DMS-MaPseq: The Complete Guide to In Vivo RNA Structure Profiling for Biomedical Research
This comprehensive guide explores DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing), a revolutionary method for probing RNA secondary and tertiary structure directly in living cells.
DMS-MaPseq: The Complete Guide to In Vivo RNA Structure Profiling for Biomedical Research
Abstract
This comprehensive guide explores DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing), a revolutionary method for probing RNA secondary and tertiary structure directly in living cells. We cover the foundational principles of chemical probing, provide a detailed walkthrough of the experimental protocol from cell treatment to computational analysis, and address common troubleshooting challenges. The article critically compares DMS-MaPseq to alternative structure-probing techniques like SHAPE-Seq and PARIS, validating its advantages for capturing native, in vivo RNA conformations. Designed for researchers, scientists, and drug development professionals, this resource aims to empower the application of this powerful technique to uncover RNA structure-function relationships, identify therapeutic targets, and advance RNA-based drug discovery.
What is DMS-MaPseq? Unraveling the Principles of In Vivo RNA Structure Analysis
Application Notes: Integrating DMS-MaPseq into Functional RNA Biology
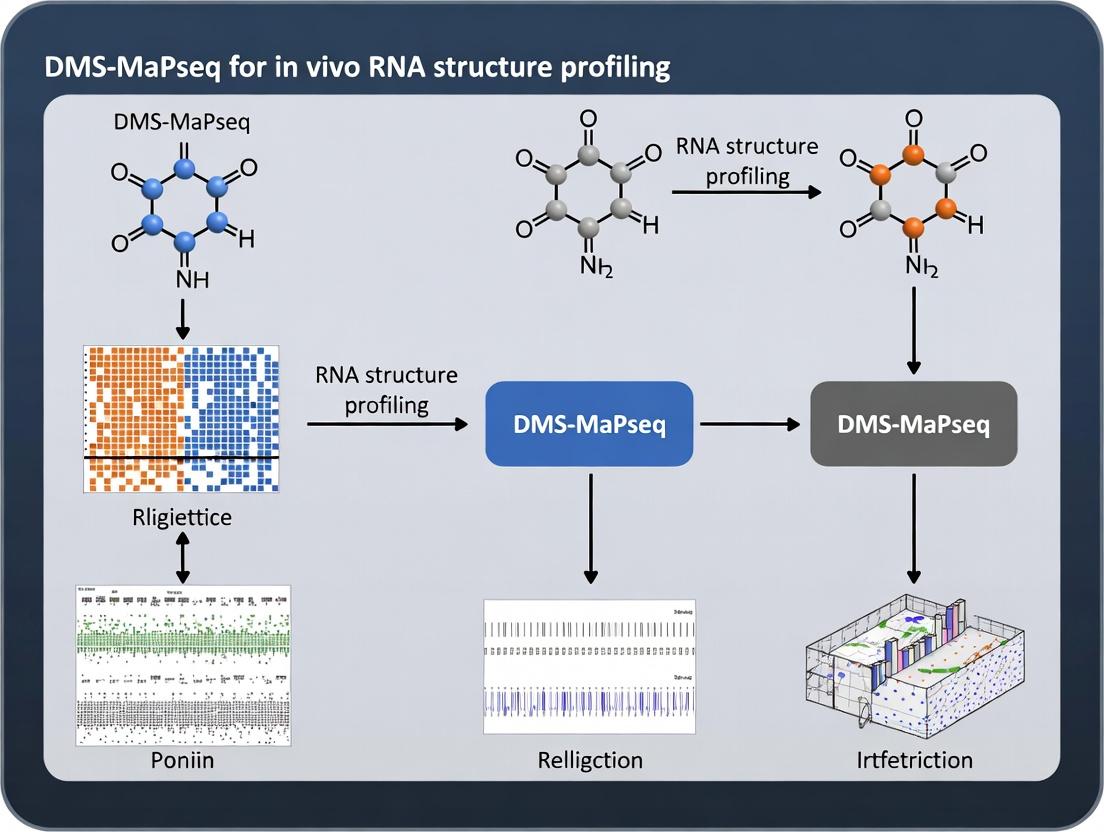
Recent advances in in vivo RNA structure probing, specifically DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing), have revolutionized our understanding of RNA's functional roles beyond sequence. DMS is a small, cell-permeable chemical that methylates unpaired adenine (A) and cytosine (C) nucleotides. In MaPseq, reverse transcriptase reads through these methylated bases, incorporating mismatches into the cDNA, which are then quantified by high-throughput sequencing to generate single-nucleotide reactivity profiles. These profiles serve as a proxy for RNA flexibility and secondary structure. This protocol suite details how to apply DMS-MaPseq to link RNA structure to biological function and disease mechanisms.
Table 1: Quantitative Insights from DMS-MaPseq Studies on Disease-Associated RNAs
| RNA Target / System | Key Structural Finding | Functional/Disease Implication | Validation Method |
|---|---|---|---|
| SARS-CoV-2 Genome in vivo | Highly conserved, structured regions in 5' and 3' UTRs | Essential for viral replication; candidate for antisense oligonucleotides (ASOs). | ASO-mediated inhibition in cell culture. |
| X-Inactive Specific Transcript (XIST) lncRNA | Specific hairpins crucial for A-repeat repeat B protein interactions. | Required for X-chromosome silencing. | CRISPR-mediated structure disruption and RNA-FISH. |
| BRCA1 mRNA 5' UTR | IRES-like element with defined structure regulates translation. | Somatic mutations alter structure, dysregulating BRCA1 oncoprotein synthesis. | Dual-luciferase reporter and ribosome profiling. |
| C9orf72 hexanucleotide repeat expansion | G-quadruplex and other structures in pathologic (GGGGCC)n repeats. | Promotes RAN translation and nucleolar dysfunction in ALS/FTD. | Small molecule G4 stabilizers & in vitro translation assays. |
| SMN2 exon 7 splicing element | A transient stem-loop structure modulates splice site recognition. | Structural stabilization can promote exon 7 inclusion, treating Spinal Muscular Atrophy. | SMN-C2 small molecule binder and RT-qPCR of splicing. |
Protocol 1: In Vivo DMS Treatment and RNA Harvest for Cultured Mammalian Cells Objective: To obtain RNA with DMS modifications reflecting native cellular structure. Materials: Adherent cells (e.g., HEK293T), growth media, fresh DMS solution (1:25 in anhydrous ethanol), ice-cold PBS, quenching buffer (1M β-mercaptoethanol in PBS), TRIzol reagent.
- Culture Cells: Grow cells to ~80% confluency in a 10cm dish.
- DMS Treatment: Aspirate media. Add 2mL of pre-warmed media containing 0.5% DMS (v/v). Incubate for 5 minutes at 37°C, 5% CO₂. (Critical: Perform in a fume hood, DMS is toxic.)
- Quench Reaction: Quickly aspirate DMS media. Immediately add 5mL of quenching buffer to inactivate residual DMS. Incubate for 2 minutes on a rocker.
- Wash & Lyse: Aspirate quenching buffer, wash cells twice with 10mL ice-cold PBS. Add 1mL TRIzol to the dish and lyse cells directly.
- RNA Isolation: Proceed with standard TRIzol-chloroform RNA extraction. Precipitate RNA with isopropanol, wash with 75% ethanol, and resuspend in RNase-free water. Determine concentration and integrity (RIN > 8.5).
Protocol 2: Library Preparation for DMS-MaPseq Objective: To generate sequencing libraries from DMS-modified RNA using mutation-prone reverse transcription. Materials: DNase I, Superscript IV reverse transcriptase (Thermo Fisher), random hexamers, dNTPs, Second Strand Synthesis enzyme mix (NEB), library prep kit (e.g., Nextera XT).
- RNA Clean-up: Treat 5-10 µg of total RNA with DNase I. Purify using RNA clean-up beads.
- Mutational Profiling RT: In a 20µL reaction, mix 1µg RNA, 50µM random hexamers, 500µM dNTPs, and 1x SSIV buffer. Heat to 85°C for 2min, then hold at 25°C. Add 200U of SSIV and incubate: 10min at 25°C, 15min at 42°C, 10min at 50°C, then 80°C for 10min. (SSIV is critical for reading through DMS modifications.)
- cDNA Purification & Second Strand Synthesis: Purify cDNA using SPRI beads. Synthesize dsDNA using the Second Strand Synthesis kit. Purify again.
- Library Construction: Fragment and tag the dsDNA using the Nextera XT kit (6 cycles of PCR). Include unique dual indices. Clean up libraries with beads.
- Sequencing: Pool libraries and sequence on an Illumina platform (Minimum 5 million 150bp paired-end reads per sample).
Visualization of Key Concepts and Workflows
Diagram Title: DMS-MaPseq Workflow & Therapeutic Pipeline
The Scientist's Toolkit: Essential Research Reagent Solutions
| Item | Function in DMS-MaPseq/RNA Structure Research |
|---|---|
| Dimethyl Sulfate (DMS) | Small cell-permeable chemical probe that methylates unpaired A and C nucleotides. |
| Superscript IV Reverse Transcriptase | Engineered to read through DMS-methylated bases, introducing mutations during cDNA synthesis. |
| β-mercaptoethanol | Quenching agent that rapidly inactivates residual DMS to halt probing. |
| Structure-Specific Small Molecules (e.g., SMN-C2) | Validates functional importance by stabilizing or destabilizing predicted RNA structures. |
| Antisense Oligonucleotides (ASOs) / Gapmers | Target accessible, single-stranded regions mapped by DMS to modulate RNA function. |
| Next-Generation Sequencing Kit (e.g., Nextera XT) | Enables preparation of multiplexed sequencing libraries from low-input cDNA. |
Computational Pipeline (e.g., dms-tools2, ShapeMapper2) |
Processes sequencing data to calculate mutation rates and model RNA secondary structure. |
The study of RNA structure has long relied on in vitro techniques, which, while informative, often fail to capture the complex realities of the cellular environment. The broader thesis of this research posits that in vivo RNA structure is fundamentally governed by trans-acting factors, macromolecular crowding, and constant metabolic activity, necessitating technologies like DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing) for accurate profiling. This application note details protocols and insights for moving structural biology from the test tube into the living cell.
The Limitations ofIn VitroData: A Quantitative Comparison
Table 1: Discrepancies Between In Vitro and In Vivo RNA Structural Metrics for a Model Riboswitch
| Structural Metric | In Vitro (SHAPE) | In Vivo (DMS-MaPseq) | Discrepancy Notes |
|---|---|---|---|
| Ligand-Bound State Occupancy | 92% ± 3% | 58% ± 7% | Cellular metabolites reduce apparent affinity. |
| Apical Loop Reactivity (mean) | 0.15 | 0.62 | Protein binding in vivo shields specific nucleotides. |
| P1 Helix Stability (ΔG) | -4.2 kcal/mol | -2.8 kcal/mol | Ionic conditions and crowding alter stability. |
| Key Tertiary Contact Formation | Yes | No | Competing RNA-RNA interactions in cell disrupt. |
| Structural Resolution (nt) | 1-2 | 1-2 | DMS-MaPseq maintains single-nucleotide resolution. |
DMS-MaPseq: A Protocol forIn VivoRNA Structure Probing
Protocol 1:In VivoDMS Treatment and RNA Harvesting
Objective: To modify accessible RNA adenines (A) and cytosines (C) in living cells.
- Culture & Treatment: Grow relevant cell line (e.g., HEK293T) to 70-80% confluency. For adherent cells, aspirate media and add fresh media containing 0.5% (v/v) DMS. Incubate for 5 minutes at 37°C, 5% CO₂.
- Quenching: Aspirate DMS media and immediately quench reaction by adding ice-cold 0.1M Tris-HCl (pH 8.0) + 0.1M β-mercaptoethanol. Wash cells twice with quench solution.
- RNA Extraction: Lyse cells directly on plate using TRIzol reagent. Perform chloroform extraction and isopropanol precipitation. Resuspend total RNA in RNase-free water. Critical: Do not use heating or denaturing conditions that will reverse DMS modifications.
Protocol 2: MaP Reverse Transcription and Library Construction
Objective: To convert DMS modifications into cDNA mutations during reverse transcription.
- Priming: Use 2 µg of total RNA. For specific RNA targets, use gene-specific primers. For transcriptome-wide studies, use random hexamers.
- Mutagenic RT: Set up reverse transcription using SuperScript II or a similar enzyme with the following conditions:
- 1X First Strand Buffer
- 5 mM MnCl₂ (replaces Mg²⁺) – Essential for mutation incorporation
- 1 mM each dNTP
- 10 mM DTT
- Primer (250 ng random hexamer or 2.5 pmol gene-specific)
- Incubate: 42°C for 3 hours.
- cDNA Purification: Clean up cDNA using SPRI beads.
- Library Amplification: Perform PCR amplification (12-16 cycles) with Illumina-compatible adapter primers. Include a unique sample index.
- Sequencing: Purify library and sequence on an Illumina platform (Minimum depth: 10-20 million reads per sample for transcriptome-wide).
Protocol 3: Data Analysis Pipeline
- Alignment: Map reads to reference genome/transcriptome using
STARorHISAT2with stringent parameters. - Mutation Calling: Use
DREEMorMapSeekersoftware to identify DMS-induced mutation rates at each A and C nucleotide, correcting for background sequencing error. - Reactivity Calculation: Calculate normalized DMS reactivity. Low reactivity indicates base-paired or protein-protected nucleotides; high reactivity indicates flexibility and accessibility.
- Structure Modeling: Feed reactivity profiles into constrained folding algorithms (
RNAstructure,ViennaRNA) to generate ensemble of probable in vivo structures.
Visualizing the Workflow and Impact
Title: DMS-MaPseq In Vivo Workflow
Title: Cellular Factors Shaping In Vivo RNA Structure
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for DMS-MaPseq In Vivo Studies
| Item & Supplier Example | Function in Protocol |
|---|---|
| Dimethyl Sulfate (DMS) (Sigma) | Small chemical probe that methylates accessible A and C nucleotides. Cell-permeable. |
| SuperScript II Reverse Transcriptase (Thermo Fisher) | Engineered RT tolerant of Mn2+, crucial for reading through DMS modifications and incorporating mismatches. |
| MnCl₂ Solution (NEB) | Divalent cation used in place of Mg2+ during RT to promote non-templated nucleotide incorporation at DMS-adducted sites. |
| TRIzol Reagent (Thermo Fisher) | Monophasic solution for simultaneous cell lysis and RNA stabilization, preserving in vivo modification state. |
| SPRI Beads (Beckman Coulter) | Magnetic beads for size selection and purification of cDNA libraries, removing primers and enzymes. |
| Random Hexamer Primers (IDT) | For unbiased, transcriptome-wide initiation of reverse transcription. |
| DREEM Analysis Software (Open Source) | Computationally extracts mutation rates from sequencing data to generate DMS reactivity profiles. |
The integration of in vivo DMS-MaPseq protocols into structural studies is non-negotiable for understanding RNA biology in its native context. The detailed protocols and tools outlined here provide a roadmap for researchers and drug developers aiming to target RNA structures with therapeutic intent, moving beyond the oversimplified models derived from test tube analyses.
Within the context of DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing) for in vivo RNA structure probing, understanding the precise chemical reactivity of DMS is fundamental. DMS is an electrophilic methylating agent that selectively modifies the Watson-Crick base-pairing faces of RNA bases only when they are not involved in canonical base pairing or tertiary interactions. This modification forms the core signal for inferring RNA secondary structure.
Key Chemical Reactions:
- With Adenine (N1): DMS methylates the N1 position of adenine. N1-methylated adenine (m1A) can base-pair with thymine/uracil but not with uracil in RNA, leading to a mutation signature during reverse transcription.
- With Cytosine (N3): DMS methylates the N3 position of cytosine. N3-methylated cytosine (m3C) blocks base pairing with guanine.
These methylations are efficiently detected in DMS-MaPseq because reverse transcriptase often misincorporates a nucleotide or terminates at the modified base. The resulting mutation patterns are quantified by high-throughput sequencing to map single-stranded, unpaired regions of RNA in vivo.
Table 1: DMS Reactivity with RNA Nucleobases
| Nucleobase | Reactive Atom | Structural Context for Reactivity | Consequence of Methylation | Detection in DMS-MaPseq |
|---|---|---|---|---|
| Adenine (A) | N1 position | Unpaired, accessible, not shielded by structure | Disrupts A-U pairing; promotes misincorporation | Mutation (A→G/C/U) or truncation |
| Cytosine (C) | N3 position | Unpaired, accessible, not shielded by structure | Disrupts C-G pairing; blocks reverse transcription | Mutation (C→T/A/G) or truncation |
| Guanine (G) | N7 position (minor) | Reactive at higher DMS concentrations; paired or unpaired. | Does not block Watson-Crick face. | Not a primary signal for pairing. |
| Uracil (U) | Not reactive | -- | -- | -- |
Key Protocols for DMS ProbingIn Vivo
Protocol 1: Standard In Vivo DMS Treatment for Bacterial or Cultured Eukaryotic Cells
Objective: To modify accessible adenines and cytosines in cellular RNA with DMS under native conditions.
Materials & Reagents: See The Scientist's Toolkit below. Procedure:
- Cell Preparation: Grow cells to mid-log phase. For a typical experiment, harvest 1-5 x 10^7 cells per condition.
- DMS Treatment: Resuspend cell pellet in pre-warmed growth media. Add DMS to a final concentration of 0.5% (v/v). Incubate for 5 minutes at the cell's growth temperature (e.g., 37°C) with gentle agitation.
- Critical: Include a no-DMS control (mock-treated with solvent only).
- Quenching: Stop the reaction by adding an equal volume of ice-cold DMS Quench Buffer (e.g., 2-Mercaptoethanol or β-mercaptoethanol in PBS). Mix immediately and pellet cells on ice.
- Washing: Wash cell pellet twice with ice-cold PBS.
- RNA Extraction: Lyse cells and perform total RNA extraction using a hot acid-phenol:chloroform method (e.g., TRIzol) to ensure recovery of all RNA species. Treat with DNase I.
- Quality Control: Assess RNA integrity (RIN > 8.5) via Bioanalyzer/TapeStation. Proceed to library preparation for MaPseq.
Protocol 2: DMS Modification of Purified RNA (In Vitro Control)
Objective: To create a fully modified control for mutation background assessment.
Procedure:
- Dilute 2-5 µg of purified, DNase-treated RNA in 100 µL of RNA Folding Buffer (e.g., 50 mM HEPES-KOH pH 8.0, 100 mM KCl).
- Denature at 95°C for 2 min, then snap-cool on ice to remove pre-existing structures.
- Add DMS to a final concentration of 2% (v/v). Incubate for 10 minutes at 30°C.
- Quench with 2-Mercaptoethanol and purify RNA via ethanol precipitation.
Experimental Workflow and Data Interpretation
Diagram Title: DMS-MaPseq Experimental Workflow from Cells to Structure
Data Interpretation:
- Reactivity Score: Per-nucleotide DMS reactivity is calculated from mutation rates, normalized to control and often to the 92nd percentile of reactivities. High reactivity = unpaired/base accessible. Low reactivity = paired/structured.
