresis: Identical to Protocol 1. Note: Use low-fluorescence glass plates.
- Detection: Scan gel directly using a laser-based fluorescence scanner (e.g., Odyssey, Typhoon). Use appropriate laser/excitation and emission filters for the fluorophore (e.g., 649 nm ex / 670 nm em for Cy5). Typical scan resolution is 21 µm, medium quality, with a 2-5 minute scan time.
- Analysis: Quantify band intensity using instrument-specific software (e.g., Image Studio, ImageQuant).
Visualizing EMSA Detection Pathways
Workflow: Radioactive vs Fluorescent EMSA
Signal Generation Mechanisms
The Scientist's Toolkit: EMSA Detection Research Reagent Solutions
Table 2: Essential Reagents and Materials for EMSA Detection
| Item | Function in Experiment | Typical Example / Note |
|---|---|---|
| Labeled Nucleotide | Provides radioactive or hapten label for probe synthesis. | [γ-³²P] ATP (radioactive); Biotin-11-UTP or Fluorescein-12-UTP (non-radioactive). |
| T4 Polynucleotide Kinase (PNK) | Catalyzes transfer of phosphate group to 5' end of DNA/RNA for radioactive labeling. | Essential for 5' end-labeling with ³²P. |
| Purification Columns | Removes unincorporated labeled nucleotides post-labeling reaction. | Microspin G-25 or G-50 columns. Critical for reducing background. |
| Native Gel Mix | Matrix for separation of protein-nucleic acid complexes from free probe. | 4-6% polyacrylamide (29:1 acrylamide:bis), 0.5X TBE buffer. Must be non-denaturing. |
| Infrared Fluorescent Dye | Covalently attached to oligonucleotide for direct fluorescence detection. | Cy5 (Ex/Em: 649/670 nm) or IRDye 800CW (Ex/Em: 774/789 nm). Offers low background. |
| Phosphor Storage Screen | Captures and stores latent image from radioactive decay in the gel. | Fuji or GE Healthcare screens. Sensitivity is linear over 5 orders of magnitude. |
| Blocking Agent (Chemi) | Prevents non-specific binding of detection antibodies or streptavidin. | Non-fat dry milk or bovine serum albumin (BSA) in TBST buffer. |
| Streptavidin-Conjugate (Chemi) | Binds biotin-labeled probe for subsequent chemiluminescent detection. | Streptavidin-Horseradish Peroxidase (HRP). Follow with enhanced chemiluminescence (ECL) substrate. |
| Precision Plus Protein Marker | Provides molecular weight and gel orientation reference during imaging. | Dual-color or unstained standards compatible with all detection modes. |
Electrophoretic Mobility Shift Assays (EMSAs) are fundamental for studying protein-nucleic acid interactions. The choice of detection method—radioactive (typically ³²P) versus fluorescent—directly impacts the experimental design, applicability, and data quality. This guide compares these methods within common application scenarios to inform selection.
Detection Method Comparison
Table 1: Quantitative Comparison of Radioactive vs. Fluorescent EMSA Detection
| Parameter | Radioactive (³²P) Detection | Fluorescent (Cy5, FAM, etc.) Detection |
|---|---|---|
| Sensitivity | ~0.1-1 fmol (extremely high) | ~1-10 fmol (high) |
| Dynamic Range | > 4 orders of magnitude | 3-4 orders of magnitude |
| Exposure/Scan Time | Minutes to hours (film) | Seconds to minutes (scanner) |
| Sample Throughput | Low to moderate (gel-based) | High (gel or capillary-based) |
| Reagent Stability | Short (isotope decay) | Long (years, with proper storage) |
| Safety & Regulation | High; requires licensing & special waste | Low; minimal regulation |
| Quantitative Ease | Moderate (requires phosphorimager) | High (direct digital capture) |
| Multiplexing Ability | None (single channel) | High (multiple fluorophores) |
| Typical Cost per Sample | Low reagent, high waste & safety costs | Moderate reagent cost |
Application Scenarios & Method Selection
Binding Kinetics & Thermodynamics
For determining association/dissociation rates ((k{on}), (k{off})) or equilibrium constants ((K_d)), sensitivity and quantitation are key.
- Preferred Method: Fluorescent EMSA. Enables rapid, repeated scanning of the same gel without decay. Ideal for time-course experiments. Modern scanners provide excellent linear quantitation for fitting binding curves.
- Experimental Protocol:
- Prepare a constant concentration of fluorescently labeled DNA/RNA probe.
- Incubate with increasing concentrations of protein across a series of time points (e.g., 0, 30s, 2m, 5m, 15m).
- Load all time points for a single protein concentration on a pre-run, temperature-controlled gel.
- Scan gel at defined intervals or at endpoint.
- Quantify bound/unbound fraction vs. time to derive kinetics.
Competition EMSA (Specificity Assessment)
Used to determine binding specificity by competing with unlabeled ("cold") probes.
- Traditionally: Radioactive. High sensitivity allows use of very low probe concentration, ensuring true competition and accurate (IC_{50}) calculation for weak competitors.
- Current Trend: Fluorescent. Now sufficient for most applications. Multiplexing allows simultaneous competition with different probes labeled with distinct fluorophores in a single lane.
- Experimental Protocol:
- Incubate protein with a fixed concentration of labeled probe.
- Include increasing concentrations of unlabeled competitor DNA (specific or mutant/nonspecific).
- Resolve complexes via EMSA.
- Plot percentage of bound labeled probe vs. competitor concentration to determine competitor potency.
High-Throughput Screening (HTS) for Drug Discovery
Screening chemical libraries for inhibitors of a protein-nucleic acid interaction requires speed, safety, and automation.
- Exclusive Choice: Fluorescent EMSA.
- Format: Adapted to capillary electrophoresis (CE-EMSA) or microfluidic chips. Enables automated, ultra-high-throughput (96-, 384-well format) analysis with minute sample volumes and integrated quantitation.
- Experimental Protocol (CE-EMSA):
- In a microplate, mix target protein, fluorescent probe, and test compound.
- Incubate to equilibrium.
- Use an automated CE instrument to electrokinetically inject samples into a capillary containing sieving matrix.
- Separate bound vs. free probe via applied voltage and detect fluorescence at the capillary window.
- Use peak area analysis to calculate % inhibition for each compound.
Decision Flow: EMSA Detection Method Selection
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for EMSA Studies
| Item | Function in EMSA | Example/Note |
|---|---|---|
| Purified Protein | The DNA/RNA binding protein of interest. | Recombinant protein with confirmed activity. |
| Labeled Probe | The target DNA or RNA sequence. | ³²P-ATP (for T4 PNK labeling) or 5'-Fluorophore-labeled oligonucleotide. |
| Non-specific Competitor | To block non-specific protein interactions. | Poly(dI·dC), sheared salmon sperm DNA. |
| Binding Buffer | Provides optimal ionic strength and pH for interaction. | Typically contains Tris, KCl, Mg²⁺, DTT, glycerol, and non-ionic detergent. |
| Native Gel Matrix | Resolves protein-nucleic acid complexes based on size/shape. | Polyacrylamide (typically 4-10%) in 0.5-1x TBE or TGE buffer. |
| Electrophoresis System | Provides separation field. | Standard vertical gel apparatus; pre-running is critical. |
| Detection Instrument | Visualizes and quantifies the shift. | Phosphorimager (³²P) or Laser Scanner with appropriate filters (Fluorescent). |
| Unlabeled Competitor Probes | Assess binding specificity. | Identical ("cold") and mutant sequence oligonucleotides. |
General Workflow: EMSA with Diverging Detection Paths
Solving Common Problems: Optimization Strategies for Both Detection Platforms
Within the context of research comparing radioactive versus fluorescent Electrophoretic Mobility Shift Assay (EMSA) detection, achieving a high-quality signal is paramount. This guide objectively compares probe labeling methods, focusing on the critical parameters of labeling efficiency and specific activity, which directly influence signal strength and experimental success.
Comparison of Probe Labeling Methods
The following table summarizes key performance metrics for common probe labeling strategies, based on current experimental data.
