Amplicon vs. Hybrid-Capture NGS: A Complete Guide for Genomics Researchers in 2024
This comprehensive guide provides researchers, scientists, and drug development professionals with a detailed comparison of two dominant targeted next-generation sequencing (NGS) approaches: amplicon-based and hybridization capture methods.
Amplicon vs. Hybrid-Capture NGS: A Complete Guide for Genomics Researchers in 2024
Abstract
This comprehensive guide provides researchers, scientists, and drug development professionals with a detailed comparison of two dominant targeted next-generation sequencing (NGS) approaches: amplicon-based and hybridization capture methods. The article explores their foundational principles, guides methodology selection based on specific applications such as oncology and microbiology, offers practical troubleshooting and optimization strategies, and presents a direct comparative analysis of performance metrics, cost, and scalability. The final synthesis provides actionable insights for choosing the optimal method to advance biomedical research and clinical diagnostics.
Understanding the Core Principles: Amplicon and Hybrid-Capture NGS Explained
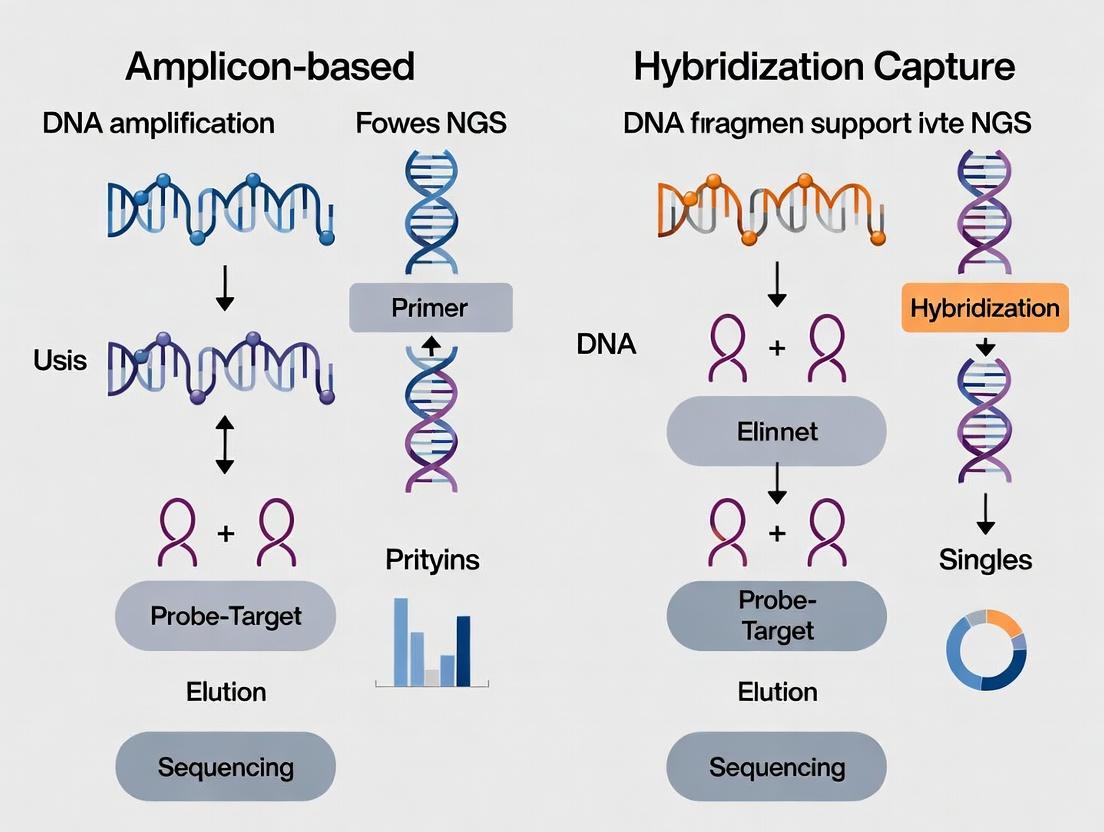
Targeted Next-Generation Sequencing (NGS) is a cornerstone of modern genomic analysis, enabling focused, cost-effective, and high-depth sequencing of specific genomic regions of interest. Unlike whole-genome sequencing, targeted NGS requires an upfront enrichment step to isolate these regions from the complex background of the entire genome. This article, framed within a thesis comparing amplicon-based and hybridization capture methods, details the application and protocols for these two dominant enrichment strategies.
The Imperative for Enrichment
Enrichment is essential for applications where deep sequencing of specific gene panels (e.g., cancer hotspots, hereditary disease genes, pharmacogenomic loci) is required. It improves sensitivity for detecting low-frequency variants, reduces per-sample costs, and simplifies data analysis. The choice between amplicon-based and hybridization capture methods is critical and depends on factors such as target size, uniformity of coverage, and sample type.
Quantitative Comparison of Enrichment Methods
Table 1: Core Characteristics of Targeted NGS Enrichment Methods
| Feature | Amplicon-Based Enrichment | Hybridization Capture |
|---|---|---|
| Typical Workflow Time | ~4-6 hours (library prep + enrichment) | ~24-48 hours (library prep + hybridization) |
| Optimal Target Size | < 1 Mb (ideal for hotspots/panels) | Any size, from panels to whole exomes (≥ 1 Mb) |
| DNA Input Requirement | Low (1-10 ng) | Moderate to High (50-200 ng) |
| Uniformity of Coverage | Lower (primer-specific bias) | Higher (more even coverage) |
| Variant Detection | Excellent for SNVs/Indels in well-amplified regions. Poor for CNVs. | Robust for SNVs, Indels, CNVs, and some fusions. |
| Multiplexing Capacity | Very High (sample-specific barcodes) | High (requires unique dual indexes) |
| Cost per Sample | Lower | Higher (reagents, hands-on time) |
| FFPE Sample Performance | Good with short amplicons (< 150 bp) | Good with optimized protocols and probe design |
Detailed Experimental Protocols
Protocol 1: Amplicon-Based Enrichment (Multiplex PCR Approach)
This protocol uses a single-tube, multiplex PCR reaction to amplify all targeted regions simultaneously.
- DNA Quantification and Dilution: Quantify genomic DNA using a fluorometric assay (e.g., Qubit). Dilute to a working concentration of 1 ng/µL in low TE buffer.
- Multiplex PCR Amplification:
- Prepare a 25 µL reaction containing:
- 1x Polymerase Master Mix (with high-fidelity, hot-start polymerase)
- 1x Primer Pool (comprising all target-specific, tailed primers)
- 10 ng (10 µL) of diluted genomic DNA.
- Cycling conditions:
- 98°C for 30s (initial denaturation)
- 98°C for 10s, 60°C for 30s, 72°C for 30s (35 cycles)
- 72°C for 5 min (final extension)
- Hold at 4°C.
- Prepare a 25 µL reaction containing:
- PCR Clean-up: Purify the amplicon pool using a magnetic bead-based clean-up system (0.8x bead-to-sample ratio) to remove primers and salts. Elute in 20 µL of nuclease-free water.
- Indexing PCR (Add Illumina Adapters):
- Prepare a 50 µL reaction containing:
- 1x Polymerase Master Mix
- 1x Indexing Primer Mix (containing i5 and i7 index sequences and full adapter)
- 10 µL of purified amplicon product.
- Cycling conditions (8-10 cycles): 98°C for 30s, followed by cycles of 98°C for 10s and 65°C for 30s, 72°C for 30s, with a final extension at 72°C for 5 min.
- Prepare a 50 µL reaction containing:
- Final Library Clean-up and Validation: Perform a double-sided magnetic bead clean-up (0.8x ratio). Quantify the final library using qPCR for accurate molarity. Check fragment size distribution on a Bioanalyzer or TapeStation (expect a single peak corresponding to amplicon size + adapters).
Protocol 2: Hybridization Capture (In-Solution Method)
This protocol involves fragmenting genomic DNA, preparing an adapter-ligated library, and capturing targets using biotinylated probes.
- DNA Shearing and Library Preparation:
- Fragment 100 ng of genomic DNA via acoustic shearing to a mean size of 200-250 bp.
- Repair DNA ends, add 'A' bases to 3' ends, and ligate Illumina-compatible stubby adapters using a commercial library prep kit.
- Clean up reactions using magnetic beads after each step.
- Library Amplification and QC:
- Amplify the adapter-ligated library with 4-6 PCR cycles using primers containing unique dual index (UDI) sequences.
- Purify with magnetic beads. Quantify by fluorometry and analyze size distribution (expected broad peak ~300-350 bp).
- Hybridization and Capture:
- Combine 200-500 ng of prepped library with blocking oligonucleotides (to suppress adapter-adapter hybridization) and a custom biotinylated probe library in hybridization buffer.
- Denature at 95°C for 5-10 minutes and incubate at 58-65°C for 16-24 hours to allow probes to hybridize to target sequences.
- Streptavidin Bead Capture and Washing:
- Add streptavidin-coated magnetic beads to the hybridization mix and incubate at room temperature for 30-45 minutes.
- Wash beads with a series of stringency buffers (SSC-based) at defined temperatures to remove non-specifically bound DNA.
- Perform all washes with the beads immobilized on a magnet.
- Post-Capture Amplification and Final QC:
- Elute captured DNA from beads in a low-salt buffer or water.
- Amplify the eluate with 10-14 PCR cycles using universal primers.
- Perform a final bead clean-up. Quantify via qPCR and assess size profile and enrichment success (e.g., via qPCR for a target vs. non-target locus).
Visualizing the Enrichment Workflows
Amplicon-Based NGS Library Preparation
Hybridization Capture NGS Library Preparation
Decision Guide for Enrichment Method Selection
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Targeted NGS Enrichment
| Item | Function in Protocol | Key Consideration |
|---|---|---|
| High-Fidelity, Hot-Start DNA Polymerase | Amplifies target regions with minimal errors and prevents non-specific amplification during reaction setup. | Essential for both amplicon and capture (pre/post-capture PCR). |
| Multiplex PCR Primer Pool | Contains all forward/reverse primers for targeted regions, often with universal adapter tails. | Design is critical for amplicon method; impacts uniformity and specificity. |
| Biotinylated RNA or DNA Probe Library | Sequence-specific baits that hybridize to and pull down targets from a fragmented library. | Probe design and tiling density impact capture efficiency for hybridization method. |
| Streptavidin-Coated Magnetic Beads | Bind biotin on captured probe-target complexes for magnetic separation and washing. | Bead size and binding capacity affect yield and background. |
| Magnetic Bead Clean-up Kits (SPRI) | Size-selectively bind and purify DNA (e.g., PCR products, fragmented libraries). | Workhorse for most NGS library prep steps; ratio determines size cut-off. |
| Dual-Indexed Adapter Kits | Provide unique barcode combinations for each sample, enabling multiplexed sequencing. | Necessary for both methods; UDIs are now standard to reduce index hopping. |
| Hybridization Buffer & Blockers | Creates optimal salt and temperature conditions for specific probe binding. Suppresses adapter dimer capture. | Critical for capture specificity and high on-target rates. |
| Library Quantification Kit (qPCR-based) | Accurately measures the concentration of adapter-ligated, amplifiable library fragments. | Essential for pooling libraries at equimolar ratios for sequencing. |
Within the broader comparison of amplicon-based versus hybridization capture Next-Generation Sequencing (NGS) methods, PCR-driven target amplification remains a cornerstone for specific, sensitive, and cost-effective genomic interrogation. This Application Note details the protocols, applications, and quantitative performance metrics of amplicon sequencing, providing a framework for researchers to select the optimal method for their needs in diagnostics, microbial ecology, and targeted mutation detection in drug development.