- Structure Modeling: Reactivity profiles are used as constraints in computational folding algorithms (e.g.,
RNAstructure,Superfold) to predict the most probable secondary structure model.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for DMS Probing Experiments
| Reagent / Solution | Function / Purpose | Critical Notes |
|---|---|---|
| Dimethyl Sulfate (DMS) | Electrophilic probe for A/C N-atoms. | Highly toxic/carcinogen. Use in fume hood with proper PPE. Purchase in small aliquots. |
| 2-Mercaptoethanol (BME) / DTT | Quenching agent. Scavenges unreacted DMS. | Must be fresh and concentrated. Quenching must be immediate. |
| Acid Phenol:Chloroform (e.g., TRIzol) | For total RNA extraction post-DMS. | Denatures proteins, inactivates RNases, recovers small RNAs. |
| DNase I (RNase-free) | Removes genomic DNA contamination. | Essential to prevent false signals in sequencing. |
| Mutational RT Enzyme (e.g., TGIRT-III, MarathonRT) | Reverse transcriptase with high processivity and misincorporation tolerance. | Key to reading through m1A/m3C and recording mutations. |
| RNA Folding Buffer (HEPES-KCl/Mg2+) | Provides physiological ionic conditions for in vitro folding/control. | Mg2+ concentration is critical for native-like folding. |
| High-Sensitivity RNA Assay Kits (Bioanalyzer) | Assess RNA integrity post-extraction. | Degraded RNA leads to noisy, unreliable reactivity data. |
Application Notes
Within the context of DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing) for in vivo RNA structure probing, the core breakthrough of MaPseq is the utilization of engineered reverse transcriptases (RTs) that continue synthesis past chemical-adduction sites (e.g., from DMS) rather than terminating. These RTs misincorporate nucleotides at and around the adducted base, creating mutation signatures in the cDNA that are directly detectable by high-throughput sequencing. This allows for single-molecule, single-nucleotide resolution of RNA structure and interactions.
Key Quantitative Findings from Recent Studies:
Table 1: Performance Metrics of MaP-Compatible Reverse Transcriptases
| Reverse Transcriptase | Read-Through Efficiency at DMS Modifications | Mutation Rate (Background) | Mutation Rate (at DMS-modified A/C) | Optimal Reaction Temperature |
|---|---|---|---|---|
| Group II Intron RT (TGIRT-III) | >95% | ~0.001% per nt | ~2-8% per modified nt | 55-60°C |
| HIV-1 RT (Mutant MMLV) | >90% | ~0.01% per nt | ~5-10% per modified nt | 42-50°C |
| Wild-type MMLV | <20% (terminates) | ~0.001% per nt | N/A | 42°C |
Table 2: DMS-MaPseq Experimental Outcomes
| Metric | Typical Result | Impact on Structure Modeling |
|---|---|---|
| Mutation Density (DMS-treated) | 0.01 - 0.05 mutations per nt | Provides sufficient signal for reactivity calculation. |
| Signal-to-Noise Ratio | 10:1 to 50:1 (DMS vs. control) | Enables high-confidence identification of paired/unpaired nucleotides. |
| Single-Molecule Coverage | 10-100x reads per RNA molecule | Allows for covariance analysis and detection of heterogeneous structures. |
| Resolution | Single nucleotide | Precise definition of RNA structural elements. |
Detailed Experimental Protocols
Protocol 1:In VivoDMS Probing for MaPseq
Function: To modify structurally accessible adenosine (N1) and cytidine (N3) atoms in native cellular RNA.
- Cell Treatment: For cultured cells, dilute pure DMS to 0.5% (v/v) in pre-warmed growth media. Incubate with cells for 5 minutes at 37°C. (Optimize concentration/time for specific cell types).
- Quenching: Aspirate DMS media and immediately add ice-cold quenching buffer (1M Tris-HCl pH 7.4, 1.2M β-mercaptoethanol). Wash cells twice with cold PBS.
- RNA Extraction: Lyse cells using a denaturing guanidinium thiocyanate-phenol-based reagent (e.g., TRIzol). Extract total RNA following manufacturer's protocol, including DNase I treatment.
- RNA Clean-up: Purify RNA using ethanol precipitation or silica-membrane columns. Quantify and assess integrity (RIN > 8.0 recommended).
Protocol 2: MaP Reverse Transcription
Function: To generate cDNA libraries with mutation signatures from DMS modifications.
- Priming: For a specific target or whole transcriptome, use 500 ng - 1 µg of total RNA. Add 2 pmol of gene-specific primer or 50 ng of random hexamers. Denature at 65°C for 5 min, then immediately place on ice.
- RT Master Mix: Prepare on ice:
- 1X First-Strand Buffer (supplied with RT)
- 1 mM each dNTP
- 5 mM DTT
- 10 U/µL RNase inhibitor
- 2 U/µL MaP-compatible RT (e.g., TGIRT-III or mutant MMLV)
- Extension: Combine RNA/primer with master mix. Incubate at the optimal temperature (55°C for TGIRT-III, 42°C for mutant MMLV) for 1-2 hours.
- RNA Degradation: Add RNase H (optional for TGIRT) or NaOH (0.1M final) and incubate at 65°C for 15 min to degrade RNA. Purify cDNA using SPRI beads.
Protocol 3: Library Preparation & Data Analysis
Function: To prepare sequencing libraries and process mutation data.
- Second Strand Synthesis & Amplification: Use PCR to add sequencing adapters and indices. Use a high-fidelity DNA polymerase for ≤12 cycles.
- Sequencing: Purify library and sequence on an Illumina platform (Paired-end 150 bp recommended).
- MaP Analysis Pipeline:
- Alignment: Map reads to reference transcriptome using sensitive aligners (e.g., HISAT2, STAR).
- Mutation Calling: Use dedicated software (e.g.,
ShapeMapper 2,dms-tools2) to identify mismatches relative to the reference, filtering PCR errors and sequencing artifacts. - Reactivity Calculation: Calculate DMS reactivity per nucleotide as (mutation rate in DMS sample) - (mutation rate in untreated control). Normalize to the 2-8% or 90-100th percentile of reactivities.
- Structure Modeling: Input reactivity profiles into folding algorithms (e.g.,
RNAstructure,Superfold) with constraints to predict secondary structure models.
Visualization
DMS-MaPseq Experimental Workflow
MaP RT Mechanism: Read-Through vs Termination
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for DMS-MaPseq
| Reagent / Material | Function & Importance | Example Product / Specification |
|---|---|---|
| Dimethyl Sulfate (DMS) | Small chemical probe that methylates accessible N1 of A and N3 of C. Penetrates cells for in vivo probing. | High-purity grade (≥99%), handle in fume hood with extreme care. |
| MaP-Compatible Reverse Transcriptase | Engineered RT that reads through DMS adducts, incorporating mismatches. Core of the MaP breakthrough. | TGIRT-III (InGex), MarathonRT (preferred for high processivity and fidelity). |
| RNase Inhibitor | Prevents RNA degradation during reverse transcription, critical for maintaining full-length templates. | Recombinant RNaseIN (40 U/µL). |
| SPRI Beads | For efficient size selection and clean-up of cDNA and sequencing libraries. Minimizes loss of material. | AMPure XP Beads. |
| High-Fidelity PCR Mix | For limited-cycle amplification of cDNA libraries. Minimizes introduction of PCR errors. | KAPA HiFi HotStart ReadyMix. |
| Bioinformatics Pipelines | Software to accurately call mutations, calculate reactivities, and model structures from sequencing data. | ShapeMapper 2, DREEM, RNAstructure. |
Application Notes
DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing) has revolutionized in vivo RNA structure probing by capturing dynamic RNA conformations within their native cellular environments. This application note details its core advantages in probing transient states, protein interactions, and contextual biology, critical for understanding RNA function and therapeutic targeting.
1. Capturing Transient and Low-Population RNA Structures Traditional chemical probing methods average signals across billions of molecules, missing short-lived intermediate folds. DMS-methylation creates covalent adducts that are recorded as mutations during reverse transcription (MaP). Single-molecule sequencing reads preserve multiple DMS modifications per RNA molecule, enabling the reconstruction of co-existing structural ensembles from a heterogeneous population.
Quantitative Data: Detection of Transient States
| Metric | DMS-MaPseq Performance | Conventional DMS-seq |
|---|---|---|
| Detection Sensitivity for Low-Population States | Can detect states at <10% abundance | Typically requires >30% abundance |
| Per-Read Modifications | 2-5 DMS modifications per 1000 nt read (enabling haplotype resolution) | Signal averaged per nucleotide across all reads |
| Data Output for Ensemble Deconvolution | 10^4 - 10^5 individual molecule read-outs per condition | Aggregate reactivity profile only |
2. Resolving Protein-Bound States and RNA-Protein Interactions DMS reactivity is attenuated at nucleotides directly protected by RNA-binding proteins (RBPs) or due to protein-induced structural remodeling. DMS-MaPseq in vivo, especially when combined with crosslinking or differential analysis in knock-out/knock-down models, identifies protein footprints and binding-induced conformational changes.
Quantitative Data: Identifying Protein-Bound Footprints
| Analysis Method | Typical Resolution | Information Gained |
|---|---|---|
| DMS Reactivity Decrease (Protection) | Single-nucleotide | Direct RBP binding site or rigidified region |
| DMS Reactivity Increase (Enhanced Accessibility) | Single-nucleotide | Protein-induced allosteric structural opening |
| Correlated Mutation Analysis (within single reads) | 2-50 nucleotide span | Coordinated protections defining larger interaction surfaces |
3. Preserving Native Cellular Context In vivo application preserves the full complement of cellular ions, metabolites, competing RNAs, and macromolecular crowding. Comparative in vivo vs. in vitro DMS-MaPseq reveals structures shaped by native cellular environment, including subcellular compartment-specific folding.
Quantitative Data: In Vivo vs. In Vitro Discrepancies
| RNA Class | Typical Nucleotide Discrepancy Rate (In Vivo vs. Denatured) | Biological Insight |
|---|---|---|
| mRNA 5' UTR | 25-40% nucleotides show reactivity change | Widespread regulation by translation machinery & RBPs |
| lncRNA | 30-50% nucleotides show reactivity change | Extensive stabilization via cellular protein partners |
| Viral RNA Genomes | 40-60% nucleotides show reactivity change | Massive reorganization induced by host cell environment |
Detailed Protocols
Protocol 1: Standard In Vivo DMS-MaPseq for Mammalian Cells
Objective: To profile RNA structural ensembles in their native cellular context.
I. Cell Treatment and RNA Extraction
- Culture & Treatment: Grow adherent cells (e.g., HEK293T) to 80% confluence in a 10 cm dish. Prepare fresh DMS solution (2% v/v in culture media or PBS). Aspirate media, wash once with PBS, and add 5 mL of DMS solution. Incubate for 5 minutes at 37°C with gentle rocking.
- Critical: Optimize DMS concentration and time to achieve ~1 modification per 200-400 nt. Include a no-DMS control.
- Quenching: Aspirate DMS and immediately quench with 10 mL of ice-cold 30% (v/v) β-mercaptoethanol in PBS. Wash cells twice with the quenching solution.
- Lysis & RNA Extraction: Lyse cells directly on plate with TRIzol reagent. Extract total RNA following manufacturer's protocol. Treat with DNase I. Purify RNA using ethanol precipitation. Assess integrity (RIN > 8.5).
II. Library Preparation for MaPseq
- Reverse Transcription (with Mutagenic MaP): For each sample (DMS-treated and control), set up 20 μL RT reactions using 2 μg total RNA, 200 U SuperScript II reverse transcriptase, and gene-specific or random primers. Use a thermocycler program: 25°C for 10 min (annealing), 42°C for 90 min (extension), 70°C for 15 min (inactivate). The RT enzyme read-through DMS-adducted bases (N1-methyladenosine and N3-methylcytidine) introduces mutations.
- cDNA Purification: Purify cDNA using 1.8x SPRI beads. Elute in 20 μL nuclease-free water.
- Second Strand Synthesis & PCR: Perform second-strand synthesis with Klenow exo- polymerase. Amplify cDNA for 12-18 cycles using Q5 Hot-Start polymerase and primers containing Illumina adaptor sequences.
- Library Purification & Sequencing: Size-select libraries (200-500 bp) via gel extraction or SPRI beads. Quantify by qPCR. Sequence on Illumina platform (Minimum: 5 million paired-end 150 bp reads per sample).
Protocol 2: Differential DMS-MaPseq to Identify Protein-Bound States
Objective: To identify RNA structural changes and protections induced by a specific RNA-binding protein (RBP).
I. Comparative Cell Line Treatment
- Isogenic Cell Lines: Use an RBP knock-out (KO) cell line and its wild-type (WT) isogenic control.
- Parallel Probing: Perform in vivo DMS treatment (as in Protocol 1) on both WT and KO cells in biological triplicate, simultaneously.
II. Data Analysis for Differential Footprinting
- Alignment & Mutation Calling: Align reads to transcriptome using HISAT2 or STAR. Call mutations relative to reference genome using tools like
dms-tools2orShapeMapper2, with the no-DMS control to establish background error rate. - Differential Reactivity Calculation: Calculate DMS reactivity per nucleotide as mutation rate. For each nucleotide, compute a differential reactivity score (Δreactivity = ReactivityKO - ReactivityWT). Use statistical testing (e.g., t-test) across replicates.
- Identification of Protected Regions: Nucleotides with significant negative Δreactivity in the KO (i.e., more reactive when protein is absent) indicate direct protection or protein-stabilized structure. Cluster adjacent significant nucleotides to define binding footprints.
The Scientist's Toolkit
| Research Reagent / Material | Function in DMS-MaPseq |
|---|---|
| Dimethyl Sulfate (DMS) | Small, cell-permeable chemical probe that methylates accessible adenine (N1) and cytosine (N3) atoms. Reactivity is inhibited by base-pairing or protein binding. |
| β-mercaptoethanol | Quenching agent that rapidly inactivates unreacted DMS, stopping the probing reaction. |
| SuperScript II Reverse Transcriptase | A retrotranscriptase with high processivity and tolerance for base modifications. Crucial for reading through DMS-methylated bases and incorporating mismatches (mutations) during cDNA synthesis. |
| Q5 Hot-Start High-Fidelity DNA Polymerase | Used in the PCR amplification of cDNA libraries. Its high fidelity ensures mutations from the MaP step are preserved and not introduced during amplification. |
| SPRI (Solid Phase Reversible Immobilization) Beads | Magnetic beads for size-selective purification and cleanup of nucleic acids (RNA, cDNA, final libraries) throughout the protocol. |
Visualizations
DMS-MaPseq Reveals RNA Structural Ensembles
Identifying RBP Footprints via Differential DMS-MaPseq
Step-by-Step Protocol: Implementing DMS-MaPseq in Your Research Workflow
This application note details experimental design principles for in vivo RNA structure profiling using DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing). Within the broader thesis on advancing in vivo RNA structural studies, robust experimental design is the critical foundation for generating reproducible, biologically relevant data. This document provides protocols and guidelines for three interdependent pillars: selecting physiologically relevant cell types, implementing rigorous controls, and optimizing the crucial parameter of DMS dosage.
Choosing Cell Types forIn VivoDMS-MaPseq
The choice of cell type directly determines the biological relevance of the captured RNA structures. Considerations must balance physiological context, experimental tractability, and RNA abundance.
Table 1: Common Cell Model Considerations for In Vivo DMS-MaPseq
| Cell Type/Category | Key Considerations & Applications | Typical Yield of Total RNA | Recommended DMS Penetration Method |
|---|---|---|---|
| Adherent Cell Lines (e.g., HEK293T, HeLa) | Easy culture, high RNA yield, ideal for method optimization and controlled perturbations. | 10-20 µg per 10⁶ cells | Direct incubation in culture medium. |
| Suspension Cell Lines (e.g., K562, Jurkat) | Easy scaling, homogeneous DMS exposure, suitable for biochemical fractionation studies. | 5-15 µg per 10⁶ cells | Direct incubation in culture medium. |
| Primary Cells (e.g., PBMCs, neurons) | High physiological relevance, more variable, may have lower RNA yield, limited expansion. | 1-5 µg per 10⁶ cells | Optimized, often lower, DMS concentration. |
| Yeast (S. cerevisiae) | Simple genetics, fundamental biology studies, robust cell wall. | 50-100 µg per OD₆₀₀ unit | Requires spheroplasting or use of DMSO as co-solvent. |
| Bacteria (e.g., E. coli) | Rapid growth, prokaryotic RNA biology, complex cell envelope. | 10-30 µg per OD₆₀₀ unit | Requires optimized permeability (e.g., Tris-EDTA buffer). |
| Stem Cells/Organoids | High relevance for development and disease; complex, heterogeneous structures. | Variable (protocol-dependent) | Careful optimization to maintain viability. |
Protocol 2.1: Culturing and Preparation of Adherent Cells for DMS Treatment
- Cell Culture: Grow HEK293T cells in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% FBS at 37°C, 5% CO₂.
- Harvesting: At ~80% confluency, aspirate media and wash cells once with 1x PBS.
- Trypsinization: Add 0.25% Trypsin-EDTA solution (1 mL per 10 cm dish) and incubate at 37°C for 3-5 minutes.
- Neutralization: Add complete media (2x volume of trypsin) to neutralize. Transfer cell suspension to a conical tube.
- Washing: Pellet cells at 300 x g for 5 minutes. Aspirate supernatant and resuspend pellet in 10 mL of 1x PBS. Repeat wash step.
- Counting & Aliquotting: Perform a cell count using a hemocytometer or automated counter. Resuspend the final cell pellet in the appropriate volume of 1x PBS or serum-free media to achieve a density of 1-2 x 10⁶ cells per 100 µL aliquot in a 1.5 mL microcentrifuge tube. Keep on ice until DMS treatment.
Essential Experimental Controls
Proper controls are non-negotiable for distinguishing DMS-induced mutations from background errors and for data normalization.
Table 2: Mandatory Controls for DMS-MaPseq Experiments
| Control Type | Purpose | Sample Preparation | Data Analysis Use |
|---|---|---|---|
| (-) DMS Control | Quantifies background mutation rate from reverse transcription and sequencing errors. | Split cell sample. Treat identical to experimental but with vehicle (e.g., ethanol) instead of DMS. | Background subtraction. Baseline for mutation rate calculation. |
| Proteinase K / Denatured Control | Identifies protein-protected RNA bases (footprinting) or defines maximum accessible signal. | Lyse cells and treat RNA with Proteinase K and/or heat denature before DMS addition. | Normalization for per-nucleotide reactivity. Calculation of protection scores. |
| (+) DMS In Vitro Control | Assesses MaP reverse transcriptase efficiency and confirms DMS activity on deproteinized RNA. | Purify total RNA from cells. Treat purified RNA with DMS in buffered solution. | Benchmarking in vivo reactivity profiles. |
| Biological Replicate | Accounts for biological variability; minimum n=3 independent experiments. | Treat independently cultured cell samples on different days. | Statistical significance testing (e.g., deltaSHAPE, Differential DMS). |
| Spike-in RNA Control | Normalizes for technical variation in DMS treatment, RNA recovery, and library prep. | Add a known amount of synthetic, structured RNA (e.g., tRNA, lncRNA fragment) to cell lysate immediately after DMS treatment. | Inter-experiment normalization and quality control. |
Protocol 3.1: Preparation of (-) DMS and Denatured Controls Part A: (-) DMS Control
- Prepare cell aliquots as in Protocol 2.1, Step 6.
- Vehicle Treatment: To the cell aliquot, add a volume of 100% ethanol equal to the volume of DMS you will use for the experimental sample (e.g., 1 µL). Mix gently.
- Incubate at the same temperature and for the same duration as the experimental sample.
- Proceed immediately to RNA extraction (see Protocol 4.2).
Part B: Denatured Control
- Prepare a cell aliquot. Pellet cells and lyse directly in 500 µL of Qiagen RLT Plus buffer (with β-mercaptoethanol) by vortexing.
- Add 1 µL of Proteinase K (20 mg/mL) and incubate at 37°C for 15 min.
- Denature RNA by heating at 95°C for 3 minutes, then immediately place on ice.
- Add DMS to the lysate (typical in vitro concentration: 0.5-2% v/v) and incubate at room temperature for 10 min.
- Proceed to RNA cleanup.
DMS Dosage Optimization Protocol
DMS methylates unpaired adenine (N1) and cytosine (N3) residues. Optimal dosage modifies a low fraction of bases (1-10%) to ensure single-hit kinetics and avoid structural perturbations or cell death.