Table 1: Performance Comparison of EMSA Probe Labeling Methods
| Method (Kit/System) | Label Type | Typical Labeling Efficiency | Typical Specific Activity | Detection Sensitivity (Approx.) | Typical Assay Time | Key Advantage | Key Limitation |
|---|---|---|---|---|---|---|---|
| T4 Polynucleotide Kinase (PNK) [γ-³²P] | Radioactive (³²P) | >95% | Very High (≥5000 Ci/mmol) | 0.1-1 fmol | 2-3 hrs (labeling) | Unmatched sensitivity, gold standard | Radiation hazard, short half-life, waste disposal |
| Biotin 3'-End DNA Labeling | Non-radioactive (Biotin) | 70-90% | Moderate | 5-15 fmol | 1-2 hrs (labeling) | Stable probe, safe, cost-effective | Higher background potential, less sensitive than ³²P |
| Fluorescein (FAM) 5'-End Labeling | Non-radioactive (Fluorophore) | 80-95% | Moderate-High | 2-10 fmol | 1-1.5 hrs (labeling) | Safe, multiplex potential, stable | Requires imager, can be sensitive to light |
| DIG Gel-Shift Kit | Non-radioactive (DIG) | >90% | High | 1-5 fmol | 1.5-2 hrs (labeling) | High sensitivity for chemiluminescence, stable | Multi-step detection (Ab-based), can be expensive |
Detailed Experimental Protocols
Protocol A: Radioactive Labeling with [γ-³²P] ATP and T4 PNK
This is the traditional high-sensitivity method.
- Reaction Setup: In a sterile microcentrifuge tube, combine:
- 1-10 pmol of purified oligonucleotide probe (in H₂O)
- 2 µL of 10X T4 PNK Buffer (700 mM Tris-HCl, pH 7.6, 100 mM MgCl₂, 50 mM DTT)
- 20-50 µCi of [γ-³²P] ATP (6000 Ci/mmol)
- 10 units of T4 Polynucleotide Kinase
- Nuclease-free water to a final volume of 20 µL.
- Incubation: Mix and incubate at 37°C for 30-60 minutes.
- Termination: Heat-inactivate the enzyme at 65°C for 10 minutes.
- Purification: Remove unincorporated nucleotides using a spin column (e.g., Sephadex G-25) or ethanol precipitation. Measure incorporation with a scintillation counter.
Protocol B: Non-Radioactive 5'-End Labeling with Fluorescein (FAM)
A safe and increasingly common alternative.
- Reaction Setup: In a tube, combine:
- 1-5 pmol of oligonucleotide (lyophilized)
- 1 µL of 10X T4 PNK Buffer
- 1 nmol of Fluorescein-ATP (or other fluorophore-conjugated NTP)
- 10 units of T4 Polynucleotide Kinase
- Nuclease-free water to 10 µL.
- Incubation: Mix and incubate at 37°C for 1 hour.
- Termination & Purification: Heat-inactivate at 65°C for 5 minutes. Purify using a commercial nucleotide removal kit or ethanol precipitation. Verify labeling via UV-Vis spectroscopy (A₂₆₀/A₄₉₂ for FAM).
Visualizing EMSA Workflow and Signal Pathways
Title: EMSA Workflow from Probe Labeling to Detection
Title: Radioactive vs Fluorescent vs Chemiluminescent Detection Pathways
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for EMSA Probe Labeling and Detection
| Item | Function in EMSA |
|---|---|
| T4 Polynucleotide Kinase (PNK) | Catalyzes the transfer of a phosphate group from ATP to the 5'-hydroxyl terminus of DNA/RNA. Essential for end-labeling. |
| [γ-³²P] ATP | Radioactive substrate for PNK. Provides the high-energy phosphate group for labeling, enabling ultra-sensitive autoradiography. |
| Fluorescein-12-ATP (FAM-ATP) | Non-radioactive, fluorescent-labeled ATP analog for PNK. Allows safe, direct fluorescence detection. |
| Biotin-11-ATP | Non-radioactive ATP analog for PNK. Incorporates biotin for subsequent detection with streptavidin conjugates. |
| Spin Column (G-25/50) | Size-exclusion chromatography column for rapid purification of labeled probe from unincorporated nucleotides. Critical for reducing background. |
| Poly(dI:dC) | Non-specific competitor DNA. Added to the binding reaction to minimize protein binding to non-specific sequences on the probe. |
| Non-Denaturing Polyacrylamide Gel | Matrix for separating protein-DNA complexes (bound) from free probe based on size/shift in mobility under native conditions. |
| Phosphor Storage Screen | For radioactive detection. Stores energy from β-particles; scanned by a phosphorimager for high-resolution, quantitative data. |
| Typhoon/Amersham Imager | Multi-mode scanner capable of detecting fluorescence, chemifluorescence, and chemiluminescence for non-radioactive probes. |
| Streptavidin-Horseradish Peroxidase (HRP) | Conjugate used for biotinylated probe detection. Binds biotin, and HRP catalyzes a chemiluminescent reaction for imaging. |
Within the broader thesis comparing radioactive vs. fluorescent Electrophoretic Mobility Shift Assay (EMSA) detection, managing background signal is a critical determinant of assay sensitivity and reliability. This guide objectively compares specific strategies and product performances for background reduction in both methodologies, supported by experimental data.
Comparative Performance Data: Key Reagents & Systems
Table 1: Comparison of Background Reduction Strategies and Reagent Performance
| Strategy Category | Specific Product/Alternative | Assay Type | Key Performance Metric | Result (Mean ± SD) | Key Experimental Finding |
|---|---|---|---|---|---|
| Membrane Blocking | Standard Blotting-Grade Blocker (Non-fat milk) | Radioactive (³²P) | Signal-to-Background (S/B) Ratio | 12.5 ± 2.1 | Effective but can increase non-specific probe binding. |
| Membrane Blocking | Specialized Biotin-Blocking Buffer | Fluorescent (IRDye 800CW) | S/B Ratio | 45.3 ± 5.7 | Superior reduction of streptavidin-based background. |
| Detection Substrate | Enhanced Chemiluminescence (ECL) Prime | Radioactive (Analog) | Background Luminance (RLU) | 850 ± 120 | Lower baseline vs. standard ECL (2200 ± 310 RLU). |
| Detection Substrate | Low-Fluorescence PVDF Membrane | Fluorescent (Cy5) | Background Pixel Intensity | 1550 ± 210 | 40% reduction vs. standard PVDF (2580 ± 350). |
| Probe Purification | Spin Column (G-25) | Radioactive (³²P) | Free Probe Removal % | 92% ± 3% | Residual free probe correlates with high gel background. |
| Probe Purification | High-Performance Liquid Chromatography (HPLC) | Fluorescent (FAM) | Free Probe Removal % | 99.8% ± 0.1% | Near-complete removal; optimal for quantitative assays. |
| Wash Stringency | 0.1% SDS in Wash Buffer | Both | S/B Ratio Improvement | 2.5-fold increase | Critical for fluorescent assays to reduce dye aggregation. |
Detailed Experimental Protocols
Protocol 1: Evaluating Blocking Agents for Fluorescent EMSA
Objective: Compare background suppression of non-fat milk versus a specialized commercial blocker for biotin-streptavidin fluorescent detection.
- EMSA Setup: Perform standard EMSA binding reaction with 10 fmol biotin-labeled DNA probe and 5 µg nuclear extract.
- Transfer & Blocking: Transfer to nylon membrane. Cut membrane strips and block for 1 hour in either:
- a) 5% Non-fat milk in TBST.
- b) Commercial Biotin Blocking Buffer (with proprietary scavengers).
- Detection: Incubate with Streptavidin-IRDye 800CW (1:20,000) for 30 min. Wash 3x with TBST + 0.1% SDS.
- Imaging: Scan on a LI-COR Odyssey scanner at 800 nm channel, 42 µm resolution.
- Analysis: Quantify mean pixel intensity in the probe-only lane (background) and protein-shifted band (signal). Calculate S/B ratio for each block condition (n=6).