Quantitative Comparison of NGS Target Enrichment Methods
Table 1: Key Performance Metrics of Amplicon vs. Hybridization Capture Sequencing
| Parameter | PCR-Driven Amplicon Sequencing | Hybridization Capture |
|---|---|---|
| Input DNA Requirement | Low (1-10 ng) | High (50-200 ng) |
| Typical Hands-on Time | Low (< 1 day) | High (1-3 days) |
| Time to Library Completion | Fast (5-8 hours) | Slow (24-72 hours) |
| Multiplexing Capacity (Samples/Run) | Very High (hundreds to thousands) | Moderate (dozens to hundreds) |
| On-Target Rate | Very High (>90%) | Moderate-High (40-80%) |
| Uniformity of Coverage | Lower (primer-dependent bias) | Higher |
| Ability to Detect CNVs | Limited (relative quantitation only) | Excellent (absolute quantitation) |
| Best for Short Targets | Excellent (up to ~500 bp amplicons) | Excellent for long/continuous regions |
| Best for Large/Gene Panels | Poor (high primer cost, complexity) | Excellent |
| Cost per Sample (excl. seq) | Very Low | High |
| Variant Allele Frequency Sensitivity | High (can detect <1% with UMIs) | Moderate (typically >1-5%) |
| Tolerance to Degraded DNA | High (short amplicons possible) | Low |
Detailed Application Notes
Primary Applications
- Microbiome 16S/ITS/18S rRNA Gene Sequencing: Profiling microbial community composition and diversity.
- Viral Genome Surveillance & Typing: Tracking pathogen evolution (e.g., SARS-CoV-2, HIV).
- Somatic Variant Detection in Cancer: Targeting known hotspots (e.g., KRAS, EGFR, BRAF) in liquid biopsy and tissue samples.
- Germline Genetic Screening: For inherited disorders (e.g., BRCA1/2, cardiomyopathy panels).
- SARS-CoV-2 Variant Detection: Rapid sequencing of the viral spike gene and other key regions.
Advantages in Context
- Sensitivity: Superior for low-input or low-abundance targets due to PCR's exponential amplification.
- Speed: Rapid turnaround from sample to sequence-ready library.
- Cost-Effectiveness: Lower reagent costs and minimal infrastructure requirements make it accessible.
- Specificity: High on-target rates minimize sequencing waste on non-relevant genomic regions.
Limitations in Context
- PCR Bias & Errors: Polymerase errors and primer bias can affect variant calling and quantitative accuracy. This is mitigated by using high-fidelity polymerases and Unique Molecular Identifiers (UMIs).
- Amplification of Contaminants: High sensitivity increases risk from sample cross-contamination or environmental DNA.
- Limited Target Multiplexing: Primer design complexity limits the number of genomic regions that can be efficiently amplified in a single reaction compared to capture.
Core Experimental Protocols
Protocol A: Two-Step PCR Amplicon Library Preparation (with UMIs)
This protocol is optimized for high-sensitivity variant detection, incorporating UMIs for error correction.
I. Materials & Equipment
- Template DNA: 1-10 ng (human gDNA) or 1-50 ng (microbial gDNA).
- High-Fidelity DNA Polymerase: e.g., Q5 Hot Start (NEB) or KAPA HiFi HotStart.
- Target-Specific Primer Pool: First-round primers with gene-specific sequences.
- Overhang Adapter Primers: Second-round primers containing partial Illumina adapter sequences (e.g., i5/i7).
- Unique Molecular Identifier (UMI) Adapters: Combinatorial dual-indexed adapters with random molecular barcodes.
- Magnetic Bead-Based Cleanup System: e.g., AMPure XP beads.
- qPCR System or Fluorometer: For library quantification (e.g., Qubit, Bioanalyzer/TapeStation).
- Thermal Cycler.
II. Procedure
Step 1: Primary Target Amplification
- Prepare the primary PCR mix on ice:
- DNA Template: 1-10 ng (in 5 µL)
- 2X High-Fidelity Master Mix: 25 µL
- Target-Specific Primer Pool (10 µM each): 2.5 µL
- Nuclease-Free Water: to 50 µL final volume.
- Thermal Cycle:
- 98°C for 30 sec (initial denaturation)
- 35 Cycles: 98°C for 10 sec, 60-65°C (primer-specific) for 30 sec, 72°C for 30 sec/kb
- 72°C for 2 min (final extension).
Step 2: Primary PCR Cleanup
- Add 1.0X volume of AMPure XP beads (50 µL) to the PCR product (50 µL). Mix thoroughly.
- Incubate 5 min at RT. Place on magnet for 2 min. Discard supernatant.
- Wash beads twice with 80% ethanol.
- Air dry for 2 min. Elute in 25 µL 10 mM Tris-HCl (pH 8.5).
Step 3: Secondary Indexing PCR (Adapter Attachment)
- Prepare the secondary PCR mix:
- Purified Primary PCR Product: 5 µL
- 2X High-Fidelity Master Mix: 25 µL
- UMI Adapter Primers (i5/i7, 10 µM each): 2.5 µL each
- Nuclease-Free Water: to 50 µL.
- Thermal Cycle (Use minimal cycles):
- 98°C for 30 sec
- 8-12 Cycles: 98°C for 10 sec, 65°C for 30 sec, 72°C for 30 sec
- 72°C for 2 min.
Step 4: Final Library Cleanup & Quantification
- Perform a double-sided size selection using AMPure XP beads (e.g., 0.6X followed by 1.2X ratios) to remove primer dimers and large artifacts.
- Quantify the final library using a fluorometric assay (Qubit dsDNA HS Assay).
- Assess library size distribution and quality via capillary electrophoresis (Bioanalyzer HS DNA or TapeStation D1000).
III. Data Analysis Note Sequencing data must be processed with a UMI-aware pipeline involving: (1) Demultiplexing, (2) UMI extraction and consensus read generation (deduplication), (3) alignment, and (4) variant calling to achieve maximal sensitivity and specificity.
Protocol B: Single-Step 16S rRNA Gene Metagenomic Sequencing (V3-V4 Region)
I. Materials & Equipment
- As in Protocol A, with the following specifics:
- Primers: 341F (5’-CCTAYGGGRBGCASCAG-3’) and 806R (5’-GGACTACNNGGGTATCTAAT-3’) with overhang adapters.
- Quantification: Use a qPCR-based kit specific for Illumina libraries (e.g., KAPA Library Quant) for accurate cluster loading.
II. Procedure
- Amplify target in a single PCR reaction using primers that already contain full Illumina adapter sequences.
- Thermocycling conditions are identical to Step 1 of Protocol A, but cycle number may be adjusted (25-30 cycles) based on template concentration.
- Cleanup with AMPure XP beads (0.9X ratio).
- Quantify via qPCR for pooling and sequencing.
Diagrams and Workflows
Title: Amplicon Sequencing Library Prep Workflow
Title: Amplicon vs Capture Selection Guide
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for PCR-Driven Amplicon Sequencing
| Item Category | Example Product | Critical Function & Notes |
|---|---|---|
| High-Fidelity Polymerase | Q5 Hot Start (NEB), KAPA HiFi HotStart | Minimizes PCR errors; essential for accurate variant calling. High processivity for GC-rich targets. |
| Magnetic Purification Beads | AMPure XP (Beckman Coulter), SPRIselect | Size-selective cleanup of PCR products; removes primers, dimers, and salts. Ratios determine size cut-off. |
| Unique Molecular Index (UMI) Adapters | IDT for Illumina UMI Adapters, Twist UMI Adapters | Adds random molecular barcodes to each original molecule for error correction and accurate quantification. |
| Library Quantification Kit | KAPA Library Quant (Roche), Qubit dsDNA HS (Thermo) | Accurate quantification is critical for optimal cluster density on the sequencer. qPCR-based kits measure amplifiable library. |
| Primer Design Software | Primer3, NCBI Primer-BLAST, Thermo Fisher AmpliSeq Designer | Designs specific, efficient primer pairs with balanced Tm and minimal off-target binding or primer-dimer formation. |
| Target-Specific Primer Pools | Illumina AmpliSeq, Qiagen QIAseq, Custom oligo pools | Pre-designed, validated primer sets for focused gene panels (e.g., cancer hotspots, pathogen detection). |
| NGS Platform | Illumina MiSeq, iSeq; Oxford Nanopore MinION | Short-read (Illumina) for high accuracy; long-read (Nanopore) for full-length amplicons (e.g., 16S). |
Application Notes
Within the comparative framework of amplicon-based vs. hybridization capture Next-Generation Sequencing (NGS) methods, probe-based hybrid-capture target enrichment is defined by its use of biotinylated oligonucleotide probes designed to hybridize to specific genomic regions of interest from a fragmented, adapter-ligated DNA library. Following hybridization, probe-target complexes are captured on streptavidin-coated magnetic beads, washed to remove non-specific fragments, and eluted to produce a sequencing-ready library. This method is central to large-scale genomic studies, including whole-exome sequencing (WES), comprehensive cancer gene panels, and complex population genetics, due to its high specificity, scalability, and superior uniformity over large, discontinuous genomic regions compared to amplicon approaches.
Quantitative Comparison of Key Performance Metrics
The following table summarizes core quantitative data differentiating hybrid-capture from amplicon-based NGS, derived from recent literature and manufacturer specifications.
Table 1: Performance Comparison of Hybrid-Capture vs. Amplicon-Based Target Enrichment
| Metric | Hybrid-Capture | Amplicon-Based | Notes |
|---|---|---|---|
| Input DNA Requirement | 50-200 ng (standard), ~10 ng (low-input) | 1-50 ng | Hybrid-capture generally requires more input. |
| Panel Design Flexibility | High; can target up to whole-exome (>50 Mb) | Moderate; optimal for < 1 Mb | Capture allows easy redesign by changing probe sets. |
| Off-Target Rate | <5-20% (depends on bait design & stringency) | <1-5% | Amplicon is highly specific but prone to primer-dimer artifacts. |
| Uniformity of Coverage | Moderate (fold-80 penalty ~2-4x) | Very High (fold-80 penalty ~1.5-2x) | Uniformity is a key strength of amplicon methods. |
| Variant Detection Sensitivity | >95% for SNVs/Indels at >100x | >99% for SNVs/Indels at >500x | Amplicon excels in ultra-deep, focused applications. |
| Tolerance to Input Quality | Moderate (FFPE-compatible with protocols) | High (works well with degraded FFPE DNA) | Both are compatible with FFPE; protocols differ. |
| Hands-on Time | High (2-3 days, complex workflow) | Low (1 day, streamlined PCR workflow) | Hybrid-capture is more labor-intensive. |
| Cost per Sample | High (reagents, probes) | Low (primers, PCR reagents) | Economical at scale for both; probe cost is upfront. |
Experimental Protocols
Protocol 1: Standard Hybrid-Capture Workflow for Whole-Exome Sequencing
This protocol is adapted from current manufacturer guidelines (e.g., Twist Bioscience, Roche NimbleGen, IDT xGen) and is suitable for 50-200 ng of high-quality genomic DNA.
Materials & Reagents:
- Fragmentation Enzyme/Instrument: Covaris sonicator or enzymatic fragmentation mix.
- Library Prep Kit: Illumina TruSeq DNA Nano or equivalent.
- Hybridization Buffer: Proprietary buffer containing SSC, SDS, formamide, and blocking agents.
- Biotinylated Probe Library: e.g., Twist Human Core Exome or custom-designed panel.
- Capture Beads: Streptavidin-coated magnetic beads (e.g., Dynabeads MyOne Streptavidin T1).
- Wash Buffers: Stringent wash buffer (e.g., SSC + SDS), non-stringent wash buffer.
- Elution Buffer: Low-salt buffer or nuclease-free water.
- Post-Capture PCR Primers & Master Mix: Indexing primers and high-fidelity polymerase.
- QC Instruments: Bioanalyzer/TapeStation, Qubit fluorometer, qPCR for library quantification.
Procedure:
- Library Preparation:
- Fragment genomic DNA to a target size of 150-350 bp.
- Perform end-repair, A-tailing, and ligation of dual-indexed sequencing adapters.
- Clean up reactions using magnetic SPRI beads and quantify library yield.
Hybridization:
- Combine 200-500 ng of purified library with hybridization buffer, blocking oligonucleotides (to suppress adapter-adapter binding), and the biotinylated probe pool.
- Denature at 95°C for 5-10 minutes and incubate at 58-65°C for 16-24 hours to allow probe-target hybridization.
Capture & Wash:
- Pre-wash streptavidin magnetic beads. Add beads to the hybridization reaction and incubate at room temperature to bind biotinylated probe-DNA complexes.
- Capture beads on a magnetic stand and discard supernatant.
- Perform a series of washes: 2x with non-stringent buffer at room temperature, followed by 2-3x with pre-heated (65°C) stringent buffer. Maintain bead pellet during washes.
Elution & Amplification:
- Elute captured DNA from beads in a low-salt buffer or water after heating to 95°C.