Table 3: Recommended Starting DMS Dosage by Cell Type
| Cell Type | Recommended Starting Dose (% v/v DMS) | Incubation Conditions | Expected Mutation Rate (Background-Subtracted) | Viability Check Post-Treatment |
|---|---|---|---|---|
| Mammalian Cell Lines | 0.5 - 2% | In PBS or serum-free media, 23-37°C, 3-10 min. | 0.5% - 3% | Trypan Blue exclusion; >80% viability for dose chosen. |
| Yeast (Spheroplasted) | 1 - 3% | In appropriate osmotically stabilized buffer, 23-30°C, 5-10 min. | 1% - 4% | Plating efficiency assay. |
| Bacteria (E. coli) | 2 - 5% | In Tris-EDTA buffer, 23°C, 5-8 min. | 2% - 6% | Monitor OD600 growth curve after dilution and recovery. |
| Primary Mammalian Cells | 0.25 - 1% | In PBS, 23-37°C, 3-5 min. | 0.2% - 2% | Flow cytometry with viability dye. |
Protocol 4.1: DMS Dosage Optimization Titration
- Prepare Cells: Harvest and wash cells as in Protocol 2.1. Prepare 8 aliquots of 1 x 10⁶ cells each in 100 µL of 1x PBS in 1.5 mL tubes. Keep on ice.
- DMS Dilution Series: Prepare a 10% (v/v) stock of DMS in 100% ethanol in a fume hood. Perform serial dilutions in ethanol to create working stocks (e.g., 5%, 2.5%, 1.25%, 0.625%, 0.312%, 0%).
- Treatment: To cell aliquots, add 2 µL of each DMS working stock (final concentrations: 0.1%, 0.05%, 0.025%, 0.0125%, 0.00625%, 0.003125%, and 0% vehicle). Mix immediately by gentle flicking.
- Incubation: Incubate tubes at 23°C (room temperature) for 5 minutes with gentle inversion every minute.
- Quenching: Add 1 mL of chilled Quenching Buffer (40% β-mercaptoethanol in 1x PBS) to each tube. Vortex thoroughly for 10 seconds.
- Pellet Cells: Centrifuge at 5000 x g for 2 minutes at 4°C. Aspirate supernatant completely.
- RNA Extraction & Library Prep: Extract total RNA using a column-based kit (e.g., Zymo RNA Clean & Concentrator). Perform DMS-MaPseq library preparation (see Protocol 4.2) for all samples in parallel.
- Sequencing & Analysis: Sequence libraries on a NextSeq or HiSeq platform. Align reads and calculate mutation rates per sample using a pipeline like
dms_tools2orShapeMapper2. - Optimal Dose Selection: Plot mutation rate vs. DMS concentration. The optimal dose is the highest concentration that maintains a linear increase in mutation rate without causing significant cell death or RNA degradation. This typically corresponds to a background-subtracted mutation rate of 1-3% for mammalian cells.
Protocol 4.2: DMS-MaPseq Workflow from Treated Cells to Sequencing Libraries
- RNA Extraction: After DMS treatment and quenching, extract total RNA using a robust, DNase I-treated protocol (e.g., Zymo Research Quick-RNA Miniprep Kit). Elute in 30-50 µL nuclease-free water. Quantify by Nanodrop/Qubit.
- Ribodepletion: Treat 1-5 µg of total RNA with a ribodepletion kit (e.g., NEBNext rRNA Depletion Kit) to enrich for mRNA and non-coding RNAs.
- MaP Reverse Transcription: Use the SuperScript II or TGIRT-III reverse transcriptase with conditions favoring read-through of DMS adducts.
- Reaction Mix: 200-500 ng ribodepleted RNA, 1x First Strand Buffer, 1 µL DTT (100 mM), 0.5 mM each dNTP, 2 µM gene-specific or random hexamer primers, 2 U/µL SuperScript II.
- Thermocycler Program: 25°C for 10 min (primer annealing), 42°C for 90 min (RT extension), 70°C for 15 min (inactivation).
- RNase A Treatment: Add 2 µL of RNase A (2 mg/mL) to the RT reaction and incubate at 37°C for 30 min to digest template RNA.
- cDNA Purification: Purify cDNA using a 1.8x ratio of AMPure XP beads. Elute in 20 µL 10 mM Tris-HCl, pH 8.0.
- Library Amplification & Barcoding: Amplify cDNA by PCR for 12-18 cycles using a high-fidelity polymerase (e.g., Q5) and primers containing Illumina adapters and sample-specific barcodes.
- Library Purification & QC: Purify the final library with a 0.9x SPRI bead clean-up. Assess size distribution on a Bioanalyzer and quantify by qPCR.
- Sequencing: Pool libraries and sequence on an Illumina platform (≥ 5 million paired-end 150 bp reads per sample is recommended).
Visualizations
DMS Experiment Design and Optimization Workflow
DMS Probing Mechanism in Live Cells
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions for DMS-MaPseq
| Reagent/Material | Supplier Examples | Function in Experiment | Critical Notes |
|---|---|---|---|
| Dimethyl Sulfate (DMS) | Sigma-Aldrich, Thermo Fisher | Small chemical probe that methylates accessible A and C residues in RNA. | Highly toxic. Use in fume hood with proper PPE. Aliquot under inert gas. |
| β-Mercaptoethanol (BME) | Sigma-Aldrich, Bio-Rad | Quenching agent; scavenges unreacted DMS to stop the probing reaction. | Must be fresh (< 2 weeks after opening) for effective quenching. |
| SuperScript II Reverse Transcriptase | Thermo Fisher | MaP enzyme. Reads through DMS adducts with high fidelity, incorporating mismatches. | Critical for mutation detection. Do not substitute with other RTs without validation. |
| TGIRT-III Enzyme | InGex, Inc. | Group II intron reverse transcriptase; alternative MaP enzyme with high processivity. | Useful for structured RNAs and full-length profiling. |
| RNase H-deficient RT Mutant | Laboratory purified | Engineered RT for ultramutational profiling; reduces bias. | Used in advanced protocols (e.g., DMS-MaPseq with ultramutagenic RT). |
| Ribonuclease A (RNase A) | Qiagen, Thermo Fisher | Digests RNA template after RT, leaving single-stranded cDNA for library prep. | Essential for removing RNA-cDNA hybrids. |
| Ribosomal RNA Depletion Kit | Illumina, NEBNext | Removes abundant rRNA to increase sequencing coverage of target RNAs. | Choice of kit depends on cell type (e.g., human, mouse, bacterial). |
| SPRIselect / AMPure XP Beads | Beckman Coulter | Magnetic beads for size selection and purification of cDNA and libraries. | 1.8x ratio post-RT; 0.9x ratio post-PCR is standard. |
| Qubit RNA HS / BR Assay Kits | Thermo Fisher | Fluorometric quantification of RNA and library concentration. | More accurate for RNA/library quant than absorbance (Nanodrop). |
| Cell Viability Stain (Trypan Blue) | Bio-Rad, Thermo Fisher | Assesses cell health before and after DMS treatment during optimization. | Quick check for gross toxicity from DMS dose. |
| DNase I (RNase-free) | Zymo Research, Qiagen | Removes genomic DNA contamination during RNA extraction. | Prevents DNA-based artifacts in sequencing libraries. |
Within the broader thesis on DMS-MaPseq for in vivo RNA structure profiling, the initial treatment stage is critical. This stage involves the controlled application of dimethyl sulfate (DMS) to living cells or tissues, requiring strategies to facilitate DMS entry and precise methods to halt the chemical probing reaction. Effective permeabilization and quenching directly impact data accuracy by ensuring consistent DMS accessibility and preventing over-modification or RNA degradation.
Permeabilization Strategies
DMS must traverse cellular membranes to modify single-stranded adenine (N1) and cytosine (N3) residues. In eukaryotic cells, the plasma membrane is a significant barrier. The choice of strategy balances modification efficiency with cell viability and structural preservation.
Quantitative Comparison of Permeabilization Methods
Table 1: Comparison of Common In Vivo DMS Permeabilization Strategies
| Method | Typical Concentration / Condition | Key Mechanism | Pros | Cons | Optimal Use Case |
|---|---|---|---|---|---|
| Detergent-based (e.g., NP-40) | 0.01% - 0.1% (v/v) | Solubilizes lipid membranes, creates pores. | Highly effective, rapid, tunable. | Can disrupt protein complexes, may over-permeabilize. | Cultured mammalian cells, standard protocols. |
| Electroporation | Specific voltage/capacitance pulses. | Electrical pulses induce transient pores. | No chemicals, applicable to many cell types. | Requires specialized equipment, optimization critical, can cause heat shock. | Cells resistant to chemical permeabilization. |
| Streptolysin O (SLO) | 50-200 U/mL | Bacterial toxin forms large pores in cholesterol-rich membranes. | Creates large pores (>30 nm), allows co-factor entry. | Cell-type specific (requires cholesterol), expensive. | Delivering large molecules alongside DMS. |
| Hypotonic Shock | Dilution in low-ionic-strength buffer. | Osmotic pressure causes swelling and membrane stress. | Mild, no added chemicals. | Inconsistent, low efficiency for many cell lines. | Preliminary screens or sensitive primary cells. |
| Native (No treatment) | N/A | Passive diffusion of DMS. | Minimally perturbing, simplest. | Very low efficiency in most eukaryotic cells. | Yeast, bacteria, or studies prioritizing native state. |
Detailed Protocol: Optimized Detergent-based Permeabilization for Adherent Cells
Objective: To achieve consistent DMS entry into adherent mammalian cells (e.g., HEK293T) while minimizing cellular disruption. Reagents: Cell culture, DMS Buffer (150 mM HEPES-KOH pH 7.5, 150 mM NaCl, 5 mM KCl, 5 mM MgCl₂), 10% NP-40 Alternative, 1M DTT, Dimethyl Sulfate (DMS, >99%), Quenching Buffer (2M β-mercaptoethanol in DMS Buffer). Procedure:
- Cell Preparation: Grow cells to ~80% confluency in a 10 cm dish. Wash twice gently with 5 mL pre-warmed DMS Buffer.
- Permeabilization: Add 4.95 mL of DMS Buffer to the dish. Add 50 µL of 10% NP-40 Alternative (final 0.1%) and 5 µL of 1M DTT (final 1 mM). Gently swirl and incubate at room temperature for 2 minutes.
- DMS Treatment: Add 5.5 µL of pure DMS (final 0.22% v/v) directly to the buffer. Swirl immediately to mix. Incubate at 37°C for 5 minutes.
- Proceed immediately to quenching (Section 3). Note: The concentration of detergent and incubation time must be empirically optimized for each cell type.
Reaction Quenching
Quenching is the rapid and irreversible termination of DMS activity. Inefficient quenching leads to continued RNA modification during sample processing, introducing artifacts.
Quenching Mechanisms & Protocols
DMS alkylation is halted by scavenging the reagent with a high concentration of thiol-containing reducing agents.
Primary Quenching Protocol:
- Rapid Removal & Addition: Immediately after DMS treatment, aspirate the DMS-containing buffer into a chemical waste container. Without delay, add 10 mL of pre-chilled Quenching Buffer (2M β-mercaptoethanol) to the dish.
- Incubation: Incubate on ice for 2 minutes with gentle rocking.
- Cell Harvesting: Aspirate quench buffer. Wash cells once with 5 mL of ice-cold PBS. Harvest cells by scraping in 1 mL of TRIzol or lysis buffer for RNA isolation.
- Secondary Quench (Optional but recommended): Include 0.5% v/v β-mercaptoethanol in the initial RNA lysis solution (e.g., added to TRIzol) to neutralize any residual DMS.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for In Vivo DMS Treatment
| Item | Function | Critical Considerations |
|---|---|---|
| Dimethyl Sulfate (DMS) | Small chemical probe that methylates accessible RNA bases (A N1, C N3). | Highly toxic and volatile. Use in a fume hood, neutralize waste with 5M NaOH. Aliquot under nitrogen. |
| NP-40 Alternative (or Igepal CA-630) | Non-ionic detergent for controlled plasma membrane permeabilization. | Less harsh than SDS; concentration is critical for balance between access and cell integrity. |
| β-mercaptoethanol (BME) | Thiol-based reducing agent used to quench DMS activity. | High molarity (2M) stock in reaction buffer is standard for efficient quenching. Alternative: DTT. |
| DMS Reaction Buffer | Provides physiological ionic conditions (e.g., Mg²⁺, K⁺) during probing. | pH must be 7.5-8.0 for optimal DMS reactivity; HEPES is standard. |
| Streptolysin O (SLO) | Protein toxin for controlled, large-pore permeabilization. | Requires pre-activation with DTT. Efficiency is cell-type dependent (cholesterol). |
| RNA Stabilization Reagent (e.g., TRIzol) | Immediately inactivates RNases upon cell lysis after quenching. | Maintains RNA integrity for subsequent MaP reverse transcription. |
Visualized Workflows and Pathways
Diagram Title: DMS Treatment and Quenching Core Workflow
Diagram Title: DMS Reaction and Quenching Chemistry
Application Notes
The accuracy of in vivo DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing) data is fundamentally constrained by the integrity of the isolated RNA. DMS modifies accessible adenosine (A) and cytidine (C) nucleotides in vivo, creating adducts that are read as mutations during reverse transcription. Degraded or fragmented RNA introduces background noise and artifacts that can obscure true structural signals, leading to erroneous downstream analysis. This stage is therefore not merely a preparatory step but a critical determinant of experimental validity.
Key Integrity Considerations:
- Rapid Inhibition of RNases: Post in vivo DMS treatment, immediate tissue homogenization in strong chaotropic denaturants (e.g., guanidinium thiocyanate) is non-negotiable to inactivate ubiquitous RNases and preserve the mutation pattern.
- Inhibition of DMS Reaction: The DMS alkylation reaction must be quenched thoroughly with β-mercaptoethanol during lysis to prevent ex vivo modifications.
- Minimizing Thermally Induced Degradation: All purification steps should be performed on ice or at 4°C. Column-based silica-membrane purification is preferred over traditional organic extraction for speed and consistency, reducing exposure to degradative conditions.
- Integrity Assessment: The RNA Integrity Number (RIN) from a Bioanalyzer or TapeStation is essential. For DMS-MaPseq, a RIN > 8.0 is typically required for long-range structural analysis. However, for highly structured or small RNAs, capillary electrophoresis traces must be inspected visually for specific peak integrity.
- Library Prep Adaptations: The reverse transcription (RT) step in library preparation must use a thermostable group II intron reverse transcriptase (e.g., TGIRT, MarathonRT) capable of reading through DMS adducts with high fidelity and low bias. Standard retroviral RTs are inadequate.
Table 1: Quantitative Benchmarks for RNA Integrity in DMS-MaPseq
| Metric | Target Value | Measurement Tool | Impact on DMS-MaPseq Data |
|---|---|---|---|
| RNA Integrity Number (RIN) | ≥ 8.0 | Bioanalyzer/TapeStation | RIN < 7 leads to increased false-positive mutation calls in 3’ regions. |
| 28S/18S rRNA Ratio | ≥ 1.8 (Eukaryotes) | Electropherogram | Lower ratios indicate degradation, increasing noise in structured regions of large RNAs. |
| DV200 (% > 200 nt) | ≥ 85% | TapeStation | Critical for long RNA structure analysis; low values necessitate targeted library prep for small RNAs. |
| A260/A280 Ratio | 1.9 - 2.1 | Spectrophotometer | Deviations indicate contaminant carryover (phenol, guanidine) that can inhibit RT. |
| A260/A230 Ratio | ≥ 2.0 | Spectrophotometer | Low values indicate salt or organic solvent contamination, affecting ligation efficiency. |
Detailed Protocols
Protocol A: Rapid RNA Extraction from DMS-Treated Mammalian Cells
Objective: To isolate high-integrity total RNA from DMS-treated cells while quenching the alkylation reaction. Reagent Solutions:
- Lysis Buffer: 4M Guanidine Thiocyanate, 1% β-mercaptoethanol, 0.1% Triton X-100.
- Wash Buffers: Standard silica-membrane kit buffers (e.g., RPE from Qiagen).
- Elution Buffer: Nuclease-free 10 mM Tris-HCl, pH 7.0 (warm to 55°C for elution).
Methodology:
- Immediately after in vivo DMS treatment and quenching, aspirate culture medium.
- Add 600 μL of ice-cold Lysis Buffer directly to the culture dish (~10⁶ cells). Lyse cells thoroughly by pipetting.
- Transfer lysate to a sterile microcentrifuge tube. Vortex for 15 seconds.
- Optional: Centrifuge at 12,000 x g for 2 min at 4°C to pellet insoluble debris. Transfer supernatant to a new tube.
- Add 1 volume of 70% ethanol to the lysate. Mix by pipetting.
- Load the mixture onto a silica-membrane column. Centrifuge at 12,000 x g for 30s. Discard flow-through.
- Wash the column with 700 μL of Buffer RW1. Centrifuge. Discard flow-through.
- Wash twice with 500 μL of Buffer RPE. Centrifuge after each wash. Perform a final empty spin.
- Transfer column to a fresh RNase-free tube. Elute RNA with 30-50 μL of pre-warmed Elution Buffer by centrifuging at full speed for 1 min.
Protocol B: DMS-MaPseq Library Preparation (RT & Adapter Ligation)
Objective: To generate sequencing libraries from DMS-modified RNA, capturing mutations via mutation-prone RT. Reagent Solutions:
- RT Primer: Gene-specific or random hexamers with 5’ adapter sequence.
- RT Mix: 1x First-Strand Buffer, 1 mM dNTPs, 5 mM DTT, 2 U/μL RNase Inhibitor, 200 U of thermostable group II intron RT (e.g., MarathonRT).
- Ligation Mix: 1x T4 RNA Ligase Buffer, 25% PEG 8000, 1 mM ATP, 20 U T4 RNA Ligase 1, 3’ DNA Adapter.
Methodology:
- RNA Primer Annealing: For 1 μg of total RNA, mix with 2 pmol of RT primer. Denature at 65°C for 5 min, then snap-cool on ice.
- Mutation-Prone Reverse Transcription: Assemble RT reaction on ice. Incubate: 10 min at 25°C, 60-120 min at 55-60°C (enzyme-dependent), 10 min at 70°C for inactivation.
- RNA Degradation: Add 1 μL of RNase A/T1 mix. Incubate at 37°C for 15 min.
- cDNA Purification: Clean up cDNA using SPRI beads at a 1.8x bead-to-sample ratio. Elute in 22 μL nuclease-free water.
- 3’ Adapter Ligation: To the purified cDNA, add Ligation Mix components. Incubate at 20-25°C for 1-2 hours.
- Ligation Clean-up: Purify with SPRI beads (1.8x ratio). Elute in 10 μL.
- PCR Amplification: Amplify with primers containing full Illumina adapter indices and sequencing primers. Use a high-fidelity polymerase. Clean up final library with SPRI beads (0.9x ratio) for size selection.
Diagrams
Title: DMS-MaPseq RNA Integrity Workflow
Title: Impact of RNA Integrity on DMS Signal
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for High-Integrity DMS-MaPseq
| Reagent / Kit | Function in Protocol | Key Feature for Integrity |
|---|---|---|
| TRIzol LS Reagent | Simultaneous lysis and inactivation of RNases. | Strong chaotropic denaturant for immediate RNase inhibition post-DMS treatment. |
| Qiagen RNeasy Mini Kit | Silica-membrane based RNA purification. | Fast, consistent recovery at 4°C; removes DMS and salts effectively. |
| β-Mercaptoethanol (BME) | Reducing agent added to lysis buffer. | Quenches residual DMS reaction during homogenization. |
| RNase Inhibitor (e.g., RNasin) | Added to RT and ligation reactions. | Protects RNA and cDNA intermediates from trace RNases. |
| Thermostable Group II RT (MarathonRT) | Mutation-prone reverse transcription. | High processivity and fidelity through DMS adducts on long RNAs. |
| SPRIselect Beads | Size-selective cleanup of cDNA and libraries. | Removes primer dimers; 0.9x ratio selects against small fragment artifacts. |
| Agilent RNA 6000 Nano Kit | RNA integrity assessment (RIN, DV200). | Provides quantitative QC metrics essential for sample triage. |
This Application Note details Stage 3 of the DMS-MaPseq workflow for in vivo RNA structure probing. Following DMS modification (Stage 1) and RNA extraction (Stage 2), this stage converts chemical adducts into heritable, sequenceable mutations during reverse transcription, enabling high-throughput sequencing to quantify RNA flexibility and structural states. This protocol is integral to a thesis on obtaining nucleotide-resolution, in vivo structural insights into functionally and therapeutically relevant RNAs.