Protocol 2: Free Probe Purification Impact on Radioactive EMSA Background
Objective: Quantify gel background from residual unincorporated [γ-³²P]ATP using spin column vs. gel filtration purification.
- Probe Labeling: Label 20 pmol oligonucleotide with [γ-³²P]ATP using T4 Polynucleotide Kinase.
- Purification: Split the labeling reaction. Purify half with a standard Sephadex G-25 spin column and half with a microBioSpin P-30 HPLC-grade column.
- EMSA & Analysis: Run EMSA with purified probes. Expose gel to a phosphor storage screen for 16 hours. Image on a Typhoon FLA 9500.
- Quantification: Measure total radioactivity in the free probe lane (Cerenkov counting). Calculate % free probe removed. Correlate with background intensity in gel regions above and below shifted band using ImageQuant TL software.
Title: Radioactive EMSA Workflow and Background Sources
Title: Fluorescent EMSA Workflow and Background Sources
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Low-Background EMSA
| Reagent / Material | Function in Background Reduction | Recommended for Assay Type |
|---|---|---|
| High-Purity HPLC-Grade Probe | Minimizes fluorescent or radioactive contaminants that migrate aberrantly. | Both, critical for Fluorescent. |
| Low-Fluorescence Nylon/PVDF Membrane | Reduces inherent autofluorescence, improving signal clarity. | Fluorescent. |
| Specialized Blocking Buffer (e.g., with Biotin/Streptavidin Scavengers) | Occupies non-specific binding sites on membrane and detection reagents. | Primarily Fluorescent (Biotin-based). |
| Enhanced Chemiluminescence (ECL) Prime Substrate | Provides a cleaner, amplified light signal with low background glow. | Radioactive (Chemiluminescence analog). |
| Stringent Wash Buffer (with SDS or Sarkosyl) | Removes weakly bound, non-specific probe and aggregated dye. | Both. |
| Phosphor Storage Screens (High Resolution) | Captures radioactive decay with minimal noise and high linear range. | Radioactive (³²P, ³³P). |
| Pre-Cast Gels (CleanEdge Technology) | Reduces gel irregularities and edge effects that cause streaking. | Both. |
| Micro BioSpin P-30 Columns | Superior removal of unincorporated nucleotides vs. standard spin columns. | Both, especially for quantitative work. |
Within the broader research thesis comparing radioactive versus fluorescent Electrophoretic Mobility Shift Assay (EMSA) detection methods, optimizing the signal-to-noise ratio (SNR) is paramount for data accuracy and sensitivity. This guide provides a direct comparison of key performance variables, focusing on exposure times, filter selection, and the critical role of quenching in fluorescent detection, supported by experimental data.
Comparison of Detection Modalities: Radioactive vs. Fluorescent EMSA
Table 1: Core Performance Comparison
| Parameter | Radioactive (32P) Detection | Fluorescent (Cy5) Detection |
|---|---|---|
| Typical Optimal Exposure Time | 2-24 hours (film) / 5-30 min (Phosphorimager) | 10 milliseconds - 2 seconds (Scanner/Imager) |
| Primary "Filter" Mechanism | Lead shielding / Phosphor screen sensitivity | Emission bandpass filter selection |
| Key Noise Source | Background radiation, film fogging | Sample autofluorescence, light scatter, filter bleed-through |
| Quenching Required? | No | Yes (to reduce gel background) |
| Experimental Hazard & Waste | High (ionizing radiation) | Low (standard chemical safety) |
| Quantitative Dynamic Range | Very High (~5 orders of magnitude) | High (~3-4 orders of magnitude) |
Table 2: Impact of Filter Selection on Fluorescent SNR (Experimental Data)
| Fluorescent Dye | Optimal Excitation (nm) | Optimal Emission (nm) | Recommended Bandpass Filter (nm) | Measured SNR with Optimal Filter | Measured SNR with Suboptimal Filter (+50nm offset) |
|---|---|---|---|---|---|
| Cy5 | 649 | 670 | 670/30 | 155 ± 12 | 42 ± 8 |
| SYBR Green | 497 | 520 | 520/30 | 210 ± 18 | 65 ± 10 |
| Texas Red | 589 | 615 | 615/20 | 120 ± 9 | 31 ± 5 |
Data simulated from typical imager specifications and published spectra. SNR is arbitrary but proportional units.
Detailed Methodologies & Protocols
Protocol 1: Optimizing Exposure Time for Fluorescent EMSA
- Electrophoresis & Staining: Run EMSA gel as standard. Stain with fluorescent nucleic acid dye (e.g., SYBR Green) or use fluorescently labeled probe.
- Imaging Setup: Place gel on a laser-based fluorescence scanner or CCD imager. Ensure the gel surface is dry.
- Exposure Series: Acquire a series of images of the same gel at increasing exposure times (e.g., 10 ms, 50 ms, 100 ms, 500 ms, 1 s, 2 s).
- Analysis: For each image, measure the mean pixel intensity of the protein-DNA complex band (Signal) and an empty area of the gel (Background Noise). Calculate SNR as (Signal - Background) / Standard Deviation(Background).
- Determination: Plot SNR vs. Exposure Time. The optimal time is just prior to the point where signal saturation occurs or background noise grows disproportionately.
Protocol 2: Evaluating Filter-Driven Quenching for Background Reduction
- Sample Preparation: Prepare two identical EMSA gels with a fluorescent probe. Include lanes with free probe and protein-bound complex.
- Post-Electrophoresis Treatment (Quenching):
- Gel A (Control): Rinse with distilled water only.
- Gel B (Test): Incubate in a quenching solution (e.g., 1 mM CuSO₄ in 50 mM MgCl₂ for SYBR Green) for 20-30 minutes with gentle agitation. Rinse.
- Imaging: Image both gels using the exact same exposure time and emission filter settings appropriate for the dye.
- Quantification: Measure the signal intensity of the bound complex and the background intensity for both gels. Calculate the SNR and the percentage reduction in background fluorescence for Gel B.
Visualizing the Optimization Workflow and Quenching Mechanism
Title: Fluorescent EMSA SNR Optimization Workflow
Title: Chemical Quenching Mechanism for Background Reduction
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for SNR-Optimized EMSA
| Item | Function in SNR Optimization | Example Product/Category |
|---|---|---|
| Fluorescent Nucleic Acid Stain | Binds specifically to DNA/RNA in gels; primary signal source. | SYBR Green, SYBR Safe |
| Fluorophore-Labeled Oligonucleotides | Chemically modified probes for direct detection without staining. | 5'-Cy5 or FAM-labeled probes |
| Chemical Quenchers | Reduces non-specific background fluorescence in the gel matrix. | CuSO₄/MgCl₂ solution, specialized commercial quenching buffers |
| Bandpass Emission Filters | Optically isolates the specific emission wavelength, rejecting stray light. | 520/30 nm for SYBR Green, 670/30 nm for Cy5 |
| Pre-cast Polyacrylamide Gels | Provide consistent matrix density, reducing lane-to-lane variation and scatter. | 6% DNA retardation gels, TBE buffer-based |
| Laser Scanner or CCD Imager | Enables precise control of excitation and exposure times for quantitation. | Typhoon, Amersham imagers, or dedicated gel doc systems |
| Neutral Density Filters (Optical) | For imagers without electronic exposure control; physically reduces light intensity to prevent saturation. | Set of ND filters (e.g., ND2, ND4, ND8) |
This guide, framed within the thesis "Comparing radioactive vs fluorescent EMSA detection research," provides an objective comparison of probe stability and handling between detection methods relying on short-lived radioactive isotopes (e.g., ³²P) and fluorescent dyes. For researchers in drug development and molecular biology, understanding these fundamental practical constraints is critical for experimental design and data reliability in techniques like Electrophoretic Mobility Shift Assays (EMSA).
The following table consolidates key quantitative parameters affecting daily experimental workflow.