- Amplify the eluted library using a limited-cycle (10-14 cycles) PCR with indexing primers.
- Clean up the final library with SPRI beads.
Quality Control & Sequencing:
- Assess library fragment size distribution (Bioanalyzer).
- Quantify library concentration via qPCR for accurate molarity.
- Pool libraries and sequence on the appropriate NGS platform (e.g., Illumina NovaSeq).
Protocol 2: Low-Input Hybrid-Capture for FFPE-Derived DNA
This variant protocol is optimized for challenging samples, a key application where hybrid-capture competes with amplicon-based methods.
Key Modifications:
- Use a library preparation kit specifically validated for FFPE DNA (e.g., Illumina TruSeq DNA Exome FFPE).
- Pre-Capture PCR: After adapter ligation, perform 4-6 cycles of PCR to amplify the limited library material before hybridization.
- Increase hybridization time to 24-48 hours to improve capture efficiency from lower-complexity libraries.
- Increase the amount of capture probes relative to library input to drive hybridization kinetics.
- Increase post-capture PCR cycles (14-18 cycles) as needed, but monitor for over-amplification artifacts.
Visualizations
Diagram Title: Hybrid-Capture Target Enrichment Workflow
Diagram Title: Decision Logic: Hybrid-Capture vs. Amplicon Selection
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Hybrid-Capture Sequencing
| Reagent/Material | Supplier Examples | Function & Importance |
|---|---|---|
| Biotinylated Probe Panels | Twist Bioscience, IDT (xGen), Roche NimbleGen, Agilent SureSelect | Core enrichment reagent. Synthetic oligonucleotides complementary to targets; biotin enables bead capture. Design dictates panel performance. |
| Streptavidin Magnetic Beads | Thermo Fisher (Dynabeads), Promega (MagneSphere) | Capture matrix. High-binding capacity streptavidin coats magnetic beads to isolate probe-bound targets during washes. |
| Hybridization Buffer & Blockers | Included with probe panels or separate kits (e.g., IDT Hybridization Buffer) | Creates optimal hybridization environment. Contains salts, detergent, and agents to block repetitive sequences and adapter-adapter interactions. |
| Library Prep Kit for FFPE | Illumina (TruSeq DNA Exome FFPE), KAPA (HyperPlus), NuGen | Adapted for degraded DNA. Includes enzymes optimized for damaged, cross-linked DNA typical of FFPE samples. |
| SPRI (Solid Phase Reversible Immobilization) Beads | Beckman Coulter (AMPure), Thermo Fisher (ProNex) | Universal cleanup. Magnetic beads for size-selective purification of DNA fragments after each enzymatic step. |
| High-Fidelity PCR Master Mix | NEB (Q5), KAPA (HiFi), Thermo Fisher (Platinum SuperFi II) | Post-capture amplification. Minimizes PCR errors and bias during the final library amplification step. |
| QC Kits (qPCR, Fragment Analyzer) | KAPA (Library Quantification), Agilent (Bioanalyzer/TapeStation) | Essential for success. Accurately quantifies functional library concentration and assesses size distribution pre-sequencing. |
Application Notes: Core Principles & Comparative Context
This document provides a technical overview and detailed protocols for two principal Next-Generation Sequencing (NGS) methods for targeted genomic analysis: Amplicon-based Sequencing and Hybridization Capture-based Sequencing. This work is framed within a broader thesis comparing these methodologies for applications in somatic variant detection, inherited germline analysis, and comprehensive genomic profiling in translational and drug development research. The amplicon approach uses PCR to directly enrich targeted regions, offering speed and simplicity for focused panels. The hybridization capture method uses biotinylated probes to pull down targets from sheared, adapter-ligated DNA, providing superior uniformity and flexibility for large panels or exome sequencing.
Table 1: Core Methodological & Performance Comparison
| Parameter | Amplicon-Based Sequencing | Hybridization Capture-Based Sequencing |
|---|---|---|
| Typical Input DNA | 10-250 ng (can be degraded, e.g., FFPE) | 50-200 ng (high-quality preferred; FFPE requires optimization) |
| Workflow Duration | ~1-1.5 days | ~2-3 days |
| Primary Enrichment Mechanism | Multiplex PCR | Solution-phase hybridization with biotinylated probes |
| Panel Size Flexibility | Low to Moderate (up to a few Mb); primer design constraints | High (from a few genes to whole exome/genome) |
| Uniformity of Coverage | Moderate to Low; prone to PCR bias and dropouts | High; more even coverage across targets |
| Variant Detection Sensitivity | High for low-frequency SNVs/Indels in focused panels | High, especially for CNVs and rearrangements in large regions |
| Ability to Detect CNVs & Rearrangements | Limited | Excellent |
| Off-Target Rate | Very Low | Moderate; manageable with probe design and bioinformatics |
| Multiplexing Capacity (Samples/Run) | High | High |
Table 2: Quantitative Performance Benchmarks (Typical Ranges)
| Performance Metric | Amplicon-Based | Hybridization Capture |
|---|---|---|
| On-Target Rate | >95% | 60-80% (exome: ~50-70%) |
| Fold-80 Base Penalty | 1.5 - 3.0 | 1.2 - 2.0 |
| Duplication Rate (100ng input) | 10-25% | 5-15% |
| Minimal Allele Frequency Detection | <1% (with UMIs) | <1% (with UMIs) |
| GC-Bias | Higher (PCR-dependent) | Lower, but present in extreme GC regions |
Detailed Experimental Protocols
Protocol 1: Amplicon-Based Target Enrichment (Multiplex PCR Workflow)
Objective: To generate sequencing-ready libraries from genomic DNA via multiplex PCR amplification of targeted regions.
Materials (Research Reagent Solutions):
- Input DNA: 10-50 ng of human genomic DNA (from blood, tissue, or FFPE).
- Multiplex PCR Master Mix: Contains a hot-start, high-fidelity DNA polymerase, dNTPs, and optimized buffer.
- Target-Specific Primer Pool: A pre-mixed, validated panel of hundreds to thousands of primer pairs targeting regions of interest.
- Library Amplification & Indexing Mix: Contains primers for amplifying the initial PCR product and attaching unique dual indices (UDIs) and full adapter sequences.
- Solid Phase Reversible Immobilization (SPRI) Beads: For size selection and purification of PCR products.
- Qubit dsDNA HS Assay Kit: For accurate library quantification.
- Bioanalyzer/Tapestation HS DNA Kit: For library fragment size distribution analysis.
Procedure:
- Library Preparation PCR:
- Set up a multiplex PCR reaction combining DNA, primer pool, and master mix.
- Cycle conditions: Initial denaturation (98°C, 30s); 15-25 cycles of [98°C for 10s, 60-65°C for 30s, 72°C for 30s]; final extension (72°C, 2 min).
- Purification:
- Clean up the PCR product using SPRI beads at a 1:1 ratio to remove primers and nonspecific products. Elute in buffer or water.
- Indexing PCR:
- Amplify the purified product with a universal primer mix containing sample-specific indices and full Illumina adapter sequences (8-10 cycles).
- Final Library Clean-up:
- Perform a double-sided SPRI bead clean-up (e.g., 0.6x followed by 1.0x ratio) to remove primer dimers and select the desired library size range (e.g., 200-500bp).
- QC and Normalization:
- Quantify the final library using the Qubit assay. Analyze size distribution on a Bioanalyzer. Pool libraries at equimolar concentrations based on QC data.
Protocol 2: Hybridization Capture-Based Target Enrichment
Objective: To generate sequencing libraries enriched for target regions via probe hybridization and streptavidin bead capture.
Materials (Research Reagent Solutions):
- Input DNA: 50-200 ng of sheared, adapter-ligated genomic DNA library.
- Biotinylated RNA or DNA Probe Library: A pool of probes complementary to the target regions (e.g., whole exome or custom panel).
- Hybridization Buffer: Contains blockers (e.g., Cot-1 DNA, adapter blockers) to suppress repetitive sequences and prevent probe-adapter binding.
- Streptavidin-Coated Magnetic Beads: For capturing the biotinylated probe-target hybrids.
- Wash Buffers (Stringent): Typically contain SSC and SDS at varying concentrations for post-capture washing.
- Capture Elution Buffer: A low-salt or alkaline buffer to release captured DNA from the beads.
- Post-Capture Amplification Master Mix: High-fidelity PCR mix for limited-cycle amplification of the captured library.
Procedure:
- Pre-Capture Library Preparation:
- Fragment genomic DNA (if not pre-sheared) and perform end-repair, A-tailing, and adapter ligation using a standard library prep kit. Amplify with 4-8 cycles of PCR.
- Hybridization:
- Mix the pre-captured library with the probe pool, hybridization buffer, and blockers. Denature at 95°C for 5-10 min, then incubate at 58-65°C for 16-24 hours to allow probes to hybridize to targets.
- Capture & Washing:
- Add streptavidin beads to the hybridization mix and incubate to bind biotinylated probe-target complexes.
- Wash beads sequentially with increasingly stringent buffers (e.g., high-SSC to low-SSC, with SDS) at the hybridization temperature to remove non-specifically bound DNA.
- Elution & Post-Capture Amplification:
- Elute the captured DNA from the beads using a low-salt or alkaline buffer. Neutralize the eluate.
- Amplify the eluted DNA with 12-16 cycles of PCR to generate sufficient material for sequencing.
- Final QC and Normalization:
- Purify the final product with SPRI beads. Quantify and assess size distribution (typically a broader profile than amplicon). Pool equimolarly.
Schematic Visualizations
Diagram 1: Amplicon vs Hybridization Capture Workflow
Title: Side-by-Side Workflow Comparison
Diagram 2: Enrichment Mechanism Logic
Title: Enrichment Core Mechanism Logic
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Targeted NGS Workflows
| Item | Function | Critical for Method |
|---|---|---|
| High-Fidelity DNA Polymerase | Minimizes PCR errors during target amplification and library indexing. Critical for both. | Amplicon & Capture |
| Unique Dual Index (UDI) Oligos | Enables high-level sample multiplexing and accurate demultiplexing, mitigating index hopping. | Amplicon & Capture |
| SPRI Magnetic Beads | For size selection and purification of nucleic acids. Used in multiple clean-up steps. | Amplicon & Capture |
| Validated Multiplex Primer Panel | Pre-optimized pool of primers for simultaneous amplification of all targets. Defines the panel. | Amplicon |
| Biotinylated Probe Library | Designed oligonucleotides (RNA or DNA) complementary to target regions for enrichment. Defines the panel. | Capture |
| Hybridization Blockers (e.g., Cot-1 DNA) | Suppress hybridization of probes to repetitive genomic elements, improving on-target efficiency. | Capture |
| Streptavidin-Coated Magnetic Beads | Bind biotin on probe-target complexes for physical separation from off-target fragments. | Capture |
| Stringent Wash Buffers | Remove loosely bound, non-specific DNA after capture, increasing specificity. | Capture |
| Fragmentation Enzyme/System | Generates dsDNA breaks to produce optimal fragment sizes for library construction. | Capture |
Historical Context and Evolution of Both Methods in Research
The development of Next-Generation Sequencing (NGS) revolutionized genomics, enabling the high-throughput analysis of DNA and RNA. Within this field, two principal target-enrichment strategies emerged: amplicon-based sequencing and hybridization capture. Amplicon sequencing, rooted in PCR techniques developed in the 1980s, leverages sequence-specific primers to amplify discrete genomic regions prior to sequencing. It gained prominence with 16S rRNA sequencing for microbial ecology (c. 2005) and for high-sensitivity variant detection in clinical oncology panels. Hybridization capture, conceptually derived from microarray technology (c. 1990s), utilizes biotinylated oligonucleotide probes to enrich for target regions from fragmented genomic libraries. Its first major NGS application was for exome sequencing (c. 2009), enabling efficient sequencing of all protein-coding genes. The evolution of both methods has been driven by the competing demands of uniformity, sensitivity, specificity, and cost-effectiveness in research and diagnostic applications.