Key Principles of MaP Reverse Transcription
The core innovation of MaP is the use of reverse transcriptases that bypass chemical adducts (e.g., DMS-methylated adenosines and cytosines) with low fidelity, incorporating mismatched nucleotides. This creates cDNA with mutations at the sites of modification. Subsequent PCR and sequencing record these mutations, whose frequency is proportional to the original DMS reactivity, a direct metric of nucleotide accessibility.
Detailed Protocol: MaP RT and Library Preparation
Part A: Mutational Profiling Reverse Transcription
Objective: Generate cDNA with mutations marking DMS-modified sites.
Reagents & Setup:
- Template: 1-500 ng of DMS-modified, purified total RNA.
- Primers: Gene-specific primers or random hexamers for whole-transcriptome analysis.
- Reverse Transcriptase: Use a thermostable, group II intron-derived RT (e.g., TGIRT-III, MarathonRT) or SuperScript II for optimal read-through and misincorporation.
- Buffer: Supplied with enzyme, supplemented with 1-6 mM MnCl₂. Note: Mn²⁺ is critical for promoting misincorporation at modified bases.
- dNTPs: High concentration (1 mM each) to support processivity.
- Conditions: Combine RNA, primer (2.5 µM), dNTPs in nuclease-free water. Heat to 65°C for 5 min, then place on ice. Add 5X RT buffer, MnCl₂, and RT enzyme. Incubate:
- For TGIRT-III: 60°C for 60-120 min.
- For SSII: 42°C for 90 min, then 52°C for 30 min.
- Clean-up: Purify cDNA using RNase H treatment followed by SPRI bead purification.
Part B: PCR Amplification and Sequencing Library Construction
Objective: Amplify cDNA and append sequencing adapters with unique molecular identifiers (UMIs).
Two-Stage PCR Approach:
- PCR 1 (cDNA Amplification): Use gene-specific or indexed primers to amplify target regions. Use high-fidelity DNA polymerase (e.g., Q5, KAPA HiFi) for 10-15 cycles to minimize PCR-induced errors.
- PCR 2 (Adapter Addition): Use 1-5 µL of purified PCR1 product in a second reaction with primers containing full Illumina adapter sequences (P5/P7). Include UMIs in the forward adapter to enable deduplication.
- Purification: Size-select and purify the final library using double-sided SPRI bead cleanup.
- QC & Sequencing: Quantify by qPCR or bioanalyzer. Sequence on Illumina platforms (MiSeq, NextSeq) with paired-end reads (2x150 bp recommended).
Table 1: Typical MaPseq Mutation Rates and Sequencing Metrics
| Parameter | Typical Value / Target | Notes / Impact |
|---|---|---|
| Mutation Rate (DMS-treated) | 0.5% - 2.0% per nucleotide | Rate correlates with DMS concentration & reactivity. |
| Mutation Rate (Untreated Control) | < 0.05% per nucleotide | Background error rate of the RT/PCR process. |
| Read Depth per Condition | > 10,000 reads per transcript | Ensures statistical power for reactivity calculation. |
| UMI Deduplication Efficiency | > 90% | Critical for removing PCR duplicates and artifact suppression. |
| Mapping Rate | > 80% of reads | Depends on genome/transcriptome complexity and quality. |
| Key Mutation Types | A>C, C>T, G>A | Primary misincorporations at DMS-modified A (N1) and C (N3). |
Table 2: Comparison of Reverse Transcriptases for MaP
| Enzyme (Vendor) | Optimal Temp. | Mn²⁺ Requirement | Processivity | Primary Use Case |
|---|---|---|---|---|
| TGIRT-III (InGex) | 60°C | 2-6 mM | Very High | Whole transcriptome, structured RNAs |
| MarathonRT (Lucigen) | 55-60°C | 1-2 mM | Very High | Standardized DMS-MaPseq protocols |
| SuperScript II (Thermo) | 42-52°C | 5-6 mM | Moderate | Targeted, well-established protocols |
| PrimeScript (Takara) | 42°C | 5 mM | Moderate | Alternative for targeted studies |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for MaP RT and Sequencing
| Item | Function & Rationale |
|---|---|
| Group II Intron RT (TGIRT/MarathonRT) | High processivity and robust misincorporation at DMS adducts under optimized Mn²⁺ conditions. |
| Manganese Chloride (MnCl₂) | Divalent cation that decreases RT fidelity, enabling misincorporation opposite modified bases. |
| Unique Molecular Identifiers (UMIs) | Random nucleotide tags added during cDNA synthesis or PCR1 to tag original molecules, enabling bioinformatic deduplication. |
| High-Fidelity DNA Polymerase (e.g., Q5) | Used for PCR amplification post-RT to minimize introduction of non-biological mutations. |
| SPRI Magnetic Beads | For size selection and clean-up of cDNA and libraries; maintains fragment size distribution. |
| Dual-Indexed Illumina Adapters | Allow multiplexing of many samples in a single sequencing run, reducing per-sample cost. |
| RNase H | Degrades RNA template after first-strand synthesis, improving cDNA yield and purity. |
Visualizing the Workflow and Analysis Logic
MaP RT to Sequencing Workflow
Bioinformatic Analysis Pipeline for MaPseq Data
Within the context of a DMS-MaPseq thesis, this stage is critical for transforming raw sequencing data into quantitative, nucleotide-resolution reactivity profiles that inform RNA structural models. This protocol details the pipeline using ShapeMapper2, the current standard for processing mutational profiling (MaP) data.
Application Notes
- Thesis Integration: This computational module directly tests the in vivo structural hypotheses posed in the introduction. The reactivity profiles generated here are the primary evidence for validating or refining predicted RNA secondary and tertiary structures.
- Key Output: The principal outputs are
.reactand.shapefiles containing normalized reactivity values for each nucleotide. High reactivity indicates DMS modification and thus, single-strandedness; low reactivity indicates base-pairing or protection. - Quality Control (QC) Metrics: Successful processing requires careful monitoring of key metrics summarized in Table 1.
Table 1: Key Quality Control Metrics for ShapeMapper2 Pipeline
| Metric | Target Value/Profile | Interpretation |
|---|---|---|
| Mutation Rate (DMS-treated) | 5-15% | Optimal for robust signal. <2% is too low; >20% may indicate over-modification or degradation. |
| Mutation Rate (Untreated Control) | 0.5-2% | Represents background error/misincorporation. Should be significantly lower than DMS-treated. |
| Read Depth per Nucleotide | >1000x (minimum) | Ensures statistical confidence in reactivity calculation. |
| Effective Depth | >80% of raw reads | Indicates high alignment efficiency. |
| Reactivity Profile | Smooth, with clear peaks & valleys | Noisy, flat profiles may indicate failed experiment or processing error. |
Experimental Protocol: ShapeMapper2 Computational Workflow
Software Prerequisites:
- ShapeMapper2 (v2.1.5 or higher)
- STAR or Bowtie2 aligner
- Python 3 with NumPy, SciPy
- R for optional downstream analysis
Step 1: Demultiplexing and FASTQ Pre-processing
- Use
bcl2fastqorguppy_basecallerto generate paired-end FASTQ files. - Trim adapter sequences using
cutadapt:
Step 2: Running ShapeMapper2
Execute the core analysis. The command below processes a DMS-treated sample (DMS) against its matched untreated control (Control).
Step 3: Normalization and Output
- ShapeMapper2 automatically performs in-line normalization to control for background noise and sequence-dependent mutability.
- The key outputs are:
RNA_target_combined.shape: Normalized reactivity profile.RNA_target_combined.react: Same data, different format.RNA_target.log: Comprehensive log file with QC metrics.
Step 4: Downstream Analysis (Thesis-Specific)
- Filter reactivities: Often, reactivities >1.0 are capped, and low-depth positions (<100 reads) are masked.
- Use
SuperfoldorVARNAto visualize reactivity on secondary structure models. - Perform differential reactivity analysis between experimental conditions using
dStructor custom R scripts.
Visualization: Computational Workflow Diagram
Title: DMS-MaPseq Computational Pipeline from FASTQ to Reactivity
The Scientist's Toolkit: Essential Research Reagents & Software
Table 2: Key Resources for Computational DMS-MaPseq Analysis
| Item | Function in Pipeline | Notes for Thesis Research |
|---|---|---|
| ShapeMapper2 Software | Core tool for mutation parsing, background subtraction, and reactivity calculation from MaP data. | Essential for reproducibility. Cite in methods. Always use the latest stable version. |
| Reference Genome & Transcriptome | FASTA file of the target RNA(s) for alignment. | For in vivo work, include flanking genomic sequence or the full transcript. |
| High-Performance Computing (HPC) Cluster | Provides necessary CPU/RAM for parallel processing of multiple samples. | Critical for thesis-scale data (dozens of libraries). |
| DMS-MaP Specific Primers | Reverse transcription primers with randomer sequences for MaP. | Sequence must be specified in the --primers file for ShapeMapper2 if not standard. |
| QC Scripts (Custom R/Python) | To parse .log files, visualize mutation rates, and filter final reactivity profiles. |
Develop or adapt scripts as part of the thesis methodology chapter. |
| Structure Visualization Software (VARNA) | Maps reactivity data onto 2D RNA structures. | Key for generating publication and thesis figures that illustrate structural findings. |
1. Introduction Within a thesis on DMS-MaPseq for in vivo RNA structure profiling, a critical challenge is translating raw chemical reactivity data into accurate secondary structure models. This protocol details the integration of experimental DMS reactivities with thermodynamic folding algorithms (RNAstructure, ViennaRNA) to generate constrained, biologically relevant RNA structural predictions.
2. Application Notes & Protocols
2.1 Protocol: Pre-processing DMS-MaPseq Reactivities for Algorithm Input
Objective: Convert sequencing-derived mutation rates into normalized reactivity profiles suitable as pseudo-free energy constraints.
Materials: DMS-MaPseq sequencing data (BAM files), reference genome/transcriptome, preprocessing pipeline (e.g., dms_tools2, ShapeMapper2).
Steps:
- Mutation Rate Calculation: Map reads and compute per-nucleotide mutation rates for DMS-treated and untreated control samples.
- Background Subtraction & Correction: Subtract the control mutation rate from the DMS-treated rate. Correct for sequence biases (e.g., using
DREEM). - Normalization: Normalize reactivities using a 2-8% or 8-12% approach. Commonly, reactivities are scaled such that the 92nd percentile value equals 1.0 for structured regions or the average of the top 10% reactivities equals 1.0.
- Profile Formatting: Output a reactivity profile in the required format for the chosen folding algorithm (see Table 1).
2.2 Protocol: Integrating Reactivities with RNAstructure (Fold & Partition) Objective: Generate a minimum free energy (MFE) and ensemble of structures using experimental constraints. Materials: RNAsequence in FASTA format, normalized reactivity profile (.txt or .shape format), RNAstructure suite (v6.4+). Steps:
- Constraint Preparation: Use the
ReactivityProfileprogram or theFoldcommand with the-shflag.
Constrained MFE Folding: Run
Foldwith pseudo-free energy constraints. The-dparameter modulates constraint strength.Generate Ensemble & Probabilities: Run
partitionto compute base-pairing probabilities.
2.3 Protocol: Integrating Reactivities with ViennaRNA (RNAfold) Objective: Perform constrained folding using the ViennaRNA Package. Materials: RNA sequence, normalized reactivity profile (.txt), ViennaRNA Package (v2.6+). Steps:
- Format Reactivities: Create a file with one reactivity per line, matching the sequence length. Use "-999" for unreactive or missing positions.
- Constrained Folding: Use the
--shapeoption inRNAfold. The--shapeMethodparameter selects the energy model (e.g., 'D', 'Z').
- Ensemble Analysis (Optional): Use
RNAsuboptwith the--shapeconstraint to sample suboptimal structures.
2.4 Data Presentation: Algorithm Comparison & Parameters
Table 1: Key Parameters for Integrating DMS Reactivities into Folding Algorithms
| Algorithm (Program) | Input File Format | Key Integration Parameter | Typical Value/Setting | Primary Output |
|---|---|---|---|---|
| RNAstructure (Fold) | .shape or .txt | -sh <file>, -d <value> |
-d 1.0 to 1.6 |
MFE structure (.ct) |
| RNAstructure (Partition) | .shape | -sh <file> |
-d 1.2 |
Pair probabilities (.pfs) |
| ViennaRNA (RNAfold) | .txt (1 col) | --shape=<file>, --shapeMethod |
--shapeMethod=D |
MFE structure (.dot-bracket) |
| Superfold (ΔΔG) | .shape, .txt | Uses Fold (RNAstructure) iteratively |
Built-in | Pseudo-free energy landscape |
Table 2: Comparative Metrics for Constrained vs. Unconstrained Folding
| Metric | Unconstrained MFE | DMS-Constrained MFE | Measurement Method |
|---|---|---|---|
| Prediction Accuracy (PPV/Sensitivity)* | 0.40 - 0.60 | 0.70 - 0.90 | Comparison to crystal/ NMR structure |
| Ensemble Shannon Entropy | Higher | Lower (by 10-30%) | Calculated from base-pair probabilities |
| Computation Time | Baseline (1X) | 1.5X - 3X Baseline | System dependent |
| Note: Accuracy gains are most significant for long (>500 nt) RNAs and in vivo data. |
3. Mandatory Visualizations
Title: DMS-MaPseq to RNA Model Workflow
Title: Reactivity to Energy Constraint Conversion
4. The Scientist's Toolkit: Research Reagent & Software Solutions
Table 3: Essential Reagents & Software for DMS-MaPseq Structure Modeling
| Item | Function/Application | Example/Note |
|---|---|---|
| DMS (Dimethyl Sulfate) | In vivo probing of A/C bases. | Highly toxic; use in controlled, ventilated setups. |
| MaP Reverse Transcriptase | Reads through DMS modifications, causing mutations. | SuperScript II, TGIRT. Critical for MaPseq. |
| Structure Prediction Suite | Core folding algorithms with SHAPE/DMS integration. | RNAstructure (v6.4+), ViennaRNA (v2.6+). |
| Normalization Scripts | Converts mutation rates to normalized reactivities. | dms_tools2, ShapeMapper2, custom R/Python. |
| Visualization Software | Visualizing structures and probability matrices. | VARNA, FORNA, PyMOL (for 3D models). |
| High-Performance Computing | For partitioning/folding long RNAs or large ensembles. | Local cluster (SLURM) or cloud (AWS, GCP). |
Within the broader thesis that DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing) is a transformative methodology for in vivo RNA structure profiling, this application note highlights its pivotal role in therapeutic discovery. The core thesis posits that accurate, nucleotide-resolution probing of RNA structural ensembles in their native cellular environment is critical for understanding functional mechanisms and identifying druggable sites. This document details how DMS-MaPseq is applied to three high-value target classes—riboswitches, viral RNAs, and long non-coding RNAs (lncRNAs)—to guide the rational design of small molecules, antisense oligonucleotides (ASOs), and other modalities.
Application Notes
Riboswitches: Targeting Metabolic Pathways
Riboswitches are structured RNA elements in the 5'-UTRs of bacterial mRNAs that regulate gene expression in response to metabolite binding. DMS-MaPseq enables the mapping of ligand-induced conformational changes in vivo, revealing dynamics crucial for inhibitor design.
Key Insight: DMS reactivity changes upon metabolite addition pinpoint nucleotides involved in binding and switching. Small molecules that mimic the native metabolite or stabilize the "off" conformation can be designed to disrupt essential bacterial metabolic pathways.
Viral RNA Genomes: Uncovering Conserved Structures
Viral genomes (e.g., SARS-CoV-2, HIV, Zika) contain highly conserved structured RNA elements essential for replication, frameshifting, and packaging. DMS-MaPseq profiling in infected cells identifies these functional, often druggable, structures.
Key Insight: Regions with low DMS reactivity (highly paired) that are conserved across strains represent attractive targets for small molecules that disrupt folding. For SARS-CoV-2, the frameshift stimulation element (FSE) has been a primary DMS-MaPseq target.
lncRNAs: Addressing Human Disease
lncRNAs play roles in gene regulation, chromatin remodeling, and disease (e.g., cancer, neurodegeneration). Their functions are tightly linked to complex 3D structures. DMS-MaPseq maps these structures in relevant cell lines, identifying domains for functional disruption.
Key Insight: Structured domains crucial for lncRNA-protein interaction or subcellular localization can be targeted with ASOs that block access, leading to functional knockdown without degradation.
Table 1: DMS-MaPseq Profiling Outcomes for Key Target Classes
| Target Class | Example Target | Key Structural Metric (DMS Reactivity Change) | Identified Druggable Regions | Potential Therapeutic Modality |
|---|---|---|---|---|
| Bacterial Riboswitch | B. subtilis glycine riboswitch | >80% reduction in reactivity at switching sequence upon glycine binding | Ligand-binding aptamer domain | Small molecule analogs |
| Viral RNA | SARS-CoV-2 Frameshift Element | Highly low-reactive stem (≤0.1 normalized reactivity) | Three-stem pseudoknot | Small molecules (e.g., MTDB) |
| Human lncRNA | MALAT1 (Metastasis-associated) | Hyper-reactive loop (≥2.5) conserved in cancer cell lines | 5' Structural motif for protein partner binding | Gapmer ASOs |
Table 2: Comparative Protocol Parameters for In Vivo DMS-MaPseq
| Step | Riboswitches (Bacteria) | Viral RNA (Infected Cells) | lncRNA (Mammalian Cells) |
|---|---|---|---|
| DMS Concentration | 5-10 mM | 0.5-1.0% (v/v) | 0.7-1.0% (v/v) |
| Treatment Time | 5 min | 5-10 min | 10 min |
| Key Control | +/- metabolite ligand | Mock-infected cells | Wild-type vs. knockout cell line |
| Seq. Depth Target | 1-5 M reads | 10-30 M reads | 20-50 M reads |
| Primary Analysis | Reactivity change (ΔΨ) | SHAPE-like reactivity profile | Correlation with protein binding data |
Detailed Experimental Protocols
Protocol 1:In VivoDMS-MaPseq for Viral RNA Genomes in Cultured Cells
Objective: To probe the structure of SARS-CoV-2 genomic RNA in infected Vero E6 cells.
Materials: Vero E6 cells, SARS-CoV-2 isolate, DMS (Sigma, D186309), DMS Stop Buffer (1M β-mercaptoethanol, 100 mM Tris pH 8.0), TRIzol LS.
Procedure:
- Infection & DMS Treatment: Infect cells at MOI=0.1 for 24h. Aspirate medium and treat with 1% DMS in PBS for 5 min at 37°C. Quench reaction with 20mL ice-cold DMS Stop Buffer.
- RNA Extraction: Wash cells with PBS. Lyse with TRIzol LS and extract total RNA following manufacturer's protocol. DNase treat.
- RNA Selection & Fragmentation: Deplete rRNA using a commercial kit. Fragment 2 µg of RNA with 0.12N NaOH on ice for 20 min. Neutralize with 1M HEPES pH 7.0.
- MaP Reverse Transcription: Use SuperScript II (Thermo) with gene-specific primers for viral RNA. Include a no-DMS control. Use a thermocycler program: 25°C for 10 min, 42°C for 90 min, 70°C for 15 min. The reaction includes Mn²⁺ to promote mutation incorporation at DMS-modified sites.