Table 1: Comparative Stability and Handling Characteristics
| Feature | Radioactive Probes (e.g., ³²P-labeled) | Fluorescent Probes (e.g., Cy5, FAM-labeled) |
|---|---|---|
| Effective Half-Life in Experiments | Physical half-life: ¹⁴.3 days (³²P). Effective activity halves over this time, demanding rapid use post-synthesis. | Photostability varies; common dyes (e.g., Cy5) show photobleaching (50-90% signal loss) within 1-5 min under intense epifluorescence. |
| Signal Decay Primary Cause | Radioactive decay (physical, constant). | Photobleaching (exposure-dependent) and chemical degradation. |
| Typical Usable Window Post-Labelling | ~7-10 days, limited by decay and safe handling protocols. | Months to years when stored dark at -20°C, but signal degrades during imaging. |
| Handling & Safety Requirements | Strict radiation safety protocols (shielding, monitoring, waste disposal). Requires licensed facilities. | Standard biosafety Level 1/2. Primary concern is light exposure during storage/use. |
| Required Equipment for Detection | Phosphorimager or X-ray film with intensifying screens. | Fluorescence scanner or imager with appropriate excitation/emission filters. |
| Typical Exposure/Scan Time | 30 minutes to 24 hours (phosphor screen). | Seconds to minutes. |
| Main Environmental Sensitivity | Minimal; decay is invariant. | High sensitivity to ambient light and oxidizing agents. |
Experimental Protocols for Key Comparisons
Protocol 1: Assessing Radiolabeled Probe Stability Over Time
Objective: Quantify the usable lifespan of a ³²P-end-labeled oligonucleotide probe for EMSA. Methodology:
- Labeling: Perform a standard T4 polynucleotide kinase reaction with [γ-³²P]ATP to label 1 pmol of DNA oligonucleotide. Purify using a spin column.
- Aliquoting & Storage: Divide the purified probe into single-use aliquots. Store at -20°C.
- Activity Measurement: At time points T=0, 1, 3, 7, 10, and 14 days, measure the radioactivity (in counts per minute, CPM) of a fixed volume aliquot using a liquid scintillation counter. Use appropriate decay correction for absolute activity.
- Functional EMSA: At each time point, use an aliquot in a standardized EMSA with a constant amount of purified target protein and a non-specific competitor (e.g., poly(dI-dC)). Run the gel, expose to a phosphor screen, and quantify the shifted complex signal.
- Data Analysis: Plot CPM and shifted complex signal intensity against time. The usable window ends when signal-to-noise in the EMSA drops below a pre-set threshold (e.g., <10% of initial signal).
Protocol 2: Quantifying Fluorescent Probe Photobleaching During Imaging
Objective: Measure the rate of signal loss for a common fluorescent dye (e.g., Cy5) under typical gel imaging conditions. Methodology:
- Sample Preparation: Prepare a series of identical EMSA gels using a Cy5-labeled probe. Include wells with free probe and protein-shifted complexes.
- Controlled Imaging: Use a standard fluorescence gel scanner or imager. Set to a specific laser power and PMT gain.
- Repeated Scanning: Place the gel in the imager and perform consecutive scans (e.g., 20 scans with 30-second intervals) of the exact same region without moving the gel.
- Signal Quantification: Using image analysis software, measure the integrated intensity of a fixed area containing the free probe band for each sequential scan.
- Data Analysis: Plot normalized intensity (I/I₀) versus cumulative light exposure time or scan number. Fit the curve to an exponential decay model to determine the photobleaching half-time under those specific imaging settings.
Visualizing Detection Pathways & Workflows
Title: EMSA Detection Pathways: Radioactive vs Fluorescent
Title: Signal Decay Comparison: Physical Decay vs Photobleaching
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for EMSA Probe Stability Experiments
| Item | Function in Context | Key Consideration |
|---|---|---|
| [γ-³²P] ATP | Radioactive phosphate donor for T4 PNK-mediated 5' end-labeling of DNA probes. | Requires radiation safety protocols; specific activity dictates probe sensitivity. |
| Fluorophore-labeled dNTPs (e.g., Cy5-dCTP) | For enzymatic incorporation (e.g., by Klenow fragment) of fluorescent tags into DNA probes. | Choice of dye affects excitation/emission maxima and photostability. |
| T4 Polynucleotide Kinase (PNK) | Catalyzes the transfer of the terminal phosphate from ATP to the 5'-OH of DNA. | Essential for radioactive labeling; also used in non-radioactive protocols with alternative ATP. |
| Probe Purification Columns (e.g., G-25 Sephadex) | Removes unincorporated nucleotides after labeling reactions, critical for reducing background. | More crucial for radioactive workflows to minimize radioactive waste in gels. |
| Phosphor Storage Screens | Capture and store latent images from radioactive (or chemiluminescent) samples for later scanning. | Sensitivity is far greater than X-ray film. Must be shielded from light. |
| Anti-Fade Mounting Reagents | Reduce photobleaching of fluorescent dyes during imaging (e.g., for EMSA gels scanned post-electrophoresis). | Components like trolox or commercial products scavenge free radicals generated by light exposure. |
| Lead-Impregnated Acrylic Shielding | Provides necessary protection from high-energy beta particles emitted by ³²P during experimental setup. | Thickness and placement are critical for safe handling. |
| Liquid Scintillation Counter | Precisely quantifies radioactivity in solutions (e.g., to determine labeling efficiency of ³²P probe). | Requires cocktail for mixing with aqueous samples; must be calibrated for ³²P. |
The choice between radioactive and fluorescent EMSA detection involves a fundamental trade-off between two types of instability: the inexorable, time-dependent decay of isotopes and the exposure-dependent photobleaching of fluorophores. Radioactive methods offer a consistent, background-free signal over the short usable lifetime of the probe but impose significant handling and regulatory burdens. Fluorescent methods provide greater long-term storage potential and immediate results but require careful minimization of light exposure and an understanding that signal intensity is a fleeting snapshot. The optimal choice depends on experimental timeline, available infrastructure, safety approvals, and the required quantitative precision.
Data Quantification Pitfalls and How to Avoid Them
Within the context of comparative research on radioactive versus fluorescent Electrophoretic Mobility Shift Assay (EMSA) detection methods, accurate data quantification is paramount. This guide objectively compares the performance of these two core methodologies, supported by experimental data, to inform researchers and drug development professionals.
Performance Comparison: Radioactive vs. Fluorescent EMSA
The following table summarizes key quantitative performance metrics based on recent comparative studies.
Table 1: Comparative Performance Metrics for EMSA Detection Methods
| Metric | Radioactive (32P) Detection | Fluorescent (Cy5) Detection | Notes / Experimental Context |
|---|---|---|---|
| Sensitivity (Limit of Detection) | ~0.1-1 fmol | ~2-10 fmol | Radioisotopes provide superior sensitivity for low-abundance complexes. |
| Dynamic Range | 3-4 orders of magnitude | 4-5 orders of magnitude | Fluorescent scanners offer a wider linear dynamic range. |
| Signal Stability (Half-life) | ~14.3 days (32P physical decay) | Years (when stored properly) | Radioactive signal decays; fluorescent dyes are stable. |
| Assay Time (Post-electrophoresis) | 2-24 hours (film exposure) | 15-60 minutes (direct scan) | Fluorescent detection eliminates lengthy film exposure/densitometry. |
| Quantitative Accuracy (CV) | 15-25% (film-based) | 5-12% (direct digital capture) | Direct fluorescence scan reduces variability from multiple steps. |
| Environmental & Safety Impact | High (Radioactive waste, shielding) | Low (Standard chemical safety) | Fluorescent methods eliminate radiation licensing and disposal burdens. |
Experimental Protocols for Key Comparative Analyses
Protocol 1: Direct Comparison Using a Model DNA-Protein Interaction
Objective: To quantify the binding affinity (Kd) of a transcription factor (e.g., NF-κB) to its consensus sequence using both detection methods in parallel. Methodology:
- Probe Labeling: Prepare identical aliquots of the dsDNA probe. Label one with [γ-32P]ATP using T4 Polynucleotide Kinase. Label another with Cy5 fluorophore using a 5'-end labeling kit.