Quantitative Comparison of Method Characteristics
Table 1: Historical Evolution and Technical Specifications
| Aspect | Amplicon-Based NGS | Hybridization Capture NGS |
|---|---|---|
| Conceptual Origin | PCR (1983), Sanger Sequencing | Southern Blot (1975), Microarray Tech (1990s) |
| First Major NGS Application | 16S rRNA profiling (~2005-2007) | Whole Exome Sequencing (~2009-2010) |
| Typical Input DNA (Human) | 1-100 ng (can use degraded FFPE) | 50-200 ng (requires higher integrity) |
| Primary Enrichment Mechanism | Multiplex PCR with target-specific primers | Solution or solid-phase hybridization to biotinylated probes |
| Key Performance Metric | Uniformity: Very high for targets. Specificity: High. | Uniformity: Moderate; requires normalization. Specificity: High with optimized wash. |
| Variant Detection Sensitivity | Excellent for low-frequency variants (down to ~1% allele frequency) | Excellent for common variants; can be ~5% for low-frequency due to capture noise |
| Off-Target Rate | Very low (<5%) | Moderate to High (10-60% depending on design) |
| Multiplexing Capacity | High (hundreds to thousands of amplicons) | Very High (entire exomes or custom panels >100 Mb) |
| Hands-on Time (Post-library) | Lower (single PCR step) | Higher (overnight hybridization, multiple wash steps) |
| Turnaround Time | ~1-1.5 days | ~2-3 days |
| Cost per Sample (2024 estimate, exome-scale) | $$ (Lower for panels < 1 Mb) | $$$ (Economical for large target regions) |
Table 2: Modern Application Domains (2020-2024)
| Application Domain | Preferred Method | Rationale |
|---|---|---|
| Liquid Biopsy & ctDNA Analysis | Amplicon-based (Digital PCR-based approaches also common) | Superior sensitivity for ultra-low frequency variants (<0.1% in some assays) |
| Infectious Disease Pathogen ID & Resistance | Amplicon-based (e.g., SARS-CoV-2 genome, microbial ITS) | Rapid, high sensitivity from low pathogen load, handles sequence divergence. |
| Hereditary Disease & Whole Exome Sequencing | Hybridization Capture | Comprehensive, unbiased coverage of all exons; scalable for large gene sets. |
| Cancer Hotspot Panels (Tissue) | Both (Amplicon for speed/sensitivity, Capture for uniformity/comprehensiveness) | Depends on required gene coverage and sample type (FFPE favors amplicon). |
| Metagenomic/ Microbiome Profiling | Amplicon (16S/ITS) for taxonomy; Capture for functional genes | Amplicon is standard for census; Capture allows tracking of specific genes across samples. |
| Structural Variant Detection | Hybridization Capture (with paired-end/long-read sequencing) | Better performance across large genomic intervals and repetitive regions. |
Detailed Experimental Protocols
Protocol 3.1: Amplicon-Based NGS for a Cancer Hotspot Panel (e.g., Illumina TruSeq Amplicon)
Objective: To detect single nucleotide variants (SNVs) and indels in 50-200 cancer-associated genes from FFPE-derived DNA. Principle: Two rounds of PCR. Round 1: Multiplex target-specific primers with overhang adapters amplify regions of interest. Round 2: Adds full Illumina sequencing adapters and sample-index barcodes.
Materials: See Scientist's Toolkit, Table 3.
Procedure:
- DNA Quantification & QC: Quantify input DNA (10-250 ng) using a fluorometric method (e.g., Qubit). Assess degradation via gel electrophoresis or genomic DNA screen tape.
- First-Stage PCR (Target Amplification):
- Prepare master mix containing: DNA, multiplex primer pool (containing target-specific sequences with 5' overhangs), and a high-fidelity, hot-start PCR polymerase.
- Thermocycler Program:
- 95°C for 2 min (initial denaturation)
- 15-25 cycles of: 95°C for 20 sec (denaturation), 60°C for 3 min (annealing/extension)
- 72°C for 5 min (final extension)
- Hold at 4°C.
- Post-PCR Cleanup: Purify the PCR amplicons using magnetic SPRI beads (e.g., AMPure XP) to remove primers, primer dimers, and salts. Elute in low TE buffer or nuclease-free water.
- Second-Stage PCR (Indexing):
- Prepare master mix containing: purified first-stage product, universal i5 and i7 index primers (containing full adapter sequences), and PCR polymerase.
- Thermocycler Program:
- 95°C for 2 min
- 8-12 cycles of: 95°C for 20 sec, 60°C for 30 sec, 72°C for 1 min
- 72°C for 5 min
- Hold at 4°C.
- Final Library Cleanup & QC: Perform a double-sided SPRI bead cleanup (e.g., 0.8X ratio to remove large fragments, then 1.2X ratio to recover the desired library). Quantify library yield via qPCR (e.g., Kapa Library Quant Kit) for accurate clustering concentration. Assess size distribution on a Bioanalyzer or TapeStation (expected peak: 200-350 bp).
- Sequencing: Normalize and pool libraries. Sequence on an Illumina platform (MiSeq, NextSeq, or NovaSeq) with paired-end reads (2x150 bp or 2x250 bp) to ensure coverage across amplicons.
Protocol 3.2: Hybridization Capture for Whole Exome Sequencing (e.g., IDT xGen Exome Research Panel)
Objective: To enrich for the ~35 Mb of human protein-coding exonic regions from genomic DNA for sequencing. Principle: Genomic DNA is fragmented, and sequencing libraries are prepared. Biotinylated DNA or RNA probes complementary to the exome are hybridized to this library. Probe-target complexes are captured on streptavidin-coated magnetic beads, washed stringently, and eluted for sequencing.
Materials: See Scientist's Toolkit, Table 4.
Procedure:
- Library Preparation (Pre-Capture):
- Fragment 50-200 ng of high-quality genomic DNA via acoustic shearing (Covaris) to a peak size of ~200-250 bp.
- Repair DNA ends, add 3' A-overhangs, and ligate platform-specific adapters containing unique dual indices (UDIs) using a kit (e.g., Illumina DNA Prep).
- Purify ligated product with SPRI beads. Perform a limited-cycle (4-8 cycles) PCR amplification to enrich for adapter-ligated fragments.
- Perform a final SPRI bead cleanup. Quantify library by fluorometry.
- Hybridization:
- For each sample or pool, combine 250-500 ng of pre-capture library, human Cot-1 DNA (to block repetitive sequences), and a universal blocker (to block adapter sequences).
- Dry down the mixture in a vacuum concentrator.
- Resuspend the pellet in hybridization buffer and add the biotinylated probe library.
- Denature at 95°C for 10 minutes and then incubate at 58-65°C for 16-24 hours in a thermocycler with heated lid to allow probes to hybridize to target DNA.
- Capture and Wash:
- Bead Preparation: Pre-wash streptavidin magnetic beads, resuspend in binding buffer, and keep at room temperature.
- Capture: Add the hybridization reaction to the prepared beads and incubate at 58-65°C for 45 minutes with gentle mixing.
- Stringent Washes: Pellet beads on a magnet and perform a series of washes at 55-65°C using pre-warmed wash buffers of increasing stringency (e.g., 2x SSC/0.1% SDS followed by 0.1x SSC/0.1% SDS) to remove non-specifically bound DNA.
- Final Washes: Perform two room-temperature washes in low-salt buffer (e.g., 0.1x SSC).
- Elution and Post-Capture Amplification:
- Elution: Resuspend beads in nuclease-free water. Denature at 95°C for 10 minutes to release the captured DNA. Immediately transfer the eluate (containing enriched library) to a fresh tube.
- Post-Capture PCR: Amplify the eluted library using primers complementary to the adapter sequences for 10-14 cycles.
- Cleanup: Purify the final library with SPRI beads (0.8X ratio).
- Final Library QC & Sequencing: Quantify by qPCR. Assess size and quality on a Bioanalyzer (peak ~250-300 bp). Normalize and pool libraries for sequencing on an Illumina platform (typically NovaSeq for exomes) with paired-end 2x150 bp reads.
Visualization: Workflows and Logical Relationships
Diagram Title: Comparative Workflow: Amplicon vs Hybridization Capture NGS
Diagram Title: Decision Logic for Amplicon vs Hybridization Capture Selection
The Scientist's Toolkit
Table 3: Key Reagents for Amplicon-Based NGS
| Item | Function | Example Product/Kit |
|---|---|---|
| High-Fidelity, Hot-Start DNA Polymerase | Catalyzes target amplification with low error rates and prevents non-specific priming at low temps. | KAPA HiFi HotStart, Q5 Hot Start, Platinum SuperFi II |
| Multiplex Primer Pool | Contains target-specific forward/reverse primer pairs, each with a universal 5' overhang sequence. | Illumina TruSeq Amplicon Assay, IDT xGen Pan-Cancer Panel |
| SPRI Magnetic Beads | Size-selective purification of DNA, removing primers, salts, and short fragments. | Beckman Coulter AMPure XP, KAPA Pure Beads |
| Library Quantification Kit (qPCR-based) | Accurately measures concentration of adapter-ligated fragments for optimal cluster density on sequencer. | Kapa Library Quant Kit (Illumina), qPCR-based Quantification |
| Dual-Indexed Adapter Primers | Contains full P5/P7 flow cell adapters and unique combinatorial barcodes for sample multiplexing. | Illumina CD Indexes, IDT for Illumina UD Indexes |
Table 4: Key Reagents for Hybridization Capture NGS
| Item | Function | Example Product/Kit |
|---|---|---|
| DNA Shearing Instrument | Fragments genomic DNA to a consistent size (~200-250 bp) for library construction. | Covaris S2/S220, Diagenode Bioruptor |
| Library Prep Kit | End-repair, A-tailing, adapter ligation, and pre-capture PCR in an optimized workflow. | Illumina DNA Prep, KAPA HyperPrep, NEBNext Ultra II |
| Biotinylated Probe Library | Pool of long (~80-120nt) DNA or RNA probes complementary to the target regions. | IDT xGen Exome Research Panel, Twist Human Core Exome, Roche SeqCap EZ |
| Human Cot-1 DNA | Blocks hybridization of probes to repetitive genomic sequences (e.g., Alu, LINE), reducing off-target capture. | Invitrogen Human Cot-1 DNA |
| Streptavidin Magnetic Beads | Binds biotin on probe-target complexes, enabling magnetic separation and washing. | Dynabeads MyOne Streptavidin C1, Streptavidin-coated Sera-Mag beads |
| Hybridization Buffer & Wash Solutions | Provides optimal ionic and chemical conditions for specific hybridization and removal of non-specifically bound DNA. | Component of commercial capture kits (IDT, Twist, Roche) |
Primary Advantages and Inherent Limitations of Each Approach
This application note, framed within a thesis comparing amplicon-based and hybridization capture Next-Generation Sequencing (NGS) methods, provides a detailed technical overview for researchers and drug development professionals. The objective is to delineate the primary advantages and inherent limitations of each approach, supported by current data, protocols, and practical resources.
Comparative Analysis of Methodologies
Amplicon-Based NGS (PCR-based Enrichment)
- Primary Advantages:
- High Sensitivity: Exceptionally effective for detecting low-frequency variants (e.g., <1% allele frequency) due to targeted amplification.
- High Efficiency with Low Input: Optimal for degraded or limited DNA samples (e.g., FFPE, liquid biopsy).
- Simpler Workflow: Fewer steps than capture, reducing hands-on time and potential handling errors.
- Lower Cost per Sample: For small, focused gene panels (< 50 genes), requires less sequencing depth to achieve high coverage.
- Rapid Turnaround: Shorter library preparation protocol enables faster results.
- Inherent Limitations:
- Limited Scalability: Difficult and costly to scale to large panels (> 500 genes) or whole exomes.
- PCR Artifacts: Risk of introducing errors during amplification, complicating variant calling.
- Amplification Bias: Uneven coverage due to primer-specific efficiency differences.
- Limited Discovery: Restricted to known, predefined targets covered by primer designs.
- Difficulty with High %GC or Repetitive Regions: Primer design challenges can lead to coverage dropouts.
Hybridization Capture NGS
- Primary Advantages:
- Unbiased, Uniform Coverage: Provides more even coverage across targets, reducing dropout regions.
- Highly Scalable: Efficiently scales from large panels to whole exomes and genomes.
- Discovery Capability: Can include non-coding regions, novel fusions, or structural variants depending on design.
- Minimal Amplification Bias: Uses fewer PCR cycles, reducing associated artifacts.
- Multiplexing Flexibility: Allows highly multiplexed sample pooling before capture.