- Library Construction: PCR amplify cDNA with Illumina adapters. Use 12-15 cycles. Purify and size-select (200-500 bp).
- Sequencing & Analysis: Sequence on Illumina NextSeq 500 (75bp single-end). Align reads to SARS-CoV-2 genome (MN908947.3). Call mutations using
dms-tools2orShapeMapper2. Normalize reactivity to no-DMS control and 8% trimmed mean.
Protocol 2: Probing Ligand-Induced Conformational Change in a Riboswitch
Objective: To map the structural change in the B. subtilis glycine riboswitch in vivo upon glycine addition.
Procedure:
- Bacterial Culture & Treatment: Grow B. subtilis strain to mid-log phase (OD600 ~0.5). Split culture. To one, add 10 mM glycine (final conc.) for 2 min. To the control, add PBS.
- In Vivo DMS Probing: Immediately add DMS to both cultures to 10 mM final concentration. Incubate with shaking for 5 min at 37°C. Quench with 30% (v/v) β-mercaptoethanol.
- RNA Extraction & Enrichment: Pellet cells, lyse with lysozyme, extract RNA. Enrich specific mRNA via bead-coupled oligonucleotide pull-down.
- MaPseq & Analysis: Proceed with fragmentation, MaP RT, and library prep as in Protocol 1. Calculate per-nucleotide reactivity (Ψ). The ΔΨ (Ψ(-glycine) - Ψ(+glycine)) identifies nucleotides protected upon glycine binding.
Visualization: Workflows and Pathways
Title: Riboswitch Targeting Workflow from DMS-MaP to Drug Design
Title: Viral RNA Structure Profiling and Target Identification Flow
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for DMS-MaPseq Drug Target Mapping
| Reagent / Solution | Vendor Examples (Catalog #) | Function in Protocol |
|---|---|---|
| Dimethyl Sulfate (DMS) | Sigma-Aldrich (D186309) | Cell-permeable chemical probe; methylates unpaired adenine (N1) and cytosine (N3). |
| β-Mercaptoethanol (BME) Stop Buffer | Thermo Fisher (21985023) | Quenches DMS reaction to halt further RNA modification. |
| SuperScript II Reverse Transcriptase | Thermo Fisher (18064014) | Engineered for high processivity; used with Mn²⁺ for mutation incorporation during cDNA synthesis. |
| MnCl₂ Solution | Sigma-Aldrich (M3634) | Divalent cation used in MaP RT to promote mis-incorporation opposite DMS-modified bases. |
| NEBNext Ultra II RNA Library Prep Kit | New England Biolabs (E7770) | For efficient construction of sequencing-compatible libraries from fragmented RNA/cDNA. |
| RiboPOOL rRNA Depletion Probes | siTOOLs Biotech | Species-specific probes to remove ribosomal RNA, enriching for target RNA species. |
| MyOne Streptavidin C1 Beads | Thermo Fisher (65001) | For pull-down enrichment of specific RNA targets using biotinylated DNA oligonucleotides. |
| ShapeMapper 2 Software | Open Source (https://github.com/Weeks-UNC/shapemapper2) | Core bioinformatics pipeline for processing sequencing data to calculate nucleotide reactivity profiles. |
Solving Common DMS-MaPseq Challenges: A Troubleshooting Handbook
Within the broader thesis on DMS-MaPseq for in vivo RNA structure profiling research, a low mutation rate in sequencing data is a critical bottleneck. It can stem from inadequate DMS modification (penetration/accessibility) or from reverse transcriptase (RT) that fails to read through DMS-adducts with high mutagenic efficiency. This application note provides a diagnostic framework and detailed protocols to distinguish between these two primary failure points.
Diagnostic Framework & Key Data
A systematic approach is required to isolate the issue. The table below outlines expected outcomes from key diagnostic experiments.
Table 1: Diagnostic Outcomes for Low Mutation Rate Scenarios
| Diagnostic Experiment | Expected if Issue is Poor DMS Penetration/Access | Expected if Issue is Low RT Fidelity/Mutagenic Efficiency |
|---|---|---|
| In vitro DMS Control on Naked RNA | Mutation rate returns to expected high levels. | Mutation rate remains low. |
| Vary DMS Concentration (in vivo) | Mutation rate increases with higher DMS dose. | Mutation rate remains low and unresponsive to dose. |
| Use a Positive Control RT (e.g., TGIRT-III) | Mutation rate may slightly improve but remains below expectation for the given DMS level. | Mutation rate shows significant improvement. |
| Measure DMS Adducts via Alternative Method (e.g., Primer Extension Halt) | Low signal of DMS modification. | Normal signal of DMS modification. |
Detailed Experimental Protocols
Protocol 3.1:In VitroDMS Control on Purified RNA
Purpose: To determine if the RNA target is intrinsically reactive to DMS and if the RT enzyme performs adequately under ideal conditions.
- Material: Purified RNA of interest (e.g., in vitro transcribed).
- DMS Reaction:
- Prepare 1-5 pmol of RNA in 100 µL of 1X DMS Structure Buffer (e.g., 50 mM HEPES-KOH pH 8.0, 100 mM KCl).
- Add 1 µL of pure DMS (diluted 1:10 in anhydrous ethanol) to the experimental tube. Add 1 µL of anhydrous ethanol only to the control tube.
- Incubate at 30°C for 5-10 minutes.
- Quench with 50 µL of 2 M 2-Mercaptoethanol (BME) and 350 µL of RNA precipitation solution (e.g., ethanol with glycogen).
- RNA Recovery: Precipitate RNA, wash with 80% ethanol, and resuspend.
- Library Preparation & Analysis: Proceed with standard DMS-MaPseq RT (using your standard and a high-fidelity control RT) and library prep. Compare mutation rates to in vivo samples.
Protocol 3.2: Titrating DMS Concentration inIn VivoExperiments
Purpose: To assess the responsiveness of the mutation rate to DMS dose, indicating penetration/access limitations.
- Cell Culture: Aliquot identical cultures of cells (or organism).
- DMS Treatment:
- Prepare DMS stock solutions in the appropriate physiological buffer (e.g., PBS) at varying concentrations (e.g., 0.2%, 0.5%, 1.0%, 2.0% v/v).
- Treat each aliquot with an equal volume of a different DMS stock for a fixed, standard time (e.g., 5 min). Include a 0% DMS control.
- Quench reaction with excess BME (final ~0.5 M).
- RNA Extraction & Processing: Extract total RNA following standard procedures. Process all samples in parallel through the identical DMS-MaPseq workflow.
- Analysis: Plot mutation rate (global or for a known single-stranded region) vs. DMS concentration. A saturating curve suggests good penetration; a flat, low line suggests an RT issue.
Protocol 3.3: Evaluating Reverse Transcriptase Fidelity
Purpose: To directly test the performance of the RT enzyme.
- RT Enzymes: Test alongside your standard RT:
- TGIRT-III (InGex): Known for high DMS-MaP efficiency.
- SuperScript IV (Thermo Fisher): Common but lower MaP efficiency.
- Standard Lab RT (e.g., Maxima H Minus).
- Template: Use a constant RNA template from a successful in vitro DMS modification (from Protocol 3.1).
- MaP Reverse Transcription:
- For each RT, set up 20 µL reactions as per manufacturer guidelines but with the provided DMS-MaP buffer conditions (e.g., 5 mM MnCl2, 1M Betaine, high dNTPs).
- Use consistent gene-specific or random primers.
- Perform cycling: 25°C for 5 min (primer annealing), ramp to 50-55°C over 2 min, hold at 55°C for 1-2 hrs.
- Analysis: After library prep and sequencing, calculate the mutation rate per read. Compare across RTs using the same RNA batch.
Visual Diagnostics & Workflows
Diagram Title: Diagnostic Decision Tree for Low Mutation Rates
Diagram Title: DMS-MaPseq Workflow & Failure Points
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for DMS-MaPseq Troubleshooting
| Reagent/Material | Function & Role in Diagnosis | Example Vendor/Product |
|---|---|---|
| Dimethyl Sulfate (DMS) | The small chemical probe that modifies unpaired A and C residues. Purity and handling are critical. | Sigma-Aldrich (D186309), Thermo Scientific |
| 2-Mercaptoethanol (BME) | Quenches unreacted DMS, stopping the modification reaction. Essential for safety and reproducibility. | Sigma-Aldrich (M6250) |
| High-Fidelity RT (TGIRT-III) | Group II intron-derived RT with superior processivity and mutant yield past DMS adducts. Positive control for RT issues. | InGex (TGIRT-III) |
| Manganese (II) Chloride (MnCl2) | Critical divalent cation for DMS-MaP. Promotes misincorporation at modification sites. Part of the RT buffer system. | Sigma-Aldrich (M3634) |
| Betaine | Additive in RT buffer that reduces secondary structure, improving RT processivity and overall mutation rate. | Sigma-Aldrich (61962) |
| RNase Inhibitor | Protects RNA templates from degradation during sample processing and RT, preserving signal. | Takara (2313A), Thermo Scientific (EO0381) |
| Glycogen (RNA Grade) | Carrier for efficient ethanol precipitation of low-concentration RNA samples post-DMS modification. | Thermo Scientific (R0551) |
| Structure Buffer (for in vitro) | Provides defined ionic conditions for in vitro DMS probing control experiments (e.g., HEPES, KCl). | In-house formulation |
1. Introduction and Background In vivo RNA structure probing using DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing) is a powerful technique for capturing RNA conformational landscapes in their native cellular context. A core challenge that directly compromises data fidelity is high background noise, primarily stemming from two sources: (1) endogenous RNA degradation, and (2) non-specific RNA modifications occurring either in vivo or during sample processing. This application note details protocols and analytical strategies to mitigate these issues, ensuring robust and interpretable DMS reactivity data for downstream thesis research on RNA structural dynamics.
2. Sources of Background Noise and Their Impact
| Noise Source | Primary Cause | Effect on DMS-MaPseq Data | Quantitative Impact (Typical Range) |
|---|---|---|---|
| RNA Degradation | Endogenous RNases, poor lysis/handling. | Introduces spurious reverse transcription (RT) stops/primer drops, misinterpreted as DMS modifications. Increases variability in per-nucleotide coverage. | >50% reduction in full-length cDNA yield; >2-fold increase in variance of per-base read depth in untreated controls. |
| Non-Specific DMS Reactivity | DMS reaction conditions (pH, temp, time) or cellular microenvironment. | Modification of adenosines and cytosines independent of RNA structural accessibility, leading to false-positive signals. | Can contribute 10-40% of total modification calls in poorly optimized experiments. |
| Chemical RNA Damage | Oxidation (e.g., from metals), hydrolysis (high temp/pH). | Creates non-DMS-mediated adducts that are read as mutations by RT. | Contributes ~5-15% background mutation rate in control samples. |
| RT/Sequencing Errors | Polymerase fidelity, sequencing platform. | Baseline error rate confounds low-level true signal detection. | Inherent RT error rate: 0.01-0.1%; Sequencing error: ~0.1% (Illumina). |
3. Protocols for Mitigation
Protocol 3.1: Rapid, RNase-Inhibiting Cell Lysis and RNA Isolation Objective: Minimize post-lysis RNA degradation. Materials: TRIzol or Qiazol Lysis Reagent, Phase Lock Gel Heavy tubes, β-mercaptoethanol, RNase-free glycogen, Acid-Phenol:Chloroform (pH 4.5), ice-cold 75% ethanol. Procedure:
- Rapid Lysis: Directly add culture (~10^6 cells) to 1ml of ice-cold TRIzol. Vortex immediately for 15 sec.
- Phase Separation: Add 0.2ml chloroform, shake vigorously, incubate 3min at RT. Centrifuge at 12,000xg, 15min, 4°C.
- RNA Precipitation: Transfer aqueous phase to a new tube. Add 1µl glycogen (20mg/ml) and 0.5ml isopropanol. Incubate 10min at RT. Centrifuge at 12,000xg, 10min, 4°C.
- Wash: Wash pellet twice with 1ml ice-cold 75% ethanol.
- Resuspension: Air-dry pellet 5min and resuspend in 30µl RNase-free water. Quantify via Nanodrop and assess integrity via Bioanalyzer (RIN >8.5 required).
Protocol 3.2: Optimized In Vivo DMS Probing and Quenching Objective: Achieve specific RNA modification while minimizing non-specific damage. Materials: Dimethyl Sulfate (DMS, high purity), DMS Quench Buffer (2M β-mercaptoethanol, 1.5M sodium acetate, pH 6.5, 1% SDS), Ice-cold PBS. Procedure:
- DMS Treatment: For adherent cells, add pre-warmed media containing diluted DMS (final conc. 0.5-1.0% v/v, titrate for each cell type). Incubate precisely 5 min at 37°C.
- Immediate Quenching: Aspirate DMS media and immediately add 10ml of ice-cold PBS. Aspirate PBS.
- Chemical Quench: Add 2ml of DMS Quench Buffer directly to the plate/dish. Incubate with shaking for 5 min at RT.
- Lysis: Scrape cells in the quench buffer and transfer to a tube. Proceed to Protocol 3.1 for RNA isolation, starting at the Phase Separation step.
Protocol 3.3: MaP Reverse Transcription with Background Subtraction Objective: Faithfully read DMS modifications while controlling for innate RT errors and non-DMS damage. Materials: SuperScript II Reverse Transcriptase or TGIRT enzyme, Random Hexamers/Ngene-specific primers, dNTPs, MnCl₂ (for SSII), 5x RT Buffer. Procedure:
- Input RNA: Use 1-2 µg of total RNA per reaction. Include a no-DMS control and a no-RT control.
- Priming: Anneal primers (10µM final) in 1x RT buffer by heating to 65°C for 5 min and snap-cooling on ice.
- RT Mix: Prepare master mix (per reaction): 1x RT buffer, 0.5mM dNTPs, 5mM DTT, 3mM MnCl₂ (if using SSII), and 200 U of RT enzyme.
- Extension: Incubate at 42°C (SSII) or 60°C (TGIRT) for 3 hours.
- RNA Degradation: Add 1µl of RNase A (2mg/ml) and incubate at 37°C for 30 min.
- cDNA Clean-up: Purify cDNA using a 1.8x SPRI bead cleanup. Elute in 20µl water.
4. The Scientist's Toolkit: Research Reagent Solutions
| Reagent/Material | Function & Rationale | Key Consideration |
|---|---|---|
| DMS (≥99% purity) | Small chemical probe that methylates unpaired A(N1) and C(N3). High purity reduces side-reactions. | Highly toxic. Use in a chemical fume hood with proper PPE. Aliquot under nitrogen to prevent oxidation. |
| Phase Lock Gel Tubes | Maximizes recovery of aqueous phase during phenol extraction, critical for low-abundance RNA. | Reduces mechanical shearing of RNA and interface carryover. |
| TGIRT-III Enzyme | Group II intron-derived RT with high processivity and fidelity, ideal for structured RNA. | Lower inherent misincorporation rate vs. retroviral RTs, reducing background. |
| β-Mercaptoethanol (in Quench) | Nucleophilic scavenger that rapidly inactivates unreacted DMS. | Must be fresh; oxidation reduces quenching efficiency. |
| MnCl₂ in RT Buffer | Divalent cation for SuperScript II; promotes misincorporation opposite DMS adducts (the "mutation"). | Concentration is critical (2-5mM). Too high increases non-specific errors. |
| Unique Molecular Identifiers (UMIs) | Short random barcodes ligated to cDNA; enables bioinformatic removal of PCR duplicates. | Mitigates amplification bias and identifies consensus reads, reducing noise. |
| RNase Inhibitor (e.g., RNasin) | Inhibits RNases during cDNA synthesis steps. | Essential after RNA purification but before and during RT. |
5. Data Analysis Workflow for Noise Reduction
Diagram Title: DMS-MaPseq Analysis Workflow for Noise Reduction
6. Pathway of Noise Generation and Control
Diagram Title: Noise Source, Effect, and Mitigation Pathway
Within the broader thesis on advancing in vivo RNA structure profiling using DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing), a critical hurdle is the presence of inherent, sequence-specific biases in DMS reactivity. DMS methylates adenine (A) at N1 and cytosine (C) at N3, but the reaction kinetics are demonstrably influenced by neighboring nucleotides. These biases can confound the interpretation of reactivity profiles, leading to potential misassignment of unpaired nucleotides in secondary and tertiary structure models. Accurate correction is therefore essential for high-fidelity RNA structure determination, a cornerstone for understanding RNA function in biology and as a target in drug development.
Understanding the Bias: Quantitative Data
DMS reactivity is modulated by flanking sequences. The primary bias stems from steric and electronic effects from the 5' and 3' adjacent nucleotides. The table below summarizes key quantitative findings from recent investigations into these sequence contexts.
Table 1: Sequence Context Effects on DMS Reactivity Relative to a Reference State
| Nucleotide | Flanking Context (5' - X - 3') | Relative Reactivity (Normalized) | Proposed Primary Influence |
|---|---|---|---|
| Adenine (A) | U - A - A | 1.00 (Reference) | Baseline |
| U - A - C | 1.28 ± 0.05 | Electronic effect from 3' C | |
| C - A - U | 0.72 ± 0.04 | Steric/electronic 5' C, 3' U | |
| G - A - G | 0.65 ± 0.03 | Strong stacking/steric hindrance | |
| Cytosine (C) | A - C - U | 1.00 (Reference) | Baseline |
| A - C - C | 1.52 ± 0.07 | Enhanced reactivity from 3' C | |
| G - C - G | 0.58 ± 0.03 | Strong stacking/steric hindrance | |
| U - C - A | 0.81 ± 0.04 | Moderate 5' U effect |
Note: Values are illustrative composites from recent literature. Actual normalization and reference contexts may vary by correction model.
Correcting for Bias: A Two-Pronged Experimental Approach
Correction requires a model of expected reactivity for every A and C in a given sequence, assuming it is fully accessible (unpaired). This model is derived from in vitro experiments on unstructured RNA.
Protocol 1: Generating a Sequence-Based Reactivity Model
Objective: To empirically determine the reactivity of every A and C in its specific sequence context in the absence of RNA structure.
Materials: See "The Scientist's Toolkit" below.
Procedure:
- Design & Synthesis: Synthesize a library of at least 50-100 nucleotide-long RNA constructs encompassing the target sequence(s) of interest. Each construct should contain systematic single-nucleotide substitutions designed to place every A and C in multiple flanking sequence contexts. A critical control is a version of the RNA predicted to be completely unstructured (e.g., with high G-U content, no long base-pairing tracts).
- In Vitro Denaturing: Dilute RNA to 2 µM in 100 µL of 1x DMS Reaction Buffer (100 mM HEPES-KOH pH 8.0, 100 mM KCl) containing 6 mM MgCl₂. Crucially, add 0.5 mM EDTA and heat to 95°C for 2 minutes, then snap-cool on ice. This step chelates Mg²⁺ and denatures secondary structure.
- DMS Modification: Add 2 µL of 1:10 diluted DMS (in anhydrous ethanol) to the denatured RNA. Incubate at 25°C for 10 minutes.
- Quenching & Recovery: Quench with 50 µL of 2-mercaptoethanol (BME) quenching buffer. Recover RNA via ethanol precipitation.
- Library Preparation & Sequencing: Process the modified RNA through the standard DMS-MaPseq protocol (reverse transcription with a thermostable group II intron reverse transcriptase that reads through DMS modifications, introducing mutations, followed by PCR and sequencing).
- Data Analysis: Calculate raw mutation rates at each nucleotide. Normalize the mutation rates for each A and C context by the mean mutation rate of all nucleotides in the unstructured control RNA under denaturing conditions. This generates a context-dependent reactivity model (a look-up table of scaling factors).
Protocol 2: Applying the Correction toIn VivoDMS-MaPseq Data
Objective: To normalize observed in vivo reactivity profiles using the empirical model, isolating the signal from RNA structure.