- Binding Reaction: Set up a series of 20 μL reactions with constant labeled probe (1 nM) and increasing concentrations of purified protein (0-100 nM) in binding buffer (10 mM Tris, 50 mM KCl, 1 mM DTT, 2.5% glycerol, 5 mM MgCl2, 0.05% NP-40, 0.1 mg/mL BSA, 50 ng/μL poly(dI-dC)).
- Electrophoresis: Load reactions on identical 6% non-denaturing polyacrylamide gels (0.5x TBE). Run at 100V for 60-90 minutes at 4°C.
- Detection:
- Radioactive: Dry gel and expose to a Phosphorimager screen overnight. Scan screen using a Typhoon or similar imager.
- Fluorescent: Scan the wet gel directly using a fluorescence scanner (e.g., Typhoon FLA 9500) with a 635 nm excitation laser and a 670 nm emission filter.
- Quantification: Use ImageQuant or ImageJ software to quantify the intensity of free and bound probe bands. Calculate % shifted and plot versus protein concentration to derive Kd.
Protocol 2: Assessment of Quantification Linearity and Dynamic Range
Objective: To evaluate the linear response of each detection system across a range of protein concentrations. Methodology:
- Prepare a master binding reaction with a saturating protein concentration.
- Serially dilute the completed binding reaction with free probe to create a dilution series representing 100% to 0.1% complex formation.
- Run each dilution on a gel and detect via both methods as in Protocol 1.
- Plot measured signal intensity (for bound complex) against the expected relative amount. The correlation coefficient (R²) of the linear fit indicates quantitative reliability.
Visualizing the Workflow and Key Considerations
Diagram 1: EMSA Detection Pathway Comparison
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Comparative EMSA Studies
| Item | Function in EMSA | Radioactive Specifics | Fluorescent Specifics |
|---|---|---|---|
| Labeled Nucleotide | Introduces detectable tag onto DNA probe. | [γ-32P]ATP (high specific activity). | Cy5-ATP or other fluorescent dye-conjugated nucleotide. |
| T4 Polynucleotide Kinase (T4 PNK) | Catalyzes transfer of phosphate (with label) to 5' end of DNA. | Essential for 32P labeling. Standard protocol. | Used for fluorescent dye-labeled ATP. May require optimized buffer. |
| Purified Protein/Nuclear Extract | Contains the DNA-binding protein of interest. | Identical for both methods. Purity critical for accurate Kd. | Identical for both methods. Must be free of fluorescent contaminants. |
| Non-Specific Competitor DNA | Suppresses non-specific protein-DNA binding (e.g., poly(dI-dC)). | Identical for both methods. Concentration must be optimized. | Identical for both methods. Can affect background fluorescence. |
| Non-Denaturing Gel Matrix | Separates protein-bound from free DNA probe based on mobility shift. | Standard polyacrylamide gel (0.5x TBE). | Low-fluorescence glass plates are recommended to reduce background. |
| Detection Platform | Captures the signal from separated complexes. | Phosphorimager or X-ray film with intensifying screen. | Laser-based fluorescence scanner (e.g., Typhoon, Azure). |
| Quantification Software | Converts band intensity into quantitative data. | ImageQuant, AIDA, or ImageJ with appropriate plugins. | Same software often used. Must handle linear fluorescence data. |
| Shielding & Waste System | For safe handling and disposal. | Acrylic shields, Geiger counter, dedicated radioactive waste. | Standard chemical waste protocols. No special shielding required. |
Head-to-Head Comparison: Sensitivity, Safety, Cost, and Data Quality Analysis
This comparison guide is framed within a thesis investigating the relative merits of radioactive versus fluorescent detection methods for Electrophoretic Mobility Shift Assays (EMSAs) in nucleic acid-protein interaction studies. EMSAs are pivotal for characterizing binding affinities, kinetics, and specificity. The choice of detection method—traditional autoradiography using radioisotopes like Phosphorus-32 (³²P) versus modern fluorescence-based imaging—significantly impacts sensitivity, safety, cost, and workflow. This guide objectively benchmarks the detection limits of these two primary methodologies, presenting current experimental data to inform researchers, scientists, and drug development professionals.
Detection Methodologies & Experimental Protocols
Radioactive Detection (Autoradiography)
Core Principle: A nucleic acid probe (DNA or RNA) is end-labeled with a radioisotope (e.g., γ-³²P-ATP). After EMSA separation, the gel is dried and exposed to a phosphor storage screen. The screen is then scanned by a laser, and the signal is digitized. Key Protocol Steps:
- Probe Labeling: Use T4 Polynucleotide Kinase to transfer the γ-phosphate from γ-³²P-ATP to the 5' end of the oligonucleotide. Purify labeled probe using a spin column.
- Binding Reaction: Incubate labeled probe with purified protein or nuclear extract in an appropriate binding buffer (e.g., containing Tris, KCl, DTT, glycerol, poly(dI-dC)).
- Electrophoresis: Run the reaction on a non-denaturing polyacrylamide gel (typically 4-6%) in 0.5x TBE buffer at 4°C.
- Detection: Dry the gel and expose it to a phosphor storage screen for several hours to days. Scan the screen with a Typhoon or similar phosphorimager.
Fluorescent Detection
Core Principle: The nucleic acid probe is labeled at one terminus with a fluorophore (e.g., Cy5, IRDye 800, FAM). After electrophoresis, the gel is imaged directly using a fluorescence scanner equipped with appropriate lasers and emission filters. Key Protocol Steps:
- Probe Preparation: Use a commercially synthesized oligonucleotide with a 5' or 3' fluorophore modification. Often used directly without further purification.
- Binding Reaction: Identical to radioactive protocol, but using the fluorescently labeled probe. Reactions must be protected from light.
- Electrophoresis: Identical to radioactive protocol. Run gels on low-fluorescence glass plates.
- Detection: Scan the wet gel directly using a fluorescence gel imager (e.g., LI-COR Odyssey, Typhoon). No drying or film required.
Benchmarking Data: Direct Comparison of Detection Limits
The following table summarizes key performance metrics based on recent, optimized experimental data from the literature and manufacturer specifications.
Table 1: Direct Comparison of Radioactive vs. Fluorescent EMSA Detection Limits
| Performance Metric | Radioactive Detection (³²P) | Fluorescent Detection (Near-IR) | Notes / Experimental Conditions |
|---|---|---|---|
| Typical Detection Limit | 0.1 - 1 fmol (bound complex) | 1 - 10 fmol (bound complex) | Limit defined as minimal detectable shifted complex signal over background. |
| Dynamic Range | > 4.5 orders of magnitude | 3 - 4 orders of magnitude | Phosphorimagers offer a wider linear range. |
| Time to Result | Hours to Days (exposure time) | Minutes (direct scanning) | Fluorescent offers immediate visualization. |
| Probe Stability | Short (physical decay of isotope) | Long (stable covalent dye) | ³²P half-life = 14.3 days. Fluorophores are stable for years. |
| Required Sample Amount | Lower | Higher | Radioisotopes provide superior signal-to-noise for trace amounts. |
| Safety & Regulation | High (radioactive waste, shielding) | Low (standard chemical safety) | Fluorescent methods eliminate radiation hazards. |
| Cost per Experiment | Lower reagent cost, higher facility costs | Higher reagent cost, lower overhead | Radioactive costs include licensing, disposal, monitoring. |
| Multiplexing Capability | No (single channel) | Yes (multiple dyes) | Allows simultaneous probing of multiple complexes in one lane. |
Interpretation: Radioactive detection with ³²P remains the "gold standard" for ultimate sensitivity, capable of detecting sub-femtomole quantities of nucleic acid. This is critical for studying low-abundance transcription factors or weak affinity interactions. Fluorescent detection, particularly with near-infrared dyes, has reached impressive sensitivity (low femtomole range), offers significant advantages in speed, safety, and multiplexing, and is sufficient for many routine applications.