- Inherent Limitations:
- Higher Input Requirements: Typically requires more DNA (e.g., 50-200 ng) of higher quality.
- Complex, Lengthy Workflow: More steps and longer hybridization times (often overnight).
- Higher Cost per Sample: Especially for smaller panels, due to reagent costs and need for greater sequencing depth.
- Off-Target Capture: Significant portion of sequencing reads may be off-target, reducing efficiency.
- Lower Sensitivity for Ultra-Low Frequency Variants: Requires higher sequencing depth to achieve similar sensitivity as amplicon for variants <1%.
Table 1: Performance Comparison of Amplicon vs. Hybridization Capture
| Metric | Amplicon-Based NGS | Hybridization Capture NGS | Notes |
|---|---|---|---|
| Typical DNA Input | 1-50 ng | 50-500 ng | FFPE can go lower for amplicon. |
| Variant Detection Sensitivity (AF) | ~0.1% - 1% | ~1% - 5% | Dependent on depth; amplicon excels at low AF. |
| Uniformity of Coverage | Low (≥90% bases at 0.2x mean coverage) | High (≥95% bases at 0.2x mean coverage) | Capture provides flatter coverage profiles. |
| On-Target Efficiency | Very High (≥90%) | Moderate-High (50%-80%) | Capture yields significant off-target reads. |
| Workflow Duration | 1-1.5 days | 2-3 days | Includes library prep and enrichment. |
| Cost per Sample (Ex-seq) | Low for panels (<$50) | High for panels ($100-$200+) | Scalable; cost reverses for whole exome. |
| Best For | Liquid biopsy, pathogen detection, hotspot/small panels, low-input FFPE. | Large panels, whole exome, discovery of novel variants, complex genomic regions. |
Table 2: Common Artifacts and Error Modes
| Approach | Common Artifacts | Mitigation Strategies |
|---|---|---|
| Amplicon-Based | PCR duplicates, chimeric reads, primer-induced errors, allele dropout. | Unique molecular identifiers (UMIs), optimized primer design, duplicate removal. |
| Hybridization Capture | Off-target reads, capture bias, non-specific hybridization, incomplete blocking. | Improved blocker design, optimized hybridization conditions, bait tiling. |
Experimental Protocols
Protocol: Amplicon-Based NGS for Liquid Biopsy cfDNA
Objective: Detect low-frequency somatic variants in circulating cell-free DNA (cfDNA). Key Materials: See "Scientist's Toolkit" (Section 6).
- cfDNA Extraction: Isolate cfDNA from 1-10 mL plasma using a silica-membrane column or magnetic bead-based kit. Elute in 20-50 µL TE buffer.
- Quantification & QC: Quantify using fluorometry (e.g., Qubit dsDNA HS Assay). Assess fragment size distribution via Bioanalyzer/TapeStation.
- Library Preparation with UMIs:
- End Repair & A-Tailing: Perform on 5-50 ng cfDNA using a master mix. Incubate at 20°C for 15 min, then 65°C for 15 min.
- Adapter Ligation: Ligate dual-indexed adapters containing UMI sequences. Use a 15:1 adapter-to-insert molar ratio. Incubate at 20°C for 15 min.
- Clean-up: Purify using magnetic beads (1.0x ratio).
- Targeted PCR Amplification:
- Perform two parallel PCRs:
- Pre-Capture PCR (5-8 cycles): Amplify the adapter-ligated library.
- Target Enrichment PCR (~25 cycles): Use a multiplexed primer panel targeting desired genomic regions.
- Clean up PCR products with magnetic beads (0.9x ratio).
- Perform two parallel PCRs:
- Library QC & Normalization: Quantify final library by qPCR. Assess size distribution.
- Sequencing: Pool normalized libraries. Sequence on an Illumina platform with paired-end 2x150 bp reads, aiming for a minimum depth of 10,000x per target.
Protocol: Hybridization Capture for Whole Exome Sequencing (WES)
Objective: Enrich and sequence the complete exome from high-quality genomic DNA. Key Materials: See "Scientist's Toolkit" (Section 6).
- DNA Shearing & QC: Fragment 100-200 ng gDNA to ~200 bp peak size using a focused-ultrasonicator. Verify size profile.
- Library Preparation:
- Perform end repair, A-tailing, and adapter ligation (as in 4.1.3) using a non-UMI kit optimized for capture.
- Perform a low-cycle (4-6 cycles) pre-capture PCR to amplify the ligated library. Clean up.
- Hybridization:
- Combine 250-500 ng of prepped library with blocking reagents (e.g., Cot-1 DNA, adapter blockers) and a whole exome biotinylated probe set in hybridization buffer.
- Denature at 95°C for 5-10 min, then incubate at 65°C for 16-24 hours in a thermal cycler with heated lid.
- Capture & Wash:
- Bead Binding: Add streptavidin-coated magnetic beads to the hybridization mix. Incubate at 65°C for 45 min with agitation.
- Stringency Washes: Perform a series of washes at 65°C (with SDS-containing buffer) and at room temperature to remove non-specifically bound DNA.
- Elution: Elute captured DNA from beads in a low-salt buffer at 95°C for 10 min.
- Post-Capture PCR:
- Amplify the captured library for 10-14 cycles to generate sufficient material for sequencing. Clean up.
- Final QC & Sequencing: Quantify by qPCR and analyze size distribution. Sequence on an Illumina platform (2x100 or 2x150 bp), targeting a mean coverage of 100-150x.
Visualizations
Title: Amplicon-Based NGS Workflow
Title: Hybridization Capture NGS Workflow
Title: Method Selection Decision Tree
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Featured Protocols
| Item | Function | Example Vendor/Kits |
|---|---|---|
| cfDNA Extraction Kit | Isolate cell-free DNA from plasma/serum with high recovery and low contamination. | Qiagen QIAamp Circulating Nucleic Acid Kit, Roche cfDNA System. |
| DNA Shearing/Covaris | Reproducibly fragment genomic DNA to a defined size distribution for library prep. | Covaris focused-ultrasonicator, Bioruptor. |
| HS DNA Quantitation Assay | Accurately quantify low concentrations and low-mass DNA samples. | Thermo Fisher Qubit dsDNA HS Assay. |
| Amplicon Panel (Multiplex PCR) | Pre-designed set of primers to amplify specific genomic targets simultaneously. | Illumina TruSeq Amplicon, Thermo Fisher AmpliSeq. |
| Hybridization Capture Probe Set | Biotinylated oligonucleotide baits designed to hybridize to genomic targets. | IDT xGen Exome Research Panel, Roche NimbleGen SeqCap EZ. |
| Streptavidin Magnetic Beads | Bind biotinylated probe-DNA complexes for separation and washing during capture. | Dynabeads MyOne Streptavidin T1, Sera-Mag SpeedBeads. |
| Library Prep Kit with UMIs | Reagents for end-prep, adapter ligation, and PCR, supporting UMI integration for error correction. | Swift Biosciences Accel-NGS, Takara Bio SMARTer. |
| Post-Capture PCR Beads | Magnetic beads for size selection and clean-up of libraries, optimizing for fragment retention. | Beckman Coulter SPRIselect, KAPA Pure Beads. |
| Library Quantification Kit (qPCR) | Precisely quantify amplifiable library molecules to ensure optimal sequencing cluster density. | KAPA Library Quantification Kit, Thermo Fisher Collibri. |
Choosing Your Method: Application-Driven Selection for Research & Diagnostics
Amplicon-based Next-Generation Sequencing (NGS) is a targeted sequencing approach that uses PCR primers to enrich specific genomic regions prior to sequencing. Within the context of comparing amplicon-based and hybridization-capture NGS methods, amplicon NGS excels in applications requiring high sensitivity, low input, rapid turnaround, and cost-effective profiling of well-defined genomic regions. This article details its ideal use cases: deep sequencing of defined cancer hotspots, comprehensive microbial profiling, and sensitive liquid biopsy detection.
Application Note 1: Ultra-Deep Sequencing of Oncology Hotspots
Context & Rationale
In clinical oncology, actionable mutations are often concentrated in specific exonic "hotspots" (e.g., in KRAS, EGFR, BRAF, PIK3CA). Amplicon NGS is ideally suited for this application due to its ability to generate ultra-deep, uniform coverage (>5,000x) from minimal DNA input, enabling reliable detection of very low variant allele frequencies (VAFs) crucial for therapy selection and resistance monitoring.
Table 1: Performance Metrics for Oncology Hotspot Panels (Amplicon vs. Hybridization-Capture)
| Metric | Amplicon-Based Panel (e.g., 50-gene hotspot) | Hybridization-Capture Panel (e.g., 500-gene exome) | Relevance to Use Case |
|---|---|---|---|
| Typical Input DNA | 1-10 ng (FFPE) / 5-30 ng (cfDNA) | 50-200 ng (FFPE) / 30-100 ng (cfDNA) | Amplicon superior for limited/degraded samples. |
| Wet-lab Time | ~8-12 hours (single-day workflow) | 24-48 hours (multi-day workflow) | Amplicon superior for rapid results. |
| On-target Rate | >95% | 60-85% | Amplicon superior for efficiency on target. |
| Uniformity of Coverage | High (low fold-80 penalty) | Moderate (higher fold-80 penalty) | Amplicon superior for consistent hotspot coverage. |
| Sensitivity (LoD) | Can reliably detect VAFs of 0.1%-1% | Typically 1-5% VAF for comparable input | Amplicon superior for low-VAF detection. |
| Ability to Call CNVs | Limited/Poor | Good to Excellent | Hybridization superior for copy-number analysis. |
| Cost per Sample | Low to Medium | Medium to High | Amplicon superior for focused queries. |
Detailed Protocol: Hotspot Mutation Detection from FFPE DNA
Title: Detection of Somatic Hotspot Mutations in FFPE Tumor Samples Using Amplicon NGS.
Objective: To identify single nucleotide variants (SNVs) and small indels in a 50-gene oncology hotspot panel with a sensitivity of 1% VAF.
Materials (Research Reagent Solutions):
- DNA Extraction: FFPE DNA extraction kit (e.g., QIAamp DNA FFPE Tissue Kit). Function: Purifies DNA from paraffin-embedded tissue, removing inhibitors.
- DNA QC: Fluorometric dsDNA assay (e.g., Qubit dsDNA HS Assay). Function: Accurately quantifies low-concentration, fragmented DNA.
- Library Prep: Targeted amplicon panel (e.g., Illumina TruSight Oncology 500 ctDNA, amplicon component). Function: Contains predesigned primer pools for co-amplification of target regions.
- Indexing: Dual-indexing plate adapters (e.g., Illumina IDT for Illumina). Function: Adds unique sample indices for multiplexing and flow cell binding sites.
- Library Clean-up: Solid-phase reversible immobilization (SPRI) beads. Function: Size-selects and purifies amplified libraries.
- Sequencing: MiSeq or iSeq Reagent Kits. Function: Provides chemistry for cluster generation and sequencing-by-synthesis.
- Analysis: Cloud-based or local bioinformatics pipeline (e.g., Illumina DRAGEN, CLC Genomics Server). Function: Aligns reads, calls variants, and generates clinical reports.
Methodology:
- DNA Extraction & QC: Extract DNA from 5-10 µm FFPE curls/sections per manufacturer's protocol. Quantify using a fluorometric assay. Assess fragmentation via agarose gel or TapeStation. Acceptable input: 5-20 ng of fragmented DNA.
- Library Preparation: Dilute DNA to 1 ng/µL in low-EDTA TE buffer. Combine 5 µL (5 ng) DNA with amplicon-specific primer pool, high-fidelity PCR polymerase, and dNTPs.
- Target Amplification: Perform thermocycling: Initial denaturation (95°C, 2 min); 20 cycles of [Denature (95°C, 20 sec), Anneal/Extend (60°C, 4 min)]; Final extension (72°C, 5 min).
- Indexing & Sample Barcoding: Add a unique pair of dual index adapters to each sample via a limited-cycle (8-10 cycles) PCR reaction. This enables multiplexing of up to 96+ samples per run.
- Library Purification & Normalization: Clean the amplified library using SPRI beads at a 0.9x bead-to-sample ratio to remove primer dimers and short fragments. Quantify libraries via qPCR (e.g., KAPA Library Quantification Kit) for accurate molarity. Pool libraries at equimolar concentrations (e.g., 4 nM each).