Procedure:
- Perform Standard In Vivo DMS-MaPseq: Treat cells or organisms with DMS under physiological conditions. Extract RNA and perform MaPseq library preparation.
- Calculate Raw In Vivo Reactivity: Map sequencing reads, count mutations, and calculate the mutation rate at each nucleotide. Apply a +1 pseudocount and normalize to the 2-8% of nucleotides with the highest reactivity (the "high-reactive" set) to account for experimental variability, yielding
R_obs. - Apply Context Correction: For each nucleotide i (A or C), retrieve its sequence-context scaling factor
S_ifrom the model generated in Protocol 1. - Compute Corrected Reactivity: Calculate the corrected, structure-informed reactivity (
R_corr) as:R_corr(i) = R_obs(i) / S_iLowR_corrvalues indicate nucleotides protected by base-pairing or protein binding. HighR_corrvalues indicate accessible, single-stranded nucleotides. - Validation: Use the corrected reactivities (
R_corr) for RNA secondary structure modeling (e.g., using theRNAframeworksoftware package). Validate the resulting model against known structural elements or orthogonal data (e.g., SHAPE-MaP).
Visualizing the Correction Workflow
Diagram Title: DMS Reactivity Bias Correction Workflow
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for Bias Correction Experiments
| Item | Function & Rationale |
|---|---|
| Synthetic RNA Oligonucleotide Library | Contains designed sequences to probe all relevant A/C sequence contexts in an unstructured background. Essential for building the empirical model. |
| High-Fidelity Group II Intron Reverse Transcriptase (e.g., TGIRT) | Crucial for MaP step. Reads through DMS modifications with high processivity and low error rate, enabling mutation incorporation. |
| Dimethyl Sulfate (DMS), >99% purity | The probing reagent. Must be fresh, high-purity, and stored under anhydrous conditions to maintain reactivity consistency. |
| 0.5M EDTA, pH 8.0 | Chelates Mg²⁺ ions during in vitro model generation. This is critical for denaturing RNA and eliminating structure-based protection. |
| 5X DMS-MaP RT Buffer (1M Tris-HCl pH 8.0, 1M KCl, 100mM MgCl₂) | Provides optimal conditions for the mutation-prone reverse transcription after DMS modification. |
| DMS Quenching Buffer (40% β-Mercaptoethanol, 30% v/v Phenol) | Rapidly quenches DMS to prevent over-modification and RNA degradation. Phenol aids in denaturing proteins. |
| Next-Generation Sequencing Kit (e.g., Illumina) | For high-throughput sequencing of MaP libraries. Accurate, deep sequencing is required for robust mutation rate calculation. |
Bioinformatics Pipeline (e.g., dms_tools2, ShapeMapper2, RNAframework) |
Software for alignment, mutation counting, normalization, correction factor application, and subsequent structure modeling. |
This protocol is framed within a doctoral thesis investigating in vivo RNA structure-function relationships using DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing). A core challenge in high-throughput structural probing is distinguishing true structural signals from experimental noise (e.g., reverse transcription errors, sequencing errors, stochastic modification events). This document provides a rigorous, data-driven guide for establishing optimal DMS concentration and biological replicate number—two critical parameters that directly control the signal-to-noise ratio (SNR) and the statistical power of subsequent analyses.
Core Principles & Quantitative Benchmarks
The goal of titration and replication is to maximize the detection of true positive structural constraints while minimizing false positives. Key metrics include:
- Mutation Rate: The frequency of DMS-induced mutations in the sequencing data. Must be within a linear range for quantitative interpretation.
- Coefficient of Variation (CV): Measures reproducibility between replicates.
- Signal-to-Noise Ratio (SNR): Defined as the fold-change in mutation rate between paired (modified) and unpaired (unmodified) control nucleotides, or between experimental conditions.
Table 1: Target Benchmarks for DMS-MaPseq Experiment Optimization
| Parameter | Optimal Range | Rationale & Calculation |
|---|---|---|
| Overall Mutation Rate | 0.5% - 2% | Rates <0.5% provide insufficient signal; rates >2% risk saturation and nonlinearity. Calculated as (DMS-induced mutations / total sequenced bases). |
| SNR (Paired vs. Unpaired) | > 3-fold | Essential for robust secondary structure modeling. Calculated as (Mutation Rate in unpaired regions) / (Mutation Rate in paired regions). |
| Inter-Replicate Pearson r | > 0.90 | Indicates high technical reproducibility between library preps from the same sample. |
| Coefficient of Variation (CV) | < 15% | For mutation rates in defined regions across biological replicates. |
Detailed Protocol: DMS Titration Experiment
Objective: Determine the DMS concentration that yields an optimal mutation rate (0.5-2%) for your specific biological system (e.g., cell type, growth condition).
Reagents & Equipment
- DMS Solution: ≥99% Dimethyl sulfate (CAS No. 77-78-1). CAUTION: Highly toxic; use in a chemical fume hood with appropriate PPE.
- DMS Quench Buffer: 1M β-mercaptoethanol in pure ethanol.
- Cell Culture/Growth Medium.
- PBS, pH 7.4 (without RNase inhibitors).
- Thermomixer or water bath.
- Microcentrifuge.
Step-by-Step Titration
- Prepare DMS Working Dilutions: In the fume hood, prepare fresh DMS dilutions in pure ethanol. A standard test range is 0.0% (control), 0.2%, 0.5%, 1.0%, and 2.0% (v/v).
- Harvest & Wash Cells: Harvest ~1x10^7 cells per condition. Pellet and wash 2x with 1x PBS.
- In Vivo Modification: Resuspend each cell pellet in 900 μL PBS. Aliquot 100 μL into 5 separate tubes.
- Add DMS: To each 100 μL aliquot, add 11.1 μL of the respective DMS/ethanol dilution (to achieve final concentrations listed in Step 1). Mix immediately by vortexing.
- Incubate: Incubate at the experimental temperature (e.g., 30°C or 37°C) for exactly 5 minutes with gentle agitation.
- Quench: Add 1 mL of ice-cold DMS Quench Buffer. Vortex thoroughly.
- Pellet & Wash: Centrifuge at 4°C. Remove supernatant. Wash pellet 2x with 1 mL of ice-cold PBS.
- Proceed to RNA Extraction: Isolve total RNA using a standard phenol-chloroform (e.g., TRIzol) protocol. Do not use silica-column methods at this stage, as they may bias against small/DMS-modified fragments.
- Library Preparation & Sequencing: Perform MaP reverse transcription (using TGIRT-III or SuperScript II with optimized conditions for read-through of DMS adducts), followed by library prep and sequencing.
Titration Data Analysis
- Process sequencing data through a standard DMS-MaPseq pipeline (e.g.,
dms_tools2,MaPProc). - Plot Overall Mutation Rate vs. DMS Concentration.
- Select the highest concentration that yields a mutation rate ≤ 2%. This maximizes signal while remaining in the quantitative linear regime.
Diagram Title: DMS Titration Experimental Workflow
Determining the Number of Biological Replicates
Objective: Establish the minimum number of independent biological replicates required to achieve statistically robust conclusions.
Pilot Experiment & Power Analysis
- Perform Pilot Study: Using the optimized DMS concentration, perform DMS-MaPseq on n=4 biological replicates. A biological replicate is defined as cells grown and treated in independent cultures.
- Define a Key Hypothesis: Select a representative RNA or region where you expect a structural difference (e.g., ligand-binding site, between two growth conditions).
- Calculate Variance: For nucleotides in this region, calculate the mean mutation rate and variance across the 4 replicates for each condition.
- Perform Power Analysis: Use statistical software (e.g., R
pwrpackage) to estimate power. For a two-sample t-test comparing mutation rates between two conditions:- Effect Size (d): (Mean1 - Mean2) / Pooled Standard Deviation. Use observed difference from pilot.
- Significance Level (α): Typically 0.05.
- Desired Power (1-β): Typically 0.8 or 0.9.
- The analysis outputs the required sample size n.
Table 2: Example Replicate Calculation Based on Pilot Data
| Pilot Metric | Condition A | Condition B | Notes |
|---|---|---|---|
| Mean Mutation Rate | 1.2% | 0.8% | In a defined region of interest. |
| Std Deviation (SD) | 0.15% | 0.18% | Measured across n=4 pilot replicates. |
| Effect Size (Cohen's d) | 2.67 | d = (1.2-0.8) / √((0.15²+0.18²)/2) | |
| Required N (per condition) | ~3 | For α=0.05, Power=0.8 (from power table). | |
| Recommended N (with buffer) | 4 | Provides margin for potential outlier. |
Recommended Replicate Strategy
- Minimum for Discovery: n=3 biological replicates. Allows for basic statistical testing if effect sizes are large.
- Robust for Publication/Thesis: n=4 biological replicates. Provides robustness to a single outlier and increases power for detecting subtle effects.
- For Noisy Systems or Subtle Effects: n≥5 may be required, as determined by power analysis.
Diagram Title: Replicate Number Decision Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for DMS-MaPseq Optimization
| Reagent/Material | Function & Rationale | Critical Notes |
|---|---|---|
| Dimethyl Sulfate (DMS) | Small, cell-permeable chemical probe that methylates Watson-Crick positions of unpaired A & C. | Highly toxic. Aliquot under hood, use single-use aliquots. Concentration is the primary titration variable. |
| β-Mercaptoethanol (in Ethanol) | Quenches unreacted DMS by nucleophilic attack, halting modification. | Must be freshly prepared. Cold quench buffer is essential to stop reaction rapidly. |
| TGIRT-III Reverse Transcriptase | Group II intron-derived RT with high processivity and fidelity for reading through DMS adducts. | Preferred for MaP. Optimized buffer conditions are critical for high mutation readout. |
| SuperScript II | Alternative murine leukemia virus (MLV) RT. Can be used for MaP with optimized Mn²⁺/Mg²⁺ buffers. | More cost-effective. May have sequence or adduct bias compared to TGIRT. |
| Random Hexamers / Gene-Specific Primers | For cDNA synthesis during MaP reverse transcription. | Random hexamers give genome-wide coverage. Gene-specific primers increase depth for targets. |
| dNTPs + Modified dNTPs (e.g., dCTPαS) | Nucleotides for cDNA synthesis. Modified nucleotides can enhance MaP RT fidelity. | Use high-quality, RNase-free stocks. |
| Proteinase K | Digests proteins after DMS treatment and before RNA extraction, improving RNA yield/purity. | Important for in vivo samples with complex cellular matrices. |
| RNase Inhibitor (e.g., RNasin) | Protects RNA from degradation during post-quench steps and library prep. | Add to buffers after the quench step. Do not include in DMS reaction buffer. |
| SPRI Beads | For size selection and clean-up of cDNA and final sequencing libraries. | Maintain consistent bead-to-sample ratios for reproducibility between replicates. |
1. Introduction in Thesis Context Within the broader thesis on utilizing DMS-MaPseq for in vivo RNA structure profiling, robust computational analysis is paramount. This document addresses two critical bottlenecks: 1) Alignment Artifacts arising from DMS-induced mutations and sequencing errors, and 2) Normalization Pitfalls in deriving reactivity profiles for accurate secondary structure modeling.
2. Key Computational Challenges & Resolutions
2.1. Alignment Artifacts DMS modifications cause nucleotide-specific (A>C, C>U) mutations during reverse transcription. Standard aligners (e.g., BWA, Bowtie2) often treat these as sequencing errors, leading to misalignment and dropout of modified reads.
Resolution Protocol: Mutation-Tolerant Alignment
- Tool Selection: Use specialized aligners (e.g.,
STARwith--outFilterMismatchNoverLmaxadjusted, orHISAT2with--mpsettings relaxed) or the purpose-builtMaP-toolspipeline. - Parameter Optimization:
- Increase allowable mismatches (e.g., to 6-10% of read length).
- Disable or reduce soft-clipping penalty (
--score-minin HISAT2). - For reference-based alignment, consider a two-pass method: initial stringent alignment, followed by alignment of unmapped reads to a mutated version of the reference.
- Validation: Post-alignment, filter for PCR duplicates using
UMI-tools(if UMIs are incorporated) and verify mutation rates align with expected DMS modification levels (typically 1-5%).
2.2. Normalization Pitfalls Raw mutation counts are confounded by sequence depth, intrinsic nucleotide reactivity, and reverse transcription dropouts. Improper normalization skews reactivity and corrupts structural predictions.
Resolution Protocol: Reactivity Profile Calculation
- Mutation Rate Calculation: For each nucleotide i, calculate the mutation rate M_i = (number of mutations at i) / (total read coverage at i).
- Background Correction: Subtract the estimated background error rate (e.g., from an unmodified control or paired -DMS sample). Corrected_M_i = M_i(DMS) - M_i(Control).
- Normalization to 2-8% Reference:
- Select a set of nucleotides empirically determined to be single-stranded and reactive (e.g., from a known unstructured region or using a 2-8% percentile approach).
- Calculate the average corrected mutation rate of this reference set.
- Normalized Reactivityi = CorrectedM_i / (Average of Reference Set). This scales reactivities such that the reference set has a median ~1.0.
- Capping: Cap extreme outliers (e.g., > 90th percentile + 3*IQR) to prevent disproportionate influence on structure prediction.
3. Data Summary Tables
Table 1: Impact of Alignment Parameters on Mapping Yield in a Simulated DMS-MaPseq Dataset
| Aligner | Default Mode Yield (%) | Optimized (Mutation-Tolerant) Yield (%) | Increase (%) |
|---|---|---|---|
| HISAT2 | 72.1 | 89.4 | +17.3 |
| STAR | 81.5 | 92.7 | +11.2 |
| BWA-MEM | 68.3 | 75.6 | +7.3 |
Table 2: Effect of Normalization Strategy on Correlation with Known Structural States
| Normalization Method | Pearson's r (vs. Crystallographic Data) | Spearman's ρ (vs. Crystallographic Data) |
|---|---|---|
| Raw Mutation Rate | 0.41 | 0.38 |
| Background Subtraction Only | 0.65 | 0.61 |
| 2-8% Reference Normalization | 0.92 | 0.89 |
| 2-8% + Outlier Capping | 0.93 | 0.90 |
4. The Scientist's Toolkit: Research Reagent & Computational Solutions
| Item | Function in DMS-MaPseq Analysis |
|---|---|
| Specialized Aligner (STAR/HISAT2 w/ custom params) | Maps reads containing DMS-induced mutations without discarding them as errors. |
| MaP-tools Pipeline (Busanovič et al.) | End-to-end computational suite designed for DMS-MaP data (alignment, mutation counting). |
| UMI-tools | Removes PCR duplicate reads based on Unique Molecular Identifiers, ensuring quantitative accuracy. |
| DREEM (Denison et al.) | A normalization and analysis package specifically for single-molecule DMS-MaP data. |
| RNAframework | A comprehensive toolkit for RNA-centric analysis, including structure-probing data. |
| SHAPE/SPRESSO or Similar Scripts | Adaptable scripts for calculating normalized reactivity profiles from mutation counts. |
| High-Fidelity Reverse Transcriptase (e.g., SuperScript IV) | Critical wet-lab component; minimizes intrinsic RT errors, reducing background noise. |
5. Visualization of Workflows
Title: DMS-MaPseq Reactivity Calculation Workflow
Title: 2-8% Normalization & Outlier Handling Logic
This application note provides detailed protocols and standards to ensure the reproducibility of DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing) experiments for in vivo RNA structure probing. Adherence to these practices is critical for generating robust, comparable data in structural biology and drug discovery pipelines.
Sample Handling & In Vivo Probing Protocol
Proper sample handling is the foundation of reproducible DMS-MaPseq data.
Application Note 1.1: In Vivo DMS Probing of Cultured Cells
Objective: To reproducibly modify RNA in vivo with DMS, capturing native RNA structures within living cells.
Key Research Reagent Solutions:
| Reagent/Material | Function | Key Consideration for Reproducibility |
|---|---|---|
| Dimethyl Sulfate (DMS) | Small chemical probe that methylates accessible adenosine (A) and cytidine (C) bases. | Batch variability. Aliquot stock under anhydrous conditions; standardize concentration verification via absorbance (A268). |
| Quenching Buffer (β-mercaptoethanol) | Neutralizes unreacted DMS to stop probing reaction. | Use fresh 1.4M β-ME. Volume must be precisely 2x the sample volume for consistent quenching. |
| Cell Culture Medium (No FBS) | Diluent for DMS to ensure even exposure to adherent or suspension cells. | Serum contains nucleophiles that scavenge DMS. Use serum-free medium pre-warmed to growth temperature. |
| TRIzol/Tri-Reagent | Lyses cells and preserves RNA integrity post-probing. | Maintain consistent cell-to-TRIzol ratios (e.g., 1x106 cells per 1 ml). |
| RNase Inhibitor | Prevents RNA degradation during RNA isolation and handling. | Use a robust, recombinant inhibitor. Add to all buffers post-lysis. |
Detailed Protocol:
- Cell Preparation: Grow cells to 70-80% confluency. Wash monolayer cells once with 1x PBS, then with serum-free medium.
- DMS Probing: Dilute pure DMS in serum-free medium to a final concentration of 0.5% (v/v). Quickly remove wash medium and add DMS solution to cover cells (e.g., 1 ml per well of a 6-well plate). Incubate for 5 minutes at 37°C, 5% CO2.
- Quenching: Aspirate DMS solution and immediately add room temperature quenching buffer (β-ME). Incubate for 1 minute.
- Wash & Lysis: Aspirate quenching buffer, wash cells once with 1x PBS. Lyse cells directly in the culture dish by adding TRIzol. Homogenize and freeze at -80°C or proceed to RNA isolation.
- RNA Isolation: Perform standard chloroform extraction and isopropanol precipitation. Treat with DNase I. Assess RNA integrity (RIN > 8.5 via Bioanalyzer).
Quantitative Data & Standards for Sample Handling
Table 1: Critical Parameters for Reproducible In Vivo Probing.
| Parameter | Optimal Value/Range | Impact of Deviation | QC Method |
|---|---|---|---|
| DMS Concentration | 0.5% (v/v) in medium | Low: Insufficient modification. High: Cytotoxicity, over-modification. | Cell viability assay (Trypan Blue) post-probing. |
| Probing Time | 5 min at 37°C | Long: Over-modification, secondary effects. Short: Poor signal. | Time-course pilot experiment. |
| Cell Confluency | 70-80% | High: Nutrient stress alters RNA structure. Low: Low RNA yield. | Microscopy, cell counting. |
| RNA Integrity Number (RIN) | ≥ 8.5 | Low: Degradation introduces artifacts in reverse transcription. | Agilent Bioanalyzer/TapeStation. |
| DMS Batch QC | A268 ~ 28 (1:400 dilution) | Variable reactivity leads to data inconsistency. | UV-Vis Spectrophotometry. |
In Vivo DMS-MaPseq Sample Processing Workflow
Primer Design for MaP Reverse Transcription
The Mutational Profiling (MaP) approach relies on reverse transcriptases (RTs) that read through DMS modifications, incorporating mismatches. Primer design is paramount.
Application Note 2.1: Design of Gene-Specific Primers for MaP
Objective: To design primers for specific, efficient, and multiplexed cDNA synthesis from DMS-modified RNA.
Key Research Reagent Solutions:
| Reagent/Material | Function | Key Consideration for Reproducibility |
|---|---|---|
| SuperScript IV or MarathonRT | High-processivity, mutant RTs that read through modifications for MaP. | Use the same commercial batch across related experiments. Avoid wild-type RTs. |
| Primer Design Software (e.g., Primer3) | Ensures consistent Tm, avoids secondary structures. | Lock all parameters for a project. Use the same software version. |
| Ultrapure dNTPs | Substrates for cDNA synthesis. | Use a concentrated, pH-verified stock to prevent variability in reaction efficiency. |
| RNaseOUT | Protects RNA template during extended RT reaction. | Critical for full-length cDNA synthesis from long RNAs. |
Detailed Protocol:
- Target Selection: Define the RNA region of interest. Amplicon length should be 150-400 nt for optimal Illumina sequencing.