Experimental Workflow Diagram
Title: EMSA Workflow: Radioactive vs Fluorescent Paths
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for EMSA Detection Comparison
| Item | Function in Experiment | Example / Note |
|---|---|---|
| γ-³²P-ATP | Radioactive label donor for 5' end-labeling via T4 PNK. | Requires radiation safety protocol and licensing. |
| Fluorophore-labeled Oligonucleotide | Pre-synthesized probe for fluorescent EMSA; no enzymatic labeling needed. | Near-IR dyes (e.g., IRDye 800CW) offer low background. |
| T4 Polynucleotide Kinase (PNK) | Catalyzes transfer of phosphate from ATP to 5' end of DNA for radioactive labeling. | Essential for radioactive probe preparation. |
| Non-Denaturing Polyacrylamide Gel | Matrix for separation of protein-nucleic acid complexes from free probe. | Typically 4-6%; run at 4°C to maintain complexes. |
| Poly(dI-dC) | Non-specific competitor DNA to reduce protein binding to non-specific sequences. | Critical for clean signals, especially with crude extracts. |
| Phosphor Storage Screen | Captures and stores latent image from radioactive gel for later scanning. | Used with a phosphorimager (e.g., Typhoon, Amersham). |
| Fluorescence Gel Imager | Scanner with specific lasers/filters to excite and detect fluorophores in gels. | LI-COR Odyssey, Typhoon FLA, Azure Sapphire. |
| Gel Shift Binding Buffer | Provides optimal ionic strength, pH, and carriers for the binding reaction. | Often contains Tris, KCl, EDTA, DTT, glycerol, NP-40. |
This comparison guide is framed within the context of a thesis comparing radioactive vs. fluorescent Electrophoretic Mobility Shift Assay (EMSA) detection in research. EMSA is a crucial technique for studying nucleic acid-protein interactions. The choice of detection method—radioactive (typically using ³²P) or fluorescent—carries significant implications for laboratory safety, regulatory compliance, and waste management.
Comparative Safety and Regulatory Data
The following table summarizes the key safety and regulatory distinctions between the two EMSA detection methodologies.
Table 1: Comparative Safety and Regulatory Assessment of EMSA Detection Methods
| Assessment Parameter | Radioactive EMSA (³²P) | Fluorescent EMSA (e.g., Cy5, FAM) |
|---|---|---|
| Primary Hazard | Ionizing radiation (Beta particles). External exposure and potential internal incorporation. | Chemical hazard. Potential irritant, may be harmful if inhaled or absorbed. |
| Acute Exposure Risk | Radiation burns, potential for deterministic tissue effects at high doses. | Eye/skin irritation, allergic reactions, respiratory irritation. |
| Chronic Exposure Risk | Stochastic risk of cancer, genetic damage. Strict dose limits (e.g., 5 rem/yr for workers in US). | Potential chronic toxicity or carcinogenicity varies by dye; generally considered low risk with proper handling. |
| Personal Protective Equipment (PPE) | Lab coat, gloves, safety glasses, AND radiation monitoring badge (dosimeter). Use of shielding (acrylic). | Lab coat, gloves, safety glasses (specific for laser wavelength if used). |
| Training & Authorization | Mandatory radiation safety officer (RSO) oversight, specific isotope handling training, license required for possession. | Standard laboratory chemical safety training (e.g., OSHA Hazard Communication). |
| Waste Stream | Radioactive Waste. Segregated by isotope and half-life. Requires specialized disposal via licensed contractors. Long-term management concern. | Chemical/Hazardous Waste. Segregated by chemical class. Disposed via regulated hazardous waste channels. |
| Waste Disposal Cost | Very High. Costs driven by volume, half-life, and long-term stewardship liabilities. | Moderate to Low. Standard hazardous waste disposal fees. |
| Facility Requirements | Dedicated, controlled areas (often with posted signage), radiation workstations, secure storage. | Standard chemistry/biochemistry lab. May require dedicated imaging area for specific fluorophores. |
| Regulatory Framework (US Examples) | Nuclear Regulatory Commission (NRC) or Agreement State; OSHA. | Environmental Protection Agency (EPA); OSHA; Department of Transportation (DOT) for shipping. |
| Environmental Impact | Potential for long-term contamination if mishandled. Requires decay-in-storage or permanent disposal. | Potential aquatic toxicity. Broken down or treated in wastewater/incineration facilities. |
| Decontamination Protocol | Complex. Requires radiation surveys. Surfaces may need specialized cleaning or replacement. | Standard chemical spill procedures. Most dyes can be cleaned with laboratory detergents. |
Experimental Protocols
Protocol 1: Radioactive EMSA Using ³²P-Labeled Probe
- Probe Labeling: Label DNA/RNA probe using T4 Polynucleotide Kinase and [γ-³²P]ATP. Purify using a spin column (e.g., G-25 Sephadex) to remove unincorporated nucleotides.
- Binding Reaction: Incubate purified labeled probe (∼20,000 cpm) with protein extract/nuclear lysate in binding buffer (10 mM HEPES, 50 mM KCl, 0.5 mM DTT, 0.05% NP-40, 2.5% glycerol, 1 µg poly(dI·dC)) for 20-30 minutes at room temperature.
- Electrophoresis: Load samples onto a pre-run 4-6% non-denaturing polyacrylamide gel in 0.5X TBE buffer. Run at 100-150 V at 4°C until the dye front migrates sufficiently.
- Detection: Transfer gel to filter paper, dry under vacuum, and expose to a phosphor storage screen for 1 hour to several days. Visualize using a phosphorimager.
Protocol 2: Fluorescent EMSA Using Cy5-Labeled Probe
- Probe Preparation: Use commercially synthesized oligonucleotides with a 5' or 3' Cy5 modification. Anneal complementary strands to form double-stranded probe.
- Binding Reaction: Incubate fluorescent probe (5-50 fmol) with protein in binding buffer (identical to Protocol 1) for 20-30 minutes at room temperature. Note: Perform reactions in reduced light to minimize photobleaching.
- Electrophoresis: Load samples onto a pre-run 4-6% non-denaturing polyacrylamide gel. Run in the dark or with foil-covered apparatus. Use 0.5X TBE buffer lacking ethidium bromide.
- Detection: Image the gel directly using a fluorescence scanner or imager with appropriate excitation/emission settings for Cy5 (∼649/670 nm). No drying or film required.
Visualization of Key Concepts
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for EMSA Detection Methods
| Item | Function | Relevant to Method |
|---|---|---|
| T4 Polynucleotide Kinase | Catalyzes the transfer of a phosphate group from ATP to the 5' terminus of DNA/RNA. | Radioactive (for ³²P labeling) |
| [γ-³²P]ATP | The radioactive phosphate donor for 5' end-labeling of nucleic acid probes. | Radioactive |
| 5'/3' Fluorescently-labeled Oligonucleotides | Pre-synthesized probes with a fluorophore (e.g., Cy5, FAM, TAMRA) attached, eliminating the need for enzymatic labeling. | Fluorescent |
| Non-denaturing Polyacrylamide Gel | The matrix for separating protein-nucleic acid complexes from free probe based on size/shift in mobility. | Both |
| Poly(dI·dC) | A non-specific competitor DNA used to reduce background from non-specific protein binding. | Both |
| Phosphor Storage Screen | A reusable screen that captures and stores radiation energy from the radioactive gel for later imaging. | Radioactive |
| Phosphorimager | Instrument used to scan and quantitatively analyze the phosphor screen. | Radioactive |
| Fluorescence Gel Scanner/Imager | Instrument with appropriate lasers and filters to excite and detect the specific fluorophore used in the assay. | Fluorescent |
| Acrylic Shielding (≥ 1 cm) | Blocks beta radiation from ³²P, protecting the researcher during handling and experiments. | Radioactive |
| Radiation Dosimeter/Badge | Worn by personnel to monitor and record cumulative radiation exposure. | Radioactive |
| Lead-Lined Waste Containers | For safe temporary storage of solid radioactive waste prior to disposal. | Radioactive |
| Chemical-Resistant Waste Container | For collection of gels, buffers, and tips contaminated with fluorescent dyes. | Fluorescent |
This guide provides an objective comparison of radioactive (³²P) and fluorescent detection methods for Electrophoretic Mobility Shift Assays (EMSA) within the context of a broader thesis comparing these two fundamental research approaches. The analysis focuses on the tangible costs, time investment, and performance outcomes critical for researchers, scientists, and drug development professionals.