- Sequencing: Denature and dilute the pooled library to 10 pM and load onto a MiSeq system using a v3 600-cycle (2x250 bp) cartridge. Aim for >5000x average coverage per amplicon.
- Bioinformatic Analysis: Demultiplex samples. Align reads to the human reference genome (hg38). Call variants using a validated somatic caller (e.g., GATK Mutect2, VarScan2). Filter variants against population databases (gnomAD) and report clinically relevant mutations with VAF >1%.
Diagram 1: Amplicon NGS Workflow for Oncology Hotspots
Application Note 2: Comprehensive Microbial Profiling (16S/ITS/18S rRNA)
Context & Rationale
For identifying and quantifying bacterial, fungal, or eukaryotic microbial communities, sequencing of conserved phylogenetic marker genes (like 16S rRNA) is standard. Amplicon NGS is the undisputed method here, as it allows for high-multiplex, cost-effective analysis of hundreds of samples, providing genus/species-level taxonomy and relative abundance data.
Table 2: Amplicon NGS for Microbial Community Analysis
| Parameter | Typical Specification | Application Implication |
|---|---|---|
| Target Regions | 16S rRNA (V1-V9 hypervariable), ITS1/2, 18S rRNA. | Enables broad or specific taxonomic profiling. |
| Read Length | 250-300 bp (paired-end). | Sufficient to cover key hypervariable regions for classification. |
| Sequencing Depth | 20,000 - 100,000 reads/sample. | Saturates diversity for most complex communities (e.g., gut). |
| Taxonomic Resolution | Genus-level (often), Species-level (with curated DB). | Accurate community composition analysis. |
| Sample Multiplexing | 96-384+ samples per MiSeq run. | Extremely high-throughput and cost-effective. |
| Key Output Metric | Relative Abundance (%), Alpha/Beta Diversity. | For comparative ecology and dysbiosis studies. |
| Limitation | Cannot profile virulence/AMR genes or strain-level variation without capture. | Functional insight requires shotgun metagenomics (capture-based). |
Detailed Protocol: 16S rRNA Gene Amplicon Sequencing of Gut Microbiota
Title: Profiling Bacterial Community Composition from Fecal DNA Using 16S rRNA Amplicon Sequencing.
Objective: To characterize the relative abundance of bacterial taxa in a fecal sample via amplification and sequencing of the V3-V4 hypervariable region of the 16S rRNA gene.
Materials (Research Reagent Solutions):
- Stool Stabilization: DNA/RNA Shield or similar preservative. Function: Preserves microbial community integrity at room temperature.
- DNA Extraction: Bead-beating based kit (e.g., DNeasy PowerSoil Pro Kit). Function: Mechanically and chemically lyses diverse cell walls for comprehensive DNA recovery.
- PCR Primers: Tailored primer pair (e.g., 341F/806R) with overhang adapters. Function: Specifically amplifies the 16S V3-V4 region and adds flow cell adapter sequences.
- High-Fidelity Polymerase: e.g., KAPA HiFi HotStart ReadyMix. Function: Provides accurate amplification with low error rates for complex templates.
- Index Adapters: Nextera XT Index Kit v2. Function: Adds dual, unique sample barcodes for multiplexing.
- Sequencing: MiSeq Reagent Kit v3 (600-cycle). Function: Standard chemistry for 2x300 bp paired-end reads ideal for 16S.
Methodology:
- Sample Preservation & DNA Extraction: Homogenize ~200 mg of fresh or stabilized stool in lysis buffer. Perform rigorous bead-beating for cell disruption. Purify DNA following kit protocol. Elute in 50 µL. Quantify via fluorometry.
- First-Stage PCR (Amplification with Adapter Overhangs): Set up reactions with 10-20 ng genomic DNA, primers containing the target-specific sequence plus Illumina adapter overhangs, and high-fidelity polymerase. Cycle: 95°C for 3 min; 25 cycles of [95°C for 30s, 55°C for 30s, 72°C for 30s]; 72°C for 5 min.
- Library Indexing (Second-Stage PCR): Use 2-5 µL of purified first-stage product as template. Add unique dual indices via a limited-cycle (8 cycles) PCR.
- Library Clean-up & Pooling: Purify indexed libraries with SPRI beads (0.8x ratio). Quantify by fluorometry. Measure fragment size (expect ~550-600 bp). Normalize and pool libraries equimolarly.
- Sequencing & Primary Analysis: Denature and dilute pool to 8 pM with 15% PhiX spike-in for low-diversity amplicon runs. Sequence on MiSeq (2x300 bp). Use on-instrument software for base calling and demultiplexing.
- Bioinformatic Processing: Use a pipeline like QIIME 2 or Mothur. Steps include: quality filtering (DADA2 for denoising), chimera removal, merging of paired-end reads, clustering into Amplicon Sequence Variants (ASVs) or Operational Taxonomic Units (OTUs), and taxonomic assignment against reference databases (Silva, Greengenes).
Diagram 2: Microbial 16S rRNA Amplicon Sequencing Workflow
Application Note 3: Sensitive Liquid Biopsy for ctDNA Analysis
Context & Rationale
Liquid biopsy analysis of circulating tumor DNA (ctDNA) is challenging due to low ctDNA concentration and fraction in plasma. Amplicon NGS, especially using unique molecular identifiers (UMIs), is optimal for this ultra-sensitive application. It supports very low DNA inputs (<10 ng cfDNA) and, via error correction with UMIs, achieves detection limits below 0.1% VAF for therapy selection and minimal residual disease (MRD) monitoring.
Table 3: Amplicon vs. Capture for Liquid Biopsy ctDNA Analysis
| Feature | UMI-Amplicon Approach (e.g., 50-gene) | Hybridization-Capture Approach (e.g., 150-gene) | Relevance to Use Case |
|---|---|---|---|
| Input cfDNA Mass | 5-30 ng | 20-100 ng | Amplicon superior for volume-limited plasma draws. |
| Effective Sensitivity | 0.1% VAF (with UMI error correction) | 0.5-1% VAF (with duplex UMIs) | Amplicon superior for ultra-low VAF detection. |
| Turnaround Time (Wet Lab) | <24 hours | 2-3 days | Amplicon superior for clinical speed. |
| Handling of FFPE Input | Excellent (short amplicons) | Good (requires longer, intact fragments) | Amplicon superior for degraded material. |
| Panel Flexibility / Scalability | Low-Moderate (new primers needed) | High (adjustable by probe design) | Hybridization superior for large/growing panels. |
| Detection of Structural Variants | Very Limited | Good (with appropriate bait design) | Hybridization superior for fusions/translocations. |
Detailed Protocol: UMI-Based ctDNA Mutation Detection from Plasma
Title: Ultra-Sensitive Detection of ctDNA Mutations in Plasma Using UMI-Amplicon Sequencing.
Objective: To detect somatic mutations at a limit of detection (LoD) of 0.1% VAF from 10 ng of plasma-derived cell-free DNA.
Materials (Research Reagent Solutions):
- Blood Collection: cfDNA Blood Collection Tubes (e.g., Streck). Function: Stabilizes nucleated blood cells to prevent genomic DNA contamination.
- cfDNA Extraction: Manual or automated cfDNA kit (e.g., QIAamp Circulating Nucleic Acid Kit). Function: Efficiently recovers short, fragmented cfDNA from plasma.
- Library Prep with UMIs: Commercial UMI-amplicon kit (e.g., ArcherDX VariantPlex, IDT xGen Prism). Function: Integrates unique molecular identifiers during initial amplification to tag original DNA molecules for error correction.
- High-Fidelity Polymerase: e.g., Platinum SuperFi II. Function: Essential for accurate pre-UMI amplification to avoid propagating early PCR errors.
- Post-Capture Beads (if used): Streptavidin-coated magnetic beads for hybrid capture of amplicon pools.
- Sequencing: High-output flow cell (e.g., NextSeq 2000 P2, 300-cycle). Function: Provides depth (>50,000x) needed for robust UMI consensus building.
Methodology:
- Plasma Processing & cfDNA Isolation: Draw blood into stabilizing tubes. Process within 72 hours: double centrifugation (1600xg, 10 min; 16000xg, 10 min) to obtain platelet-poor plasma. Extract cfDNA from 2-5 mL plasma per kit protocol. Elute in 20-30 µL. Quantify by highly sensitive qPCR (e.g., targeting 80-100 bp ALU repeats).
- Initial UMI Tagging & Target Amplification: Use 5-20 ng of cfDNA. Perform an initial limited-cycle PCR (4-6 cycles) using gene-specific primers that contain a random UMI sequence and a partial adapter sequence. This uniquely tags each original DNA molecule.
- Library Completion Amplification: Clean up the initial product. Perform a second PCR (14-18 cycles) to add the full Illumina adapters and sample-specific dual indices.
- Library Purification & QC: Purify with SPRI beads (0.9x). Quantify by qPCR. Assess fragment size distribution via Bioanalyzer (peaks ~200-350 bp).
- Deep Sequencing: Pool libraries and sequence on a high-output platform to achieve a raw depth >50,000x per amplicon. Use 2x150 bp reads.
- Bioinformatic Analysis with UMI Consensus: Use dedicated software (e.g., Archer Analysis, smCounter2). Steps include: a) Group reads by UMI family, b) Create a consensus sequence for each family to eliminate PCR and sequencing errors, c) Align consensus reads, d) Call variants against a matched normal or population baseline. Report variants above a statistically defined threshold (e.g., ≥3 supporting consensus reads, VAF ≥0.1%).
Diagram 3: UMI-Amplicon Sequencing for ctDNA Analysis
Amplicon-based NGS demonstrates distinct advantages in three critical applications: profiling defined oncology hotspots with high sensitivity and speed, conducting cost-effective and high-throughput taxonomic surveys of microbial communities, and enabling ultra-sensitive liquid biopsy assays via UMI-based error correction. In the broader methodological comparison, its strengths lie in efficiency, sensitivity, and speed for focused genomic queries, while hybridization-capture remains preferable for large gene panels, copy number analysis, and discovery-oriented sequencing. The choice between methods is thus fundamentally driven by the specific clinical or research question.
Within the broader methodological comparison of Amplicon-based versus Hybrid-Capture Next-Generation Sequencing (NGS), this document details specific, ideal applications for the hybridization capture approach. While amplicon methods excel in sensitivity for low-variant-allele-frequency detection in small, predefined genomic regions, hybrid-capture NGS demonstrates superior utility for larger, more complex targets. This application note frames its content within this thesis, highlighting scenarios where the capture-based method's strengths—including off-target probe binding, uniform coverage across difficult sequences, and ability to target non-contiguous regions—are paramount.
Ideal Use Case 1: Whole Exome & Large Genomic Subsets
Hybrid-capture is the established method for whole exome sequencing (WES) and large, targeted panels (>1 Mb). Its efficiency in capturing thousands of discrete exons spread across the genome is unmatched by amplicon-based approaches, which struggle with primer design and multiplexing at this scale.
Key Advantages:
- Comprehensive Coverage: Efficiently targets the entire coding region (~1-2% of the genome).
- Design Flexibility: Easy to update panels by adding or removing probes without re-validating the entire assay.
- Uniformity: Provides more even coverage across regions with varying GC content compared to many amplicon schemes.
Quantitative Performance Data (Representative):
| Metric | Hybrid-Capture WES (150bp PE) | Typical Amplicon Panel (≤ 500 genes) |
|---|---|---|
| Target Region Size | ~35-60 Mb | 0.1 - 2 Mb |
| Mean Fold Coverage | 100x - 200x | 500x - 2000x |
| Uniformity (% >0.2x mean) | >90% | Varies (80-95%) |
| DNA Input Required | 50-200 ng | 10-50 ng |
| Preparation Time | 1.5 - 2 days | 6 - 8 hours |
| SNV/Indel Sensitivity | High at ≥5% VAF | Very High at ≥1% VAF |
| Best For | Discovery, unknown etiology | Profiling known hotspots |
Detailed Protocol: Hybrid-Capture Whole Exome Sequencing
A. Library Preparation (Illumina Compatible)
- DNA Shearing: Fragment 50-100 ng of high-quality genomic DNA to 150-200 bp using a focused-ultrasonicator (e.g., Covaris).
- End Repair & A-Tailing: Use a bead-based library prep kit. Perform end-repair to generate blunt ends, followed by 3' adenylation.