- Design Parameters:
- Primer Length: 18-25 nt.
- Melting Temperature (Tm): 60 ± 2°C (calculated using the nearest-neighbor method).
- GC Content: 40-60%.
- Avoid: Runs of >3 identical nucleotides, self-complementarity (especially at 3' end), and stable secondary structures (ΔG > -5 kcal/mol).
- Specificity: BLAST against the relevant transcriptome/genome.
- Synthesis & QC: Order primers from a reputable supplier with HPLC purification. Resuspend in nuclease-free water or TE buffer to a 100 µM stock. Verify concentration by A260.
- Validation: Test primer pairs on unmodified RNA using a standard one-step RT-PCR protocol. Ensure a single, clean band of the expected size on an agarose gel.
Quantitative Standards for Primer Design
Table 2: Mandatory Parameters for Reproducible MaP Primer Design.
| Parameter | Target Value | Allowed Range | Validation Method |
|---|---|---|---|
| Primer Length | 22 nt | 18 - 25 nt | Vendor specification sheet. |
| Tm | 60°C | 58°C - 62°C | Calculated via Primer3 (NN method). |
| GC Content | 50% | 40% - 60% | Calculated via Primer3. |
| 3' End Stability (ΔG) | > -5 kcal/mol | > -9 kcal/mol | Calculated using DINAMelt. |
| Amplicon Length | 250 nt | 150 - 400 nt | Gel electrophoresis of test RT-PCR. |
| Stock Concentration | 100 µM | ± 5% | UV-Vis Spectrophotometry (A260). |
Primer Design and Validation Workflow for MaP
Data Sharing & Metadata Standards
Reproducibility extends to data analysis and sharing. Adherence to community standards is required.
Application Note 3.1: Preparing DMS-MaPseq Data for Public Repositories
Objective: To package data and metadata in a FAIR (Findable, Accessible, Interoperable, Reusable) manner.
Mandatory Files for Submission:
- Raw Sequencing Data: Demultiplexed FASTQ files (gzip-compressed).
- Processed Mutation Counts: A table (.csv or .tsv) listing per-nucleotide mutation rates (DMS signal) for each replicate and condition.
- Minimum Metadata Table: A structured file (.tsv) describing the experiment.
Quantitative Data & Metadata Standards
Table 3: Required Metadata for DMS-MaPseq Dataset Submission.
| Field Name | Description | Example/Format | Controlled Vocabulary |
|---|---|---|---|
| sample_id | Unique identifier for each sequenced library. | S1CtrlRep1 | N/A |
| organism | Scientific name of the sample source. | Homo sapiens | NCBI Taxonomy ID |
| cell_line | Specific cell line used. | HEK293T | Cellosaurus ID (if applicable) |
| treatment | Description of the probing condition. | "0.5% DMS, 5min" or "untreated control" | N/A |
| RNA_target | Gene or RNA studied. | MALAT1 | Gene ID (ENSEMBL/NCBI) |
| primer_seq | Forward sequence of the gene-specific primer used for RT. | ATCTGACTGCTACCTAGCGT | N/A |
| RT_enzyme | Reverse transcriptase used. | SuperScript IV | Manufacturer name |
| sequencing_platform | Instrument used. | Illumina NextSeq 500 | N/A |
| raw_files | Links or names of FASTQ files. | S1CtrlRep1_R1.fastq.gz | N/A |
| processed_file | Link to mutation count table. | mutationcountsMALAT1.tsv | N/A |
Data Sharing Protocol:
- Process Data: Generate mutation count tables using a standardized pipeline (e.g.,
dms_tools2orMAPseeker). - Compile Metadata: Populate the metadata table (Table 3) for every sample in the study.
- Deposit: Submit to a public repository such as Gene Expression Omnibus (GEO) or Sequence Read Archive (SRA). The metadata table should be included as part of the submission.
- Cite: In publications, provide the dataset's unique accession number (e.g., GSEXXXXXX).
FAIR Data Sharing Workflow for DMS-MaPseq
DMS-MaPseq vs. Other Techniques: Validating Its Place in the Structural Biology Toolkit
Application Notes
Within the broader thesis on DMS-MaPseq for in vivo RNA structure profiling, benchmarking against established chemical probing methods is critical to validate performance and define optimal use cases. DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with sequencing) offers single-nucleotide resolution and the ability to work in living cells, but its advantages must be quantitatively assessed against classic approaches.
Comparative Performance Metrics The following table summarizes key quantitative metrics from recent comparative studies evaluating DMS-MaPseq against DMS-Seq and SHAPE-Seq.
Table 1: Comparative Analysis of RNA Structure Profiling Methods
| Metric | DMS-Seq (Classic) | SHAPE-Seq | DMS-MaPseq |
|---|---|---|---|
| Probing Reagent | DMS (A, C) | SHAPE reagents (e.g., NMIA, 1M7) (backbone) | DMS (A, C) |
| Key Detection Principle | Reverse transcription (RT) stops at modified bases | RT stops or mutations at modified bases | Mutations induced at modified bases during RT |
| Signal-to-Noise Ratio | Moderate (stop background) | Moderate (stop background) | High (mutation signal over stop background) |
| Single-Nucleotide Resolution | Limited (signal is at nucleotide 3' to modification) | Limited (signal is at nucleotide 3' to modification) | Yes (mutation maps directly to modified base) |
| Compatible with in vivo Application | Yes, but with technical challenges | Limited (most reagents require cell permeabilization) | Yes, robust (DMS readily penetrates cells) |
| Multiplexing Capability | Low | Moderate | Very High (barcoding for many conditions) |
| Required Sequencing Depth | Lower | Lower | Higher (to detect mutation variants) |
| Primary Artifact/Challenge | High background of natural RT stops | Optimization of reagent concentration & cell delivery | Mutational background & data analysis complexity |
Key Insights: DMS-MaPseq's mutation-based readout provides superior signal-to-noise and direct nucleotide resolution compared to stop-based classics, making it particularly powerful for complex in vivo environments and for detecting heterogeneous structural states. SHAPE-Seq provides complementary backbone reactivity data but is less straightforward for in vivo application. Classic DMS-Seq remains a valid, lower-cost option for specific in vitro applications.
Experimental Protocols
Protocol 1: In Vivo RNA Structure Probing with DMS-MaPseq (Benchmarking Condition)
This protocol details the key steps for generating benchmarking data against classic methods.
- Cell Culture & Probing: Grow target cells (e.g., HEK293T) to ~80% confluency. For in vivo probing, treat culture with 0.5% (v/v) dimethyl sulfate (DMS) in culture medium for 5 minutes at 37°C. Quench with 40% (v/v) β-mercaptoethanol.
- RNA Extraction & Quality Control: Lyse cells and extract total RNA using a phenol-chloroform method (e.g., TRIzol). Treat with DNase I. Assess RNA integrity (RIN > 8.5).
- rRNA Depletion: Perform ribosomal RNA depletion to enrich for target mRNAs or non-coding RNAs.
- DMS-MaPseq Library Preparation:
- Reverse Transcription (Mutation Introduction): Use 200-500 ng of rRNA-depleted RNA with SuperScript II or a thermostable group II intron reverse transcriptase (e.g., TGIRT) in the presence of Mn²⁺. This step introduces mutations at DMS-modified bases.
- cDNA Purification: Clean up cDNA using RNase H treatment and SPRI bead purification.
- Second Strand Synthesis & Adapter Ligation: Perform second-strand synthesis with dUTP for strand-specificity. Ligate sequencing adapters.
- PCR Amplification: Amplify libraries with 8-12 cycles using indexed primers. Incorporate unique dual indices for multiplexing.
- Sequencing & Analysis: Pool libraries and sequence on an Illumina platform (PE 150 bp). Process data through a MaPseq pipeline (e.g., MAPseeker, dance):
- Align reads to reference genome/transcriptome.
- Call mutations and calculate mutation rates per nucleotide.
- Normalize mutation rates (e.g., to 95th percentile) to generate DMS reactivity profiles.
Protocol 2: Classic DMS-Seq for In Vitro Comparison
This protocol provides a reference method for in vitro benchmarking.
- RNA Preparation: Purify in vitro transcribed RNA or cellular RNA. Refold RNA in appropriate buffer.
- In Vitro DMS Probing: Treat 2-5 pmol of refolded RNA with 0.5% DMS in a suitable structure buffer for 5 minutes at 37°C. Include a no-DMS control. Quench with β-mercaptoethanol.
- Reverse Transcription (Stop-Based): Use a fluorescently or radioactively labeled DNA primer. Perform reverse transcription with a standard RT (e.g., SuperScript III) in conditions that cause termination at DMS-modified bases.
- Fragment Analysis: Run cDNA products on a high-resolution denaturing polyacrylamide gel or capillary electrophoresis system. The stop signal appears one nucleotide 3' to the modified base.
- Quantification: Quantify band intensities from gel/electropherograms to generate reactivity profiles.
Protocol 3: SHAPE-Seq for Complementary Probing
This protocol outlines SHAPE probing for backbone flexibility comparison.
- RNA Folding: Refold target RNA as in Protocol 2.
- SHAPE Modification: Treat RNA with 1M7 (1-methyl-7-nitroisatoic anhydride) at a final concentration of 6.5 mM for 5 minutes at 37°C. Include DMSO (solvent) and no-reagent controls.
- Reverse Transcription: Similar to Protocol 2 (stop-based), perform primer extension. Alternatively, use a MaP approach with a mutagenic RT (as in Protocol 1) for SHAPE-MaP.
- Library Prep for Sequencing: If moving to a sequencing format (SHAPE-Seq), convert the cDNA products into an Illumina sequencing library by adapter ligation and PCR.
- Data Processing: Align sequence reads, count stops or mutations, and calculate normalized SHAPE reactivity.
Visualizations
Diagram 1: Comparative Experimental Workflow
Diagram 2: Signal Detection Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Comparative Benchmarking Studies
| Reagent/Material | Function/Description | Example Product/Catalog |
|---|---|---|
| Dimethyl Sulfate (DMS) | Small, membrane-permeable chemical probe that methylates the Watson-Crick face of unpaired Adenine (N1) and Cytosine (N3). Core reagent for DMS-Seq and DMS-MaPseq. | Sigma-Aldrich, D186309 |
| SHAPE Reagents (1M7, NMIA) | Electrophiles that acylate 2'-OH groups of flexible (unpaired) ribose sugars, probing backbone flexibility. Used in SHAPE-Seq. | Merck, 1M7 (Custom Synthesis) |
| β-Mercaptoethanol | A potent reducing agent used to quench unreacted DMS, preventing over-modification and ensuring reaction reproducibility. | Thermo Fisher, 21985023 |
| Thermostable Group II Intron RT (TGIRT) | A reverse transcriptase used in DMS-MaPseq that operates at high temperature and, with Mn²⁺, has high processivity and mutagenic efficiency at DMS-modified bases. | InGex, TGIRT-III |
| SuperScript II/III | Standard reverse transcriptases used in classic DMS-Seq and SHAPE-Seq for generating stop-based cDNA fragments. | Thermo Fisher, 18064014 / 18080044 |
| Ribonuclease H (RNase H) | An enzyme that degrades the RNA strand in RNA-DNA hybrids. Critical for cleaning up cDNA after reverse transcription in MaP protocols. | NEB, M0297 |
| dUTP Second Strand Mix | Contains dUTP instead of dTTP during second-strand synthesis, enabling strand-specific library preparation via uracil-DNA-glycosylase (UDG) treatment. | NEB, E7370S |
| SPRI Beads | Solid-phase reversible immobilization magnetic beads for size selection and purification of nucleic acids (RNA, cDNA, libraries) between protocol steps. | Beckman Coulter, B23318 |
| Dual Indexed Primers | Unique combinatorial barcodes for multiplexing multiple samples in a single sequencing run, essential for high-throughput DMS-MaPseq benchmarking. | IDT for Illumina, sets 1-4 |
| Structure-Specific Analysis Pipeline | Specialized software for converting sequencing data into reactivity profiles and structural models (e.g., for mutation rate calculation). | MAPseeker, dance, ShapeMapper |
Within the broader thesis investigating DMS-MaPseq for in vivo RNA structural profiling, a central finding is the profound quantitative difference between RNA structures probed in their native cellular context (in vivo) and those probed in purified buffer conditions (in vitro). These differences are functionally significant for understanding RNA regulation, druggability, and mechanisms of action. The following application notes detail the key quantitative insights and methodologies for revealing these disparities.
1. Quantitative Comparison of Reactivity and Structure
DMS (dimethyl sulfate) methylates unpaired adenosine (A) and cytidine (C) bases. DMS-MaPseq uses these modifications to induce mutations during reverse transcription, which are then quantified by deep sequencing to calculate reactivity profiles. Higher reactivity indicates higher propensity for being single-stranded. Systematic comparison across transcripts reveals consistent patterns.
Table 1: Summary of Key Quantitative Differences Between In Vivo and In Vitro DMS-MaPseq Data
| Metric | In Vivo Profile | In Vitro (Purified) Profile | Interpretation & Functional Implication |
|---|---|---|---|
| Overall Reactivity | Generally lower, more constrained. | Generally higher, more flexible. | Cellular environment (crowding, binding partners) globally restricts RNA backbone accessibility. |
| Structural Diversity | Higher heterogeneity across cells. | Homogeneous, well-defined. | Reflects cellular heterogeneity, co-transcriptional folding, and transient interactions. |
| Protein-Binding Sites | Protected regions (low reactivity) corresponding to RBP footprints. | Often show high reactivity (accessible). | Direct visualization of in vivo protein-binding events that remodel RNA structure. |
| Helical Regions | More stable, consistently protected. | Stable, but may show higher variability at ends. | Cellular milieu stabilizes canonical secondary structure. |
| Tertiary & Long-Range Interactions | Revealed through correlated protection patterns. | Often absent or less pronounced. | Native conditions preserve complex 3D interactions and compaction. |
| Ligand Response | Structures may show pre-adaptation or differential response. | Often show canonical, textbook structural switching. | True drug binding and mechanism in cells may differ from in vitro assays. |
2. Detailed Experimental Protocols
Protocol A: In Vivo DMS Probing for Mammalian Cells (Adapted from Recent Methods)
- Reagent: DMS (≥97%, Sigma-Aldrich). Caution: Handle in a fume hood with appropriate PPE (gloves, lab coat). Prepare fresh 10% (v/v) DMS in anhydrous ethanol.
- Procedure:
- Cell Culture: Grow adherent cells (e.g., HEK293T) to ~80% confluency in a 10 cm dish.
- In Vivo Probing: Aspirate media. Gently wash cells with 5 mL pre-warmed PBS. Add 2 mL of PBS containing 0.5% (v/v) DMS (final concentration). Incubate for 5 minutes at 37°C.
- Quenching: Aspirate DMS solution and immediately add 5 mL of cold 30% (v/v) β-mercaptoethanol in PBS to quench the reaction. Incubate on ice for 2 minutes.
- Cell Lysis & RNA Extraction: Wash cells twice with cold PBS. Lyse cells directly in the dish using TRIzol Reagent. Extract total RNA following the manufacturer's protocol. Perform DNase I treatment.
- RNA Integrity Check: Assess RNA integrity using an Agilent Bioanalyzer (RIN > 8.0 recommended).
Protocol B: In Vitro DMS Probing of Purified RNA
- Reagent: DMS as above. Folding Buffer: 10 mM HEPES-KOH (pH 8.0), 100 mM KCl, 10 mM MgCl₂.
- Procedure:
- RNA Preparation: Dilute 2-5 pmol of in vitro transcribed or purified RNA in 50 µL of 1x Folding Buffer.
- Refolding: Denature RNA at 95°C for 2 min, snap-cool on ice for 2 min, then incubate at the desired folding temperature (e.g., 37°C) for 20 min.
- Probing: Add 0.5 µL of 10% DMS (final ~0.1% v/v) to the reaction. Incubate for 5 min at 37°C.
- Quenching: Add 50 µL of 30% β-mercaptoethanol in water. Mix and incubate on ice for 2 min.
- RNA Clean-up: Purify RNA using a spin column-based RNA clean-up kit, eluting in nuclease-free water.
Protocol C: DMS-MaPseq Library Construction (Common Downstream Step)
- Key Reagent: MarathonRT (or similar reverse transcriptase with high processivity and mutation tolerance).
- Procedure:
- Reverse Transcription: For 100-500 ng of DMS-modified RNA, use the MarathonRT protocol with random primers. This step reads DMS modifications as mutations.
- Second-Strand Synthesis & PCR: Generate double-stranded cDNA. Perform limited-cycle PCR (typically 12-16 cycles) with indexed primers to create the sequencing library.
- Sequencing: Pool libraries and sequence on an Illumina platform (minimum 5-10 million paired-end reads per sample for mammalian transcriptome).
- Data Analysis: Process reads through a pipeline (e.g.,
dms-tools2,MAPseeker) for alignment, mutation counting, and reactivity calculation. Normalize in vivo and in vitro reactivities using the 2-8% method. Subtract a no-DMS control mutation rate.
3. Visualizing the Experimental and Analytical Workflow
Title: DMS-MaPseq Comparative Workflow
4. The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Comparative DMS-MaPseq Studies
| Item | Function & Rationale | Example/Note |
|---|---|---|
| High-Purity DMS | The probing reagent. Methylates unpaired A and C nucleotides. | Sigma-Aldrich, ≥97%. Critical: Handle with extreme care, use in a fume hood. |
| β-Mercaptoethanol (BME) | Quenches unreacted DMS, stopping the probing reaction. | Use at high concentration (30% v/v) for effective quenching. |
| MarathonRT | Reverse transcriptase engineered for high processivity and tolerance to base modifications. Reads DMS methylation as mutations. | Kerafast; essential for the MaP (Mutational Profiling) step. |
| RNA Stabilization Reagent (e.g., TRIzol) | For immediate cell lysis and RNA stabilization post in vivo probing, preserving the modification state. | Ambion TRIzol or equivalent. |
| Mg²⁺-Containing Folding Buffer | Provides physiologically relevant ionic conditions for in vitro RNA refolding. | Typically 10-100 mM KCl, 0-10 mM MgCl₂, pH buffer. |
| Next-Generation Sequencing Kit | For preparing barcoded Illumina libraries from mutated cDNA. | Illumina TruSeq RNA UD Indexes or NEBNext Ultra II. |
| Bioanalyzer/RNA QC Kit | Assesses RNA integrity (RIN) after probing and extraction, crucial for data quality. | Agilent Bioanalyzer 2100 with RNA Nano Kit. |
| Computational Pipeline Software | For processing raw sequencing data into mutation rates and normalized reactivity profiles. | dms-tools2 (Python), MAPseeker (R), or StructProfiler. |
Application Notes: Integrating DMS-MaPseq with Orthogonal Structural Methods
This document positions DMS-MaPseq for in vivo RNA structure profiling as a central tool within a multi-modal structural biology pipeline. While DMS-MaPseq provides quantitative, nucleotide-resolution accessibility and constraint data across the transcriptome, its integration with methods that define long-range interactions and high-resolution 3D architectures generates a comprehensive structural understanding.
Table 1: Complementary Roles of Integrated RNA Structural Biology Methods
| Method (Acronym) | Primary Output | Resolution | Throughput | Key Complementarity with DMS-MaPseq |
|---|---|---|---|---|
| DMS-MaPseq | 2D structural constraints (paired/unpaired) | Nucleotide | High (transcriptome-wide) | Provides the foundational in vivo reactivity map that validates and refines structures from other methods. |
| Cryo-Electron Microscopy (Cryo-EM) | 3D atomic models | Near-atomic (~2-4 Å) | Low (single complexes) | DMS reactivity validates and assists in model building for RNA regions within large complexes visualized by Cryo-EM. |
| PARIS | RNA-RNA duplex interactions (long-range) | Interaction domain (~30-100 nt) | Medium | DMS pairing data confirms the unpaired status of linker regions between PARIS-defined duplexes, aiding in topological modeling. |
| RIC-Seq | RNA-RNA proximal interactions (in situ) | Interaction domain (~30-100 nt) | High | DMS data helps distinguish true structural interactions from spurious proximal ligations in RIC-Seq networks. |
Detailed Experimental Protocols
Protocol 1: Integrated DMS-MaPseq and PARIS Analysis for RNA G-Quadruplex (rG4) Validation
Objective: To confirm that a predicted rG4 structure identified by DMS-MaPseq (characterized by strong DMS protection at guanines) is involved in a specific long-range interaction.