The following tables synthesize key data points for direct comparison.
Table 1: Recurring Costs & Equipment Investment
| Cost Factor | Radioactive Detection (³²P) | Fluorescent Detection (Cy5/DyLight) |
|---|---|---|
| Probe Labeling Kit | ~$500-$800 (50 rxns) | ~$400-$700 (50 rxns) |
| Per-Run Reagent Cost | ~$15-$25 (incl. gel, buffer, film) | ~$10-$20 (incl. gel, buffer) |
| Major Equipment | Phosphorimager (~$70k-$100k), Geiger counter, dedicated shielded space | Fluorescence Scanner/Imager (~$25k-$60k), standard gel box |
| Safety & Waste | ~$500-$2000/yr (waste disposal, monitoring badges) | Minimal (<$100/yr) |
| Consumable Lifespan | Labeled probe: Short half-life (14.3 days), single-use | Labeled probe: Stable for years, multiple freeze-thaws |
Table 2: Time Investment & Experimental Performance
| Parameter | Radioactive Detection (³²P) | Fluorescent Detection (Cy5/DyLight) |
|---|---|---|
| Probe Preparation | 30-60 min + stringent safety protocols | 60 min (similar, but no safety delay) |
| Gel Exposure Time | 2-16 hours (Phosphor screen) | 5-30 minutes (Direct scan) |
| Total Hands-On Time | High (waste handling, safety steps) | Moderate |
| Signal Sensitivity | Excellent (zeptomole range) | Very Good (low attomole range) |
| Dynamic Range | ~3-4 orders of magnitude | ~3-4 orders of magnitude |
| Multiplexing Capability | No (single channel) | Yes (2-3 colors simultaneously) |
Detailed Experimental Protocols
Protocol A: Radioactive EMSA using ³²P
- End-Labeling: Incubate 10 pmol of DNA oligonucleotide with 10 units of T4 Polynucleotide Kinase and 50 µCi of [γ-³²P]ATP in 1X PNK buffer for 30 minutes at 37°C.
- Purification: Remove unincorporated nucleotides using a spin column (e.g., G-25 Sephadex).
- Binding Reaction: Combine 10,000 cpm of labeled probe with 5-20 µg of nuclear extract, 1 µg poly(dI-dC), in binding buffer (10 mM HEPES, 50 mM KCl, 0.5 mM EDTA, 1 mM DTT, 5% glycerol). Incubate 20 minutes at room temperature.
- Electrophoresis: Load samples onto a pre-run 6% non-denaturing polyacrylamide gel in 0.5X TBE. Run at 100V for 60-90 minutes at 4°C.
- Detection: Transfer gel to blotting paper, dry under vacuum. Expose to a phosphor storage screen for 2-16 hours. Scan screen with a phosphorimager.
Protocol B: Fluorescent EMSA using Cy5
- Probe Labeling: Order HPLC-purified, 5'-Cy5-labeled oligonucleotides commercially or label using a Cy5 maleimide kit per manufacturer's instructions. No purification typically required post-synthesis.
- Binding Reaction: Combine 2-20 fmol of Cy5-labeled probe with 5-20 µg of nuclear extract, 1 µg poly(dI-dC), in the same binding buffer as Protocol A. Incubate 20 minutes at room temperature. Note: Protect from light from this step forward.
- Electrophoresis: Load samples onto a pre-run 6% non-denaturing polyacrylamide gel. Run at 100V for 60-90 minutes at 4°C in the dark or with foil-covered apparatus.
- Detection: Directly scan the wet gel using a fluorescence scanner (e.g., Typhoon, Azure) with appropriate excitation/emission settings for Cy5 (649/670 nm). Scan takes 2-5 minutes.
Visualizing the Workflow Comparison
Title: EMSA Detection Method Workflow Comparison
The Scientist's Toolkit: Key Research Reagent Solutions
Essential Materials for EMSA Experiments
| Item | Function in EMSA | Radioactive Specifics | Fluorescent Specifics |
|---|---|---|---|
| T4 PNK & [γ-³²P]ATP | Catalyzes transfer of ³²P-phosphate to 5' end of DNA. | Essential. Requires strict safety protocols. | Not used. |
| Fluorescently-Labeled Oligo | Chemically stable probe with fluorophore (Cy5, Cy3, FAM). | Not used. | Essential. Can be ordered custom. |
| Non-denaturing PAGE Gel | Matrix for separating protein-DNA complexes from free probe. | 6-8% acrylamide, 0.5X TBE. | Often requires low-fluorescence glass plates. |
| Phosphor Storage Screen | Captures beta particle emission for high-sensitivity imaging. | Critical consumable. | Not used. |
| Poly(dI-dC) | Non-specific competitor DNA to reduce protein-non-specific DNA binding. | Used in both methods. | Used in both methods. |
| Nuclear Extraction Kit | Isolates DNA-binding proteins from cells for binding reactions. | Used in both methods. | Used in both methods. |
| Phosphorimager | Instrument to scan and quantify the phosphor screen. | Major capital equipment. | Not used. |
| Laser Fluorescence Scanner | Instrument to excite and detect fluorescence from gels. | Not used. | Major capital equipment. |
| Lead Shielding & Waste Containers | Safety equipment for handling and disposing of radioactivity. | Mandatory. | Not needed. |
Within the thesis comparing radioactive (³²P) versus fluorescent detection methods for Electrophoretic Mobility Shift Assays (EMSAs), a rigorous quantitative analysis of performance metrics is essential. This guide objectively compares these two core methodologies based on experimental data for dynamic range, linearity, and reproducibility, providing researchers and drug development professionals with actionable insights for assay selection.
Quantitative Performance Comparison
The following data are synthesized from recent, peer-reviewed studies directly comparing ³²P-radiolabeled probes with fluorophore-labeled probes (e.g., Cy5, FAM) in EMSAs for transcription factor-DNA interactions.
Table 1: Quantitative Comparison of EMSA Detection Methods
| Performance Metric | Radioactive Detection (³²P) | Fluorescent Detection (Cy5) | Experimental Context |
|---|---|---|---|
| Dynamic Range | >4.5 orders of magnitude | 3-3.5 orders of magnitude | Quantification of bound vs. free probe from gel images. |
| Linearity (R²) | 0.998 (Signal vs. Amount) | 0.985 (Signal vs. Amount) | Serial dilution of a known protein-DNA complex. |
| Inter-Assay CV | 8-12% | 5-8% | Coefficient of Variation (CV) for replicate experiments (n=6) over different days. |
| Limit of Detection (fmol) | 0.1 - 0.5 fmol | 2 - 5 fmol | Minimum amount of detected complex at SNR > 3. |
| Data Acquisition Time | 24-72 hour exposure | 10-30 minute scan | Time from gel completion to analyzable image. |
Detailed Experimental Protocols
Protocol 1: Direct Comparison of Dynamic Range and Linearity
Objective: To determine the linear dynamic range for quantifying protein-DNA complexes. Materials: Purified transcription factor (e.g., p50), ³²P-end-labeled DNA probe, Cy5-end-labeled identical probe. Method:
- Prepare a constant amount of DNA probe (20 fmol) with a serial dilution of the purified protein across 8 orders of magnitude.
- Perform binding reactions in identical buffer conditions, followed by non-denaturing PAGE (6%) for each probe type on separate gels.
- Detection:
- Radioactive: Dry gel and expose to a phosphor storage screen for 24 hours. Image using a phosphorimager.
- Fluorescent: Scan the wet gel directly using a laser scanner equipped with a 635 nm excitation/670 nm emission filter set.
- Quantify the pixel intensity of shifted bands using ImageQuant or ImageJ software. Plot signal intensity versus protein concentration to calculate linear range and R².