- Adapter Ligation: Ligate indexed, flow-cell-compatible adapters to the fragments. Clean up with magnetic beads.
- Library Amplification: Perform 4-8 cycles of PCR to enrich adapter-ligated fragments. Quantify using fluorometry (Qubit).
B. Target Enrichment by Hybridization
- Probe Hybridization: Combine 200-500 ng of pooled library with a biotinylated oligonucleotide probe library (e.g., IDT xGen, Roche SeqCap, Twist Bioscience) in a hybridization buffer. Denature at 95°C for 5-10 minutes and incubate at 58-65°C for 12-16 hours.
- Capture with Streptavidin Beads: Bind the biotinylated probe-DNA hybrids to streptavidin-coated magnetic beads. Wash away non-specifically bound DNA with stringent buffers.
- Elution & Post-Capture PCR: Elute the captured DNA from the beads. Perform a final amplification (10-14 cycles) to generate the sequencing-ready enriched library. Purify with magnetic beads.
C. Sequencing & Analysis
- QC & Pooling: Quantify final libraries by qPCR (for molarity) and analyze size distribution (Bioanalyzer/TapeStation). Pool libraries equimolarly.
- Sequencing: Load onto Illumina NovaSeq 6000, NextSeq 2000, or equivalent for 2x150 bp paired-end sequencing to a mean depth of >100x.
- Data Analysis: Align to reference genome (hg38) using BWA-MEM or Dragen. Call variants with GATK or Dragen, and annotate using databases like ClinVar, gnomAD.
Diagram Title: Hybrid-Capture Whole Exome Sequencing Workflow
Ideal Use Case 2: Fusion & Structural Variant Detection
Detection of gene fusions, translocations, and other structural variants (SVs) requires sequencing across breakpoints that can occur in introns or intergenic regions. Hybrid-capture panels using "tiling" probes across large genomic segments or introns are ideal for this discovery-based application.
Key Advantages:
- Breakpoint Agnostic: Can identify novel fusion partners without prior knowledge of exact breakpoints.
- Intronic Coverage: Probes can be designed to cover introns and known partner gene loci.
- DNA/RNA Compatibility: Can be applied to both DNA (for rearrangements) and RNA (for expressed fusion transcripts).
Quantitative Performance Data (Representative):
| Metric | Hybrid-Capture DNA Fusion Panel | Hybrid-Capture RNA Fusion Panel | Amplicon (RNA-based) |
|---|---|---|---|
| Primary Target | Genomic breakpoints | Expressed fusion transcripts | Known expressed fusion isoforms |
| Novel Partner Discovery | Yes | Yes | Limited/No |
| Probe Design Strategy | Tiling across introns | Exon/transcript-based | Span known breakpoints |
| Input Material | 50-100 ng DNA | 10-100 ng RNA | 10 ng RNA |
| Complexity (Library Duplicates) | Moderate | Moderate-High | Low |
| Best For | Discovery, complex SVs | Discovery, expressed fusions | High-sensitivity for known fusions |
Detailed Protocol: RNA-based Hybrid-Capture for Fusion Detection
A. RNA Library Preparation
- RNA QC & rRNA Depletion: Assess RNA integrity (RIN >7). Deplete ribosomal RNA using probe-based methods (e.g., Illumina RiboZero, QIAseq FastSelect).
- cDNA Synthesis: Convert RNA to double-stranded cDNA using random hexamers and reverse transcriptase.
- Library Construction: Fragment cDNA (~200 bp), perform end-repair, A-tailing, and adapter ligation as per the DNA protocol.
B. Enrichment for Fusion Transcripts
- Custom Probe Design: Design probes tiling across full exons and known breakpoint hotspots of genes of interest (e.g., ALK, ROS1, RET, NTRK1/2/3).
- Hybridization & Capture: Follow the same hybridization and bead-capture steps as the WES protocol, using the custom fusion-focused probe set.
- Amplification & QC: Perform post-capture PCR. Validate library quality and check for enrichment of target genes via qPCR before sequencing.
C. Sequencing & Bioinformatics
- Sequencing: Sequence with 2x100 bp or 2x150 bp paired-end reads to high depth (~10-20M on-target reads).
- Specialized Analysis: Use fusion-aware aligners (STAR, BWA-MEM) and dedicated callers (Arriba, STAR-Fusion, Manta) to identify chimeric reads spanning breakpoints.
Diagram Title: RNA Hybrid-Capture Workflow for Fusion Detection
Ideal Use Case 3: Complex Genomic Regions
Regions with high GC content, pseudogenes, or segmental duplications (e.g., SMN1/SMN2, CYP2D6, HLA) pose significant challenges for amplicon design due to primer misalignment and amplification bias. Hybrid-capture, with its longer probes and post-capture PCR, often provides more accurate representation.
Key Advantages:
- Mitigates Amplification Bias: Avoids PCR competition inherent in large amplicon multiplexes.
- Superior Mapping: Longer reads from hybrid-capture allow more confident alignment in repetitive regions.
- Haplotype Resolution: When combined with long-read or linked-read technologies, can resolve complex phasing.
Quantitative Performance Data (Representative):
| Metric | Hybrid-Capture for Complex Loci | Amplicon for Complex Loci |
|---|---|---|
| Example Target | HLA Locus, CYP2D6 | EGFR T790M, KRAS G12/13 |
| Coverage Uniformity | Good (Can use GC-balanced probes) | Often Poor (High variability) |
| Specificity in Pseudogenes | High (With careful probe design) | Low (Risk of co-amplification) |
| Ability to Phase Variants | Possible with long fragments | Very Limited |
| Best For | Highly homologous regions, copy number variation | Simple, non-repetitive hotspots |
Detailed Protocol: Targeting a Complex Region (e.g., CYP2D6)
A. Custom Panel Design
- Identify Homologous Regions: Map the entire CYP2D6 locus and its pseudogenes (CYP2D7, CYP2D8).
- Design Discriminatory Probes: Using tools like BLAST, design 80-120 bp biotinylated probes with unique sequences specific to CYP2D6, avoiding shared homology with pseudogenes. Tile across the entire gene and structural variation breakpoints.
B. Enrichment & Analysis
- Standard Hybrid-Capture: Follow the standard library prep and hybridization capture protocol using the custom CYP2D6 probe set.
- Long-Read Sequencing Option: For phased haplotyping, use a long-read compatible hybrid-capture protocol (e.g., PacBio HiFi or Oxford Nanopore). This involves creating SMRTbell or ligation sequencing libraries before capture, then performing capture on the long-molecule library.
- CNV & SV Analysis: Use depth of coverage algorithms (e.g., CNVkit) and split-read analysis to determine gene copy number, identify hybrid CYP2D6/D7 genes, and call star alleles.
Diagram Title: Hybrid-Capture Advantages for Complex Regions
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function & Application |
|---|---|
| Biotinylated Oligo Probe Libraries (xGen, SeqCap, Twist) | Designed oligonucleotides that hybridize to target sequences; biotin enables streptavidin capture. Fundamental to the method. |
| Streptavidin Magnetic Beads (Dynabeads, Sera-Mag) | Solid support for capturing biotin-probe:DNA complexes. Critical for post-hybridization purification. |
| Hybridization Buffer & Enhancers (Cot-1 DNA, Blocking Oligos) | Suppresses non-specific binding of repetitive sequences and adapter oligos, improving on-target efficiency. |
| NGS Library Prep Kit with Bead Cleanup (KAPA, Illumina) | Provides enzymes and buffers for end-prep, ligation, and PCR; magnetic beads enable rapid, clean size selection. |
| Targeted Hybridization Kit (IDT xGen Hybridization Kit, Nimblegen SeqCap) | Optimized buffers and protocols for the hybridization and wash steps, ensuring high specificity and yield. |
| QC Instruments (Qubit Fluorometer, Bioanalyzer) | Quantifies DNA/RNA concentration and assesses library fragment size distribution pre- and post-capture. |
| qPCR Quantification Kit (KAPA Library Quant) | Accurately measures the molar concentration of adaptor-ligated libraries for precise pooling before sequencing. |
In the comparative analysis of amplicon-based and hybridization capture Next-Generation Sequencing (NGS) methods for genomic research and diagnostic applications, panel design is a critical determinant of success. This document outlines key application notes and protocols for designing target enrichment panels, with a focus on the trifecta of flexibility, scalability, and updateability. These considerations directly impact the feasibility, cost, and longevity of studies comparing the inherent biases, uniformity, and off-target rates of amplicon versus capture methodologies.
Quantitative Comparison of Panel Design Attributes
The following table summarizes the core quantitative and qualitative differences in design considerations between the two methods, which influence flexibility, scalability, and updateability.
Table 1: Panel Design Considerations for Amplicon vs. Hybridization Capture
| Design Attribute | Amplicon-Based Panels | Hybridization Capture Panels | Impact on Flexibility/Scalability/Updateability |
|---|---|---|---|
| Optimal Panel Size | Typically < 500 targets; practical limit ~10-20 kb. | Virtually unlimited; routinely 0.1 - 50 Mb. | Scalability: Capture excels for large, genome-scale targets. |
| Design & Synthesis Time | Fast (days), once primers are designed. | Slow (weeks), due to complex oligo synthesis and validation. | Updateability: Amplicon allows rapid iterative design. |
| Initial Design Cost | Low to moderate. | High (custom oligo pool synthesis). | Scalability: Higher upfront cost for capture scale-up. |
| Per-Sample Cost | Low. | Moderate to High. | Scalability: Amplicon is more cost-effective for high sample numbers on small panels. |
| Ease of Adding Targets | Low; requires re-optimization of multiplex PCR. | High; new probes can be spiked into existing pools. | Updateability & Flexibility: Capture panels are inherently more modular. |
| Compatibility with Sample Types | High for high-quality DNA. Challenging for FFPE/degraded DNA. | Robust for FFPE and degraded DNA. | Flexibility: Capture offers greater application flexibility. |
| Variant Type Flexibility | Best for SNVs, small Indels. Poor for CNVs, fusions, large rearrangements. | Excellent for SNVs, Indels, CNVs, fusions, rearrangements. | Flexibility: Capture supports a broader range of genomic alterations. |
Experimental Protocols for Panel Validation
Protocol 1: Assessing Panel Uniformity and Coverage
Objective: To quantify the evenness of target coverage, a critical metric for comparing amplicon and capture performance. Materials: Validated NGS panel, reference genomic DNA (e.g., NA12878), NGS library prep kit, sequencer. Procedure:
- Prepare libraries from 100 ng input gDNA using the manufacturer's protocol for the chosen panel type.
- Sequence libraries to a mean target coverage of >500x on an appropriate NGS platform.
- Align reads to the reference genome (hg38) using BWA-MEM or STAR.
- Calculate depth at each base in the target bed file using
samtools depthorGATK DepthOfCoverage. - Analysis: Compute the percentage of bases covered at >100x and >500x. Calculate the uniformity as the percentage of targets within ±20% of the mean coverage. Generate a cumulative coverage plot.
Protocol 2: Evaluating Off-Target Rate
Objective: To measure the fraction of sequencing reads mapping outside target regions, indicative of panel specificity. Procedure:
- Using the aligned BAM file from Protocol 1, separate reads into "on-target" and "off-target" groups based on overlap with the panel's target coordinates.
- Calculate: Off-Target Rate (%) = (Off-target reads / Total mapped reads) * 100.
- Comparison: Expect amplicon panels to have very low off-target rates (<1-5%) while hybridization capture typically yields 10-40% off-target reads, which can be leveraged for copy number analysis.
Protocol 3: Sensitivity and Specificity for Variant Detection
Objective: To establish the limit of detection (LoD) for SNVs/Indels using a validated reference standard. Materials: Seraseq FFPE Tumor DNA Reference Material or similar, with known variant allele frequencies (VAFs down to 1-5%). Procedure:
- Prepare libraries in triplicate from the reference material using both panel types.
- Sequence to appropriate depth (e.g., >1000x mean).
- Call variants using a standardized pipeline (e.g.,
GATK Mutect2for capture,Illumina DRAGENfor amplicon). - Analysis: For each known variant, calculate:
- Sensitivity = (True Positives) / (True Positives + False Negatives)
- Specificity = (True Negatives) / (True Negatives + False Positives)
- Plot sensitivity vs. VAF to determine LoD for each technology.