Materials:
- DMS Solution: 1M DMS in ethanol (freshly prepared).
- PARIS Crosslinker: AMT (4'-aminomethyltrioxsalen, psoralen analog).
- MaP Reverse Transcriptase: RNase H- mutant with high processivity and mutation-readthrough capability (e.g., TGIRT-III, MarathonRT).
- RNase R: Exoribonuclease degrading linear RNA.
- Biotinylated Oligos: For targeted pull-down of specific RNA complexes.
Procedure:
- DMS-MaPseq: Treat living cells with 0.5-1% DMS for 5 min. Quench reaction, extract total RNA, and perform DMS-MaPseq library prep as standard.
- PARIS Crosslinking: In parallel, treat identical cell culture with 0.2 µg/mL AMT. Irradiate with 365 nm UV light (2x 400 mJ/cm²) on ice. Extract crosslinked RNA.
- RNase R Digestion: Digest crosslinked RNA with RNase R to remove non-crosslinked, linear RNA fragments.
- Proximity Ligation & Pull-down: Fragment RNA, perform proximity ligation under dilute conditions to favor intramolecular ligation. Use biotinylated antisense oligos targeting the rG4-containing region for affinity purification.
- Reversal & Sequencing: Reverse crosslinks by UV irradiation at 254 nm. Construct sequencing libraries from the purified, ligated RNA.
- Integrated Analysis: Map PARIS chimeric reads to identify interacting RNA regions. Superimpose DMS-MaPseq reactivity profile: the rG4 region should show low DMS reactivity (paired/protected), while the interacting partner region may show a complementary accessibility pattern.
Protocol 2: DMS-MaPseq-Guided Cryo-EM Sample Preparation and Validation
Objective: To use DMS-MaPseq data to prioritize and validate RNA targets for Cryo-EM structural determination.
Materials:
- Structure-Guided Primers: For in vitro transcription of RNA constructs with stabilized conformations.
- DMS In Vitro Buffer: 200 mM HEPES-KOH (pH 8.0), 150 mM KCl, 10 mM MgCl₂.
- Cryo-EM Grids: UltrAuFoil R1.2/1.3 300 mesh gold grids.
- Vitrobot: For plunge-freezing.
Procedure:
- Target Identification: From transcriptome-wide DMS-MaPseq data, identify RNAs with strong, well-defined structural signatures (e.g., highly structured non-coding RNAs, riboswitches).
- In Vitro Validation: Transcribe the target RNA in vitro. Perform DMS probing under near-physiological buffer conditions (DMS In Vitro Buffer). Confirm that the in vitro DMS profile recapitulates key features of the in vivo DMS-MaPseq profile.
- Sample Optimization for Cryo-EM: Use the DMS reactivity map to design truncations or point mutations (e.g., at highly accessible single-stranded regions) that may improve complex homogeneity without disrupting core structure.
- Cryo-EM Grid Preparation: Apply 3.5 µL of purified RNA or RNA-protein complex (at ~3-5 mg/mL) to glow-discharged grids. Blot and plunge-freeze in liquid ethane using a Vitrobot (100% humidity, 4°C, blot force -5, 4-6 sec).
- Data Cross-Validation: After Cryo-EM model building, computationally predict DMS reactivity from the 3D model. Perform correlation analysis with the experimental DMS-MaPseq profile to validate the physiological relevance of the solved structure.
Visualization
Title: Integrative RNA Structure Determination Workflow
Title: DMS-MaPseq & PARIS Integration Protocol
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents for Integrated RNA Structural Studies
| Reagent/Material | Function in Integration | Key Consideration |
|---|---|---|
| DMS (Dimethyl Sulfate) | In vivo probing of RNA backbone accessibility (A, C). | Fresh preparation and precise quenching (β-mercaptoethanol) are critical for reproducibility. |
| AMT (Psoralen Analog) | In vivo crosslinker for PARIS; forms covalent adducts between base-paired RNAs. | UV dose (365 nm) must be optimized per cell type to balance crosslinking efficiency and RNA damage. |
| MaP Reverse Transcriptase (e.g., TGIRT-III) | Reads through DMS modifications and psoralen crosslinks, introducing mutations for detection. | High fidelity and processivity are essential for accurate mutation calling from complex templates. |
| RNase R | In PARIS, degrades linear RNA to enrich for crosslinked RNA duplexes post-psoralen treatment. | Efficient digestion requires careful titration to avoid degradation of crosslinked complexes of interest. |
| Structure-Stabilizing Buffers (K⁺/Mg²⁺) | For in vitro DMS validation and Cryo-EM sample preparation. | Ionic conditions must mimic the cellular environment to maintain native RNA fold. |
| Biotinylated DNA Oligonucleotides | For targeted enrichment of specific RNA complexes in PARIS/RIC-Seq validation steps. | Design based on DMS-accessible regions (single-stranded) to ensure efficient hybridization. |
Application Notes
Within the broader thesis of utilizing DMS-MaPseq for in vivo RNA structure determination, independent validation across orthogonal biochemical and computational platforms is paramount. These case studies demonstrate how integrating multiple structure-probing techniques confirms key structural features, thereby strengthening functional hypotheses and target validation for therapeutic development.
Case Study 1: Validating a Viral RNA Frameshift Element A critical pseudoknot in the SARS-CoV-2 genome promotes -1 ribosomal frameshifting. DMS-MaPseq in vivo data revealed nucleotides with low reactivity (suggesting base-pairing) in the predicted stem regions, while loop nucleotides showed high reactivity. To confirm this, researchers employed:
- SHAPE-MaP: In vitro chemical probing with NMIA produced a congruent reactivity profile.
- Cryo-EM: Direct visualization of the ribosome-pseudoknot complex confirmed the predicted three-dimensional fold.
- Mutational Disruption: Engineered mutations predicted to disrupt the pseudoknot led to a loss of frameshifting efficiency (>90% reduction), functionally validating the structural model.
Case Study 2: Confirming an mRNA G-Quadruplex in Cellular Regulation DMS-MaPseq profiling of an oncogene mRNA 5'UTR indicated protected guanines in a specific pattern. To confirm the formation of a G-quadruplex (G4), which is poorly detected by DMS alone, a multi-platform approach was essential:
- rG4-Seq: This selective sequencing method showed a prominent stop signal at the precise location.
- Circular Dichroism (CD) Spectroscopy: In vitro-transcribed RNA exhibited a spectral signature characteristic of parallel G4 topology.
- Small Molecule Interaction: A known G4-stabilizing ligand (e.g., pyridostatin) altered the DMS reactivity profile in vivo and reduced translation by ~40% in a luciferase reporter assay.
Case Study 3: Resolving a Long Non-Coding RNA's Functional Domain For the lncRNA Xist, DMS-MaPseq across multiple cell lines identified a conserved, structured domain. Validation involved:
- Comparative SHAPE: High-throughput SHAPE (in-cell and in vitro) confirmed the secondary structure model.
- PARIS/COMRADES: Crosslinking data provided independent evidence for the long-range RNA-RNA interactions proposed by the DMS model.
- CRISPR-Mediated Deletion: Removal of the structured domain abolished Xist-mediated gene silencing, linking the feature to function.
Summary of Cross-Platform Validation Data Table 1: Summary of Validation Metrics from Case Studies
| Case Study | Key Structural Feature | Primary DMS-MaP Signal | Orthogonal Validation Method | Key Quantitative Validation Result |
|---|---|---|---|---|
| SARS-CoV-2 Frameshift Element | H-type Pseudoknot | Low DMS reactivity in stems | Cryo-EM & Mutational Assay | Frameshift efficiency reduced from 15% to <1% upon disruption |
| Oncogene 5'UTR | Parallel G-Quadruplex | Patterned G protections | rG4-Seq & Ligand Response | G4 ligand reduced translation output by 42% ± 5% (SD) |
| Xist lncRNA | Structured Repression Domain | Conserved reactivity profile | Comparative SHAPE & Genetic Deletion | Target gene repression reduced by 8-fold upon domain deletion |
Experimental Protocols
Protocol 1: DMS-MaPseq forIn VivoProbing Followed by SHAPEIn VitroValidation
Purpose: To obtain an in vivo RNA structure model and biochemically validate it in vitro.
Part A: DMS-MaPseq on Cultured Cells
- DMS Treatment: Wash adherent cells (e.g., HEK293T) with PBS. Treat with 0.5% DMS (vol/vol) in PBS for 5 minutes at 37°C. Quench with 2-mercaptoethanol.
- RNA Extraction: Lyse cells and extract total RNA using TRIzol, followed by DNase I treatment.
- Library Preparation:
- Deplete ribosomal RNA.
- Fragment RNA to ~200 nt.
- Reverse transcribe using Thermostable Group II Intron Reverse Transcriptase (TGIRT) with Mn2+ to promote DMS-adduct-induced misincorporation.
- PCR amplify cDNA libraries.
- Sequencing & Analysis: Sequence on an Illumina platform. Map mutations and calculate DMS reactivity profiles using the
dms-tools2orShapeMapper2pipelines.
Part B: In Vitro SHAPE-MaP Validation
- RNA Preparation: In vitro transcribe the RNA region of interest using T7 RNA polymerase, followed by purification via gel electrophoresis.
- SHAPE Probing: Fold 3 pmol of RNA in buffer. Treat one sample with 1M NMIA (in DMSO) and a control with DMSO only for 45 minutes at 37°C.
- Library Preparation & Analysis: Use the SHAPE-MaP protocol (Siegfried et al., 2014) for reverse transcription and sequencing. Compute normalized SHAPE reactivities.
Protocol 2: Functional Validation via Mutagenesis and Reporter Assay
Purpose: To test the functional necessity of a validated RNA structure.
- Plasmid Design: Clone the wild-type RNA sequence of interest into the 5'UTR of a luciferase reporter plasmid (e.g., psiCHECK-2).
- Mutagenesis: Introduce point mutations designed to disrupt key base pairs (e.g., stem region) using site-directed mutagenesis PCR. Create a compensatory mutant that restores pairing.
- Cell Transfection: Transfect HEK293T cells in triplicate with wild-type, disruptive, and compensatory mutant plasmids.
- Measurement: Harvest cells 48h post-transfection. Measure Firefly and Renilla luciferase activities using a dual-luciferase assay kit.
- Analysis: Normalize the reporter signal (Renilla/Firefly). Compare relative expression levels across constructs.
Visualization
Title: Cross-Platform RNA Structure Validation Workflow
Title: Viral Frameshift Element Mechanism & Inhibition
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for DMS-MaPseq and Validation Studies
| Reagent/Material | Function in Validation Workflow | Example Product/Catalog |
|---|---|---|
| Dimethyl Sulfate (DMS) | Small chemical probe that methylates accessible adenine (N1) and cytosine (N3) in vivo. Reactivity inversely correlates with base pairing. | Sigma-Aldrich, 99% purity |
| TGIRT-III Enzyme | Reverse transcriptase used in MaP protocols. Efficiently reads through DMS adducts, incorporating mismatches during cDNA synthesis. | InGex, LLC |
| NMIA (1-methyl-7-nitroisatoic anhydride) | SHAPE reagent for in vitro validation. Modifies flexible nucleotides at the 2'-OH, confirming unpaired regions. | Santa Cruz Biotechnology |
| Pyridostatin (PDS) | High-affinity G-quadruplex stabilizing ligand. Used in functional assays to test G4-dependent phenotypes. | Tocris Bioscience |
| Dual-Luciferase Reporter Vector | Plasmid system for cloning RNA sequences of interest into a UTR to measure translational output via luciferase activity. | Promega, psiCHECK-2 |
| Ribonuclease Inhibitor | Critical for maintaining RNA integrity during all stages of sample preparation and library construction. | New England Biolabs, Murine RNase Inhibitor |
| Next-Generation Sequencing Kit | For preparation of barcoded cDNA libraries compatible with Illumina sequencing platforms. | Illumina, Stranded Total RNA Prep |
Application Notes and Protocols
Within the broader thesis on in vivo RNA structure profiling using DMS-MaPseq (Dimethyl Sulfate Mutational Profiling with Sequencing), selecting an appropriate experimental and computational pipeline is critical. The choice is governed by a trade-off between resolution (nucleotide-level accuracy), throughput (number of samples/conditions), and accessibility (cost, expertise, and infrastructure). This document provides a comparative assessment and detailed protocols for implementing different lab-scale approaches.
Data Presentation: Platform Comparison
Table 1: Comparative Analysis of DMS-MaPseq Implementation Platforms
| Platform / Approach | Resolution (Key Limitation) | Throughput (Samples/Week) | Accessibility (Cost & Expertise) | Ideal Use Case |
|---|---|---|---|---|
| High-Fidelity Long-Read Sequencing (e.g., PacBio HiFi, Oxford Nanopore) | High. Enables full-length RNA structure analysis, identification of alternative conformations, and direct detection of modifications. | Low (10-20). Longer library prep and sequencing run times. | Low. Very high sequencing cost per sample; requires specialized bioinformatics for raw signal analysis. | Profiling long, non-coding RNAs or studying isoform-specific structural dynamics. |
| Short-Read Illumina Sequencing (Standard) | Medium-High. Excellent single-nucleotide resolution for regions <~300 nt. Challenged by repetitive sequences and determining long-range interactions. | High (50-100+). Mature, parallelized library prep and ultra-high-throughput sequencing. | Medium. Moderate sequencing cost; requires standard NGS and DMS-MaP specific computational pipelines. | Genome-wide profiling or condition-compare studies of structured regions under ~300 nt. |
| Benchtop Sequencer (e.g., Illumina iSeq, MiniON) | Medium. Sufficient for targeted applications. Lower raw data quality may reduce signal-to-noise. | Medium-Low (10-30). Faster turnaround but lower multiplexing capacity. | High. Lower capital investment; suitable for individual labs. Bioinformatic expertise still required. | Validating hits from larger studies or focused investigation on a specific RNA target. |
| Capillary Electrophoresis (Legacy) | Low. Very limited throughput and quantitative capability. Primarily for single RNA probes. | Very Low (1-5). | Medium. Low instrumentation cost but very labor-intensive and low-resolution. | Not recommended for MaPseq. Historical context only. |
Experimental Protocols
Protocol 1: Standard In Vivo DMS Treatment and RNA Harvesting for Cultured Cells Objective: To chemically probe the RNA structurome in living cells.
- Grow cells in appropriate medium to ~80% confluence.
- DMS Treatment: Prepare fresh DMS buffer (200 mM HEPES-KOH pH 8.0, 150 mM KCl). For treatment, dilute DMS to a final concentration of 0.5% (v/v) in pre-warmed culture medium. Aspirate old medium, add DMS-medium, and incubate for 5 min at 37°C. Note: DMS is highly toxic; use a chemical fume hood.
- Quenching: Quickly aspirate DMS-medium and immediately add ice-cold quenching buffer (30% β-mercaptoethanol in PBS). Incubate on ice for 2 min.
- RNA Extraction: Wash cells twice with PBS. Lyse cells using TRIzol or a similar guanidinium-based reagent. Isolate total RNA according to the manufacturer's protocol. Perform DNase I treatment.
- Quality Control: Assess RNA integrity (RIN > 8.5) using an Agilent Bioanalyzer.
Protocol 2: DMS-MaPseq Library Preparation for Illumina Platforms Objective: To convert DMS-modified RNA into a sequencing library for mutational profiling.
- Reverse Transcription (MaP): For 1 µg of total RNA, set up a 20 µL reaction using a SuperScript II or similar RT enzyme with thermostable reverse transcriptase capabilities. Use random primers or gene-specific primers. Use a thermal profile that includes extended incubation at 42°C for 45 min, then heat-shock at 55°C for 10 min, and finally 65°C for 20 min. This promotes read-through and misincorporation at DMS-adducted bases.
- cDNA Purification: Purify cDNA using SPRI beads (e.g., AMPure XP).
- Second Strand Synthesis & Adapter Ligation: Perform standard second-strand synthesis. Use a kits such as NEBNext Ultra II FS DNA Library Prep for Illumina for end-repair, dA-tailing, and adapter ligation.
- PCR Enrichment: Amplify the library with 8-12 cycles of PCR using indexed primers compatible with your sequencer.
- Library QC: Validate library size distribution (peak ~300-500 bp) using a Bioanalyzer and quantify by qPCR.
Protocol 3: Computational Processing of DMS-MaPseq Data Objective: To calculate DMS reactivity per nucleotide from raw sequencing data.
- Alignment: Trim adapters (using Cutadapt) and align reads to the reference genome/transcriptome using a splice-aware aligner (e.g., STAR or HISAT2). For long-read data, use minimap2.
- Mutation Calling: Use a dedicated DMS-MaPseq analysis pipeline (e.g.,
dms-tools2,ShapeMapper2). The pipeline will:- Identify mismatches in aligned reads relative to the reference.
- Filter mutations based on quality scores and sequence context.
- Calculate a mutation rate (M) for each nucleotide: M = (number of mismatches at position i) / (total read coverage at position i).
- Reactivity Calculation: Subtract the background mutation rate (from a no-DMS control sample) and normalize the per-nucleotide DMS reactivity, often to a scale where the 92nd percentile of reactivities is set to 1.0.
Mandatory Visualization
Diagram 1: DMS-MaPseq Experimental Workflow
Diagram 2: Decision Logic for Platform Selection
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for DMS-MaPseq
| Item | Function in DMS-MaPseq |
|---|---|
| Dimethyl Sulfate (DMS) | Small, cell-permeable chemical probe that methylates unpaired adenine (N1) and cytosine (N3). Reactivity indicates nucleotide accessibility. |
| Thermostable Group II Intron Reverse Transcriptase (e.g., SuperScript II) | Critical for the Mutational Profiling (MaP) step. Promotes misincorporation at DMS-modified bases during cDNA synthesis, encoding the structural signal. |
| SPRI Magnetic Beads (e.g., AMPure XP) | For size-selective purification and cleanup of cDNA and sequencing libraries, removing enzymes, primers, and adapter dimers. |
| Ultra II FS DNA Library Prep Kit (NEB) | A robust, widely-used kit for preparing high-quality, Illumina-compatible sequencing libraries from double-stranded cDNA. |
| DMS-MaPseq Analysis Software (ShapeMapper2) | The core computational tool that processes aligned sequencing data, calls mutations, and outputs per-nucleotide DMS reactivity profiles. |
| RNA Structure Modeling Package (e.g., RNAfold, ΔΔG) | Uses DMS reactivity constraints to predict the minimum free energy secondary structure or calculate folding energy changes between conditions. |
Conclusion
DMS-MaPseq has fundamentally transformed our ability to interrogate RNA structure within the native, complex environment of the cell, moving beyond the limitations of in vitro analyses. By mastering its foundational principles, meticulous protocol, and optimization strategies outlined here, researchers can reliably generate high-resolution structural maps of diverse RNAs. This capability is pivotal for deciphering the regulatory codes embedded in RNA architecture, from understanding viral replication mechanisms to identifying novel, druggable structural pockets in non-coding RNAs. The future of DMS-MaPseq lies in its integration with single-cell technologies, spatial transcriptomics, and dynamic time-course studies, promising to unveil the four-dimensional landscape of RNA structure in health and disease. This will accelerate the development of RNA-targeted small molecules and antisense oligonucleotides, solidifying RNA structure as a cornerstone of next-generation therapeutic development.