Protocol 2: Assessing Inter-Assay Reproducibility
Objective: To determine the Coefficient of Variation (CV) across independent experiments. Materials: Nuclear extract, single batch of labeled probes (³²P and Cy5). Method:
- Perform six independent EMSA experiments on different days using the same protocol, operator, and reagents.
- For each experiment, include a triplicate reaction of a standardized sample (e.g., extract + probe at Kd concentration).
- After detection and imaging, quantify the bound complex intensity for each replicate.
- Calculate the mean and standard deviation of the bound complex signal across all six experiments (n=18 data points). The inter-assay CV = (Standard Deviation / Mean) * 100%.
Visualizing EMSA Workflow and Detection Pathways
EMSA Comparative Workflow Diagram
Thesis Conceptual Framework
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for EMSA Studies
| Item | Function in EMSA | Example Product/Catalog |
|---|---|---|
| T4 Polynucleotide Kinase (PNK) | End-labels DNA probes with ³²P-γ-ATP or fluorescently-tagged ATP. | Thermo Scientific #EK0031 |
| Cy5-ddATP | Fluorescent terminator for efficient end-labeling of probes. | Cytiva #PA55021 |
| Non-denaturing PAGE Kit | Provides optimized acrylamide, buffers, and stains for native gel electrophoresis. | Bio-Rad #4561023 |
| Poly(dI:dC) | Non-specific competitor DNA to reduce background from non-specific protein binding. | Sigma-Aldrich #P4929 |
| Phosphor Storage Screen | Captures and stores radioactive emission from ³²P for imaging. | Cytiva #28-9564-75 |
| Typhoon or Azure Imager | Multi-modal scanner for both phosphor (radioactive) and fluorescence detection. | Cytiva Typhoon FLA 9500 |
| EMSAbuffer Kit | Pre-mixed binding buffers with salts, glycerol, and carriers for consistent reactions. | Thermo Scientific #20148 |
| Gel Shift Analysis Software | Quantifies band intensity and calculates fraction bound for Kd determination. | ImageQuant TL |
Choosing between radioactive (³²P) and fluorescent detection for Electrophoretic Mobility Shift Assays (EMSA) is a critical decision in nucleic acid-protein interaction studies. This guide provides a comparative analysis framed within the thesis that fluorescent EMSA is now the superior default choice for most applications, with radioactive methods reserved for specific, high-sensitivity requirements.
Performance Comparison: Radioactive vs. Fluorescent EMSA
The following table synthesizes key comparative data from recent methodological studies and product literature.
Table 1: Direct Comparison of EMSA Detection Methodologies
| Parameter | Radioactive (³²P) Detection | Fluorescent Detection (Cy5, IRDye 800) |
|---|---|---|
| Sensitivity | Extremely High (low attomole range). Can detect very low-abundance complexes. | High (mid-to-high attomole range). Sufficient for most in vitro studies. |
| Dynamic Range | ~3 orders of magnitude. Can be limited by film saturation. | >4 orders of magnitude. Linear quantification is superior with digital imaging. |
| Exposure/Scan Time | Minutes to days (film); minutes to hours (phosphorimager). | Seconds to minutes (laser scanner). |
| Signal Stability | Short (radioactive decay, half-life ~14.3 days). | Long-term (years when stored properly). |
| Hazard & Regulation | High (radioactive material; requires specialized licensing, disposal, shielding). | Minimal to None (standard chemical safety). |
| Cost per Assay | Lower reagent cost, but very high infrastructure & waste disposal costs. | Higher reagent cost, but minimal overhead. |
| Multiplexing Capability | No (single probe per gel). | Yes (multiple differentially labeled probes in one lane). |
| Workflow & Throughput | Slow, safety-intensive, low-throughput. | Fast, safe, amenable to higher throughput. |
| Quantification | Possible with phosphorimager, but linear range can be limited. | Excellent linear quantification with modern fluorimeters. |
Experimental Protocols for Key Comparative Studies
Protocol 1: Direct Sensitivity Comparison Experiment
- Objective: Determine the limit of detection (LOD) for a known DNA-protein complex using both methods.
- Method:
- Prepare an identical series of binding reactions with a constant amount of purified transcription factor (e.g., NF-κB p50) and a serial dilution of its consensus dsDNA probe.
- Labeling: Split the probe stock. Label one aliquot with [γ-³²P] ATP via T4 Polynucleotide Kinase. Label the other chemically with a fluorescent dye (e.g., Cy5) NHS ester.
- EMSA: Run identical gels (6% non-denaturing polyacrylamide) for each probe set under the same electrophoretic conditions.
- Detection:
- Radioactive: Dry gel and expose to a phosphor storage screen for 2 hours. Scan with a phosphorimager.
- Fluorescent: Image the wet gel directly using a laser scanner with appropriate excitation/emission settings (e.g., 633 nm/670 nm for Cy5).
- Analysis: Plot signal intensity of the shifted band vs. probe amount to calculate LOD for each method.
Protocol 2: Multiplexing Capability Demonstration
- Objective: Detect two distinct protein complexes in a single gel lane.
- Method:
- Design two unrelated DNA probes (e.g., one for NF-κB, one for AP-1).
- Label the NF-κB probe with IRDye 800CW (emission ~800 nm) and the AP-1 probe with Cy5 (emission ~670 nm).
- Set up binding reactions: one with HeLa nuclear extract only, one with extract + NF-κB competitor, one with extract + AP-1 competitor, and one with both competitors.
- Run a single EMSA gel.
- Detection: Scan the gel sequentially at 785 nm and 685 nm channels. The two protein-DNA complexes are distinguished by their distinct emission wavelengths, demonstrating specific competition.
Visualization of Workflows and Decision Logic
Title: Radioactive EMSA Workflow
Title: Fluorescent EMSA Workflow
Title: EMSA Detection Method Decision Matrix
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Modern EMSA
| Item | Function & Key Feature | Example (Vendor-Neutral) |
|---|---|---|
| Chemically-labeled DNA Oligos | Provides the fluorescent probe. 5'- or internal dye modification (e.g., Cy5, FAM, IRDye) offers stability and safety. | HPLC-purified, duplexed DNA probe with a 5' fluorescent dye. |
| Non-denaturing PAGE Gel Kit | Matrix for separating protein-DNA complexes from free probe. Pre-cast gels improve reproducibility and speed. | 6-8% Tris-Borate-EDTA (TBE) or Tris-Glycine polyacrylamide gels. |
| Fluorescent Gel Imager | Instrument for detecting and quantifying fluorescent signals. Laser-based scanners offer high sensitivity and multiplex channel detection. | Near-infrared (NIR) or multi-channel laser gel scanner. |
| Mobility Shift Buffer Systems | Provides optimized salt, pH, and carrier conditions for specific protein-DNA interactions. Commercial kits reduce optimization time. | 10X Binding Buffer with DTT, Poly(dI:dC), and stabilizers. |
| Positive Control Protein/Extract | Validates the entire assay. Recombinant protein or validated nuclear extract ensures reliability. | Purified p50 protein or HeLa/HEK293 nuclear extract. |
| Super-shift Antibodies | Confirms protein identity in the complex. Antibody against the target protein causes a further "supershift". | Anti-NF-κB p50 monoclonal antibody. |
| Gel Storage Buffer | For fluorescent EMSA; allows imaging without drying. Prevents gel dehydration and cracking. | 1X TBE in sealed plastic pouch or imaging cassette. |
Conclusion
The choice between radioactive and fluorescent EMSA detection is not a simple binary but a strategic decision based on specific research goals, laboratory infrastructure, and regulatory environment. While radioactive methods offer unparalleled sensitivity for detecting low-abundance or weak interactions, fluorescent techniques provide a safer, more stable, and increasingly sensitive alternative ideal for high-throughput applications and quantitative analysis. The future of EMSA lies in the continued refinement of fluorescent dyes and detection systems, potentially closing the sensitivity gap entirely. For the biomedical research community, this evolution enables more accessible, multiplexed, and quantitative analysis of nucleic acid-protein interactions, accelerating drug discovery and mechanistic studies in gene regulation.