Visualization of Panel Design and Analysis Workflows
Title: NGS Panel Design and Validation Workflow
Title: Post-Sequencing Data Analysis and Comparison
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents & Materials for Panel Comparison Studies
| Item | Function & Relevance | Example Products/Brands |
|---|---|---|
| Reference gDNA Standards | Provides ground truth for validating panel sensitivity, specificity, and variant calling accuracy. Critical for benchmarking. | Seraseq Tumor DNA, Horizon Discovery Multiplex I, NIST Genome in a Bottle (GIAB) |
| FFPE-mimetic DNA Controls | Evaluates panel performance on degraded samples, a key differentiator between amplicon and capture. | Seraseq FFPE Tumor DNA, Horizon Discoverys FFPE DNA |
| Hybridization & Wash Buffers | For capture panels: Stringent washing post-hybridization is crucial for specificity and low off-target rates. | IDT xGen Hybridization & Wash Kit, Roche KAPA HyperCapture Beads |
| Multiplex PCR Enzyme Master Mix | For amplicon panels: Specialized polymerases capable of unbiased, high-plex amplification are essential. | Takara Bio PrimeSTAR GXL, QIAGEN Multiplex PCR Plus |
| Target-Specific Oligo Pools | The core panel component. Design dictates flexibility and updateability. | IDT xGen Lockdown Probes, Twist Bioscience Custom Panels, Agilent SureSelect XTHS |
| Magnetic Beads (SPRI) | For universal library cleanup, size selection, and bead-based normalization. | Beckman Coulter AMPure XP, MagBio HighPrep PCR |
| Unique Dual Index (UDI) Kits | Enables sample multiplexing, prevents index hopping artifacts, and is essential for scalable sequencing. | Illumina TruSeq UD Indexes, IDT for Illumina UDI |
| NGS Library Quantification Kits | Accurate quantification (qPCR-based) is vital for achieving optimal sequencing cluster density. | KAPA Library Quantification Kits, Thermo Fisher TaqMan |
| Bioinformatics Pipeline Software | For standardized alignment, coverage analysis, and variant calling to ensure comparable results. | Illumina DRAGEN, GATK, QIAGEN CLC Genomics Server |
This application note is part of a broader thesis comparing amplicon-based and hybridization capture-based Next-Generation Sequencing (NGS) methods. A critical factor influencing method selection is the quality and quantity of input DNA. This document details protocols and performance data for analyzing low-quantity, low-quality, and Formalin-Fixed Paraffin-Embedded (FFPE) DNA samples using both NGS approaches, providing a framework for informed platform selection.
Table 1: Comparative Performance of NGS Methods Across Challenging Sample Types
| Sample Type | Input DNA | Amplicon-Based | Hybridization Capture |
|---|---|---|---|
| Standard Control | 50 ng, intact DNA | 99.9% Uniformity, >99% on-target | 98.5% Uniformity, 95-98% on-target |
| Low Quantity | 1-10 ng | Robust: Reliable down to 1 ng with dedicated kits. High duplication rates. | Challenging: Requires ≥10-50 ng for efficiency. Poor capture below 10 ng. |
| Low Quality (Fragmented) | 50 ng, DV200: 30-50% | Tolerant: Works on short fragments; risk of primer dropout. | Moderate: Efficiency drops with fragmentation; requires optimization. |
| FFPE-Derived | 50-100 ng, DV200: 20-40% | Resilient: Short amplicons (<150bp) perform best. High C>T artifacts. | Variable: Long probes fail; short probes recommended. High duplication, lower complexity. |
| Key Metric Impact | High Sensitivity, Lower Specificity: Prone to PCR bias/artifacts. Lower complexity. | Higher Specificity, Lower Sensitivity: Better for large panels/CNV; requires more input. |
Table 2: Recommended Use Case Summary
| Application Priority | Recommended Method | Rationale |
|---|---|---|
| Small panels (<50 genes) from FFPE/Low-DNA | Amplicon-Based | Sensitivity with minimal input, rapid workflow. |
| Large panels/Exomes, intact DNA | Hybridization Capture | Superior uniformity & specificity for larger targets. |
| Detecting low-frequency variants (<1%) | Amplicon-Based (with UMI) | Ultimate sensitivity with unique molecular identifiers. |
| Copy number variation (CNV) analysis | Hybridization Capture | More uniform coverage provides reliable log2 ratios. |
Experimental Protocols
Protocol 3.1: Library Preparation from Low-Quantity/Low-Quality DNA A. Amplicon-Based (Multiplex PCR)
- DNA Quantification & QC: Quantify using fluorometry (e.g., Qubit dsDNA HS Assay). Assess fragmentation via TapeStation/ Bioanalyzer (DV200 metric).
- Input Normalization: For low-quantity samples, use 1-10 ng input in a reaction volume ≤ 10 µL. Include a no-template control.
- Multiplex PCR (2-Step):
- Primer Pool 1 (Target Amplification): Combine DNA with a multiplex primer pool (designed for short amplicons, ~80-150bp for FFPE), polymerase, and PCR master mix.
- Cycling Conditions: Initial denaturation: 95°C, 2 min; 20-25 cycles of [95°C, 20 sec; 60°C, 4 min]; Final extension: 72°C, 5 min.
- Clean-up: Use AMPure XP beads (1.0x ratio) to purify amplicons.
- Primer Pool 2 (Indexing): Attach sample-specific indices and sequencing adapters via a limited-cycle (5-10 cycles) PCR.
- Final Library Clean-up & QC: Perform a double-sided AMPure bead cleanup (e.g., 0.6x then 1.0x ratios). Quantify via qPCR (library quantification kit) for accurate pooling.
B. Hybridization Capture
- Pre-Capture Library Prep: Convert 10-100 ng input DNA into a sequencing library using a ligation-based kit designed for degraded samples (incorporating repair steps).
- Fragmentation & End-Prep: If DNA is not pre-fragmented, use a sonication or enzymatic method. Repair ends and add an ‘A’ base.
- Adaptor Ligation: Ligate unique dual-indexed adaptors. Use a higher adaptor:insert ratio for low-input samples.
- Limited-Cycle Pre-Capture PCR: Amplify libraries with 4-10 cycles to generate sufficient mass for capture.
- Target Capture:
- Pool up to 8 libraries equimolarly (based on qPCR).
- Hybridize with biotinylated DNA or RNA probes (designed as short, tiled probes for FFPE) for 16-24 hours at 65°C.
- Capture with streptavidin magnetic beads. Perform stringent washes.
- Elute captured DNA.
- Post-Capture PCR: Amplify captured libraries for 10-14 cycles.
- Final Clean-up & QC: Purify with AMPure beads (1.0x). Validate size distribution and quantify by qPCR.
Protocol 3.2: In-silico Analysis for Cross-Platform Comparison
- Raw Data Processing: Demultiplex reads (bcl2fastq). Record total reads per sample.
- Primary Alignment: Map reads to the human reference genome (hg38) using optimized aligners (BWA for capture, specialized tools for amplicons).
- Duplicate Marking: Use tools that recognize UMIs (for amplicon) or coordinate-based marking (for capture). Report duplicate rates.
- Target Coverage Metrics: Calculate mean depth, uniformity (% bases at >100x or >500x), and on-target rate.
- Variant Calling: Use a single, high-confidence callset (e.g., using GAT4K or VarScan2) for both datasets. Compare variant allele frequencies (VAFs), especially for known low-frequency variants.
Visualizations
Decision Workflow for NGS Method Selection
NGS Method Pros and Cons Comparison
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 3: Key Reagents for NGS of Challenging Samples
| Reagent/Material | Function | Example Product Types |
|---|---|---|
| Fluorometric DNA Quant Kit | Accurately quantifies low-concentration, fragmented DNA. Critical for input normalization. | Qubit dsDNA HS Assay; PicoGreen. |
| DNA Integrity Assessment | Evaluates fragmentation level (DV200), guiding method choice. | Agilent TapeStation (Genomic DNA ScreenTape); Bioanalyzer. |
| Library Prep Kit for FFPE | Enzymatic mixes for repair, ligation, and amplification of damaged DNA. | Illumina DNA Prep; KAPA HyperPlus; Swift Biosciences Accel-NGS. |
| Multiplex PCR Panels (Short) | Primer pools generating amplicons <150bp for degraded samples. | Qiagen GeneRead; Illumina AmpliSeq Cancer Panels. |
| Hybridization Capture Probes | Biotinylated, tiled DNA/RNA probes; short designs for FFPE. | IDT xGen; Agilent SureSelect; Twist Bioscience. |
| Streptavidin Magnetic Beads | Solid-phase capture of biotinylated probe-target complexes. | Dynabeads MyOne Streptavidin C1. |
| Unique Molecular Indices (UMI) | Molecular barcodes to correct for PCR/sequencing errors and duplicates. | IDT UMI adapters; Twist UMI kits. |
| SPRI/AMPure Beads | Size-selective purification and cleanup of libraries. | Beckman Coulter AMPure XP. |
| Library Quantification Kit (qPCR) | Accurate molar quantification for optimal pooling and loading. | KAPA Library Quant Kit; Illumina Library Quant. |
Analysis Pipelines and Computational Demands for Each Method
Within a comprehensive thesis comparing amplicon-based and hybridization capture next-generation sequencing (NGS) methods, a critical component is the analysis of the bioinformatic workflows and their associated computational burdens. The choice of wet-lab methodology inherently dictates the required data processing steps, the tools employed, and the infrastructure needed. This application note details the standard pipelines, their key steps, and a quantitative comparison of computational demands, providing protocols for researchers embarking on such comparisons or implementing these methods in drug development and diagnostics.
Analysis Pipelines: Workflow and Tools
The fundamental difference in library preparation propagates through the bioinformatic analysis. Amplicon sequencing, targeting specific loci via PCR, requires stringent removal of PCR duplicates and careful handling of primer sequences. Hybridization capture, which enriches for broader genomic regions via probe hybridization, demands more sophisticated read alignment and duplicate marking due to its off-target reads and larger target space.
Amplicon-Based NGS Analysis Pipeline
The primary goal is to generate accurate variant calls from targeted PCR products, with a focus on sensitivity for low-frequency variants.
Detailed Protocol: Amplicon Variant Calling
- Demultiplexing: Use
bcl2fastq(Illumina) orbcl-convertto generate FASTQ files, assigning reads to samples based on index sequences. - Quality Control & Trimming: Run
FastQCfor quality assessment. UsecutadaptorTrimmomaticto remove adapter sequences and primer sequences (critical step). Provide the exact primer sequences used in the assay to the tool. - Alignment: Map reads to a reference genome (e.g., hg38) using a fast aligner like
BWA-MEMorBowtie2. - PCR Duplicate Marking: Use
Picard MarkDuplicatesorsamtools markdupto identify and tag reads originating from the same PCR template, preventing false-positive variant calls. - Variant Calling: Call variants (SNVs, indels) using a tool optimized for amplicon data, such as
VarScan2,GATK Mutect2(with careful panel-of-normals setup), orLoFreq. These tools are sensitive to low-allele-frequency variants common in amplicon data. - Variant Annotation & Filtering: Annotate variants using
SnpEfforVEP. Filter based on depth, strand bias, and population frequency (e.g., gnomAD).
Diagram Title: Amplicon NGS Analysis Pipeline
Hybridization Capture NGS Analysis Pipeline
This pipeline is more complex due to the nature of capture data, requiring robust handling of off-target reads and often incorporating copy number variation (CNV) analysis.
Detailed Protocol: Hybridization Capture Analysis
- Demultiplexing & QC: Identical to amplicon Step 1.
FastQCis run, but adapter trimming is typically less critical if library prep kits with robust adapters are used. - Alignment: Map reads using
BWA-MEMto a reference genome. This step is more computationally intensive due to higher total reads and a larger effective genomic search space. - Post-Alignment Processing: This is a critical multi-step refinement:
- Sort & Index: Use
samtools sortand index. - Duplicate Marking: Use
Picard MarkDuplicatesto mark optical and library duplicates (not PCR-driven). - Base Quality Score Recalibration (BQSR): Use
GATK BaseRecalibratorandApplyBQSRto correct systematic errors in base quality scores.
- Sort & Index: Use
- Variant Calling: Use a suite of tools for comprehensive calling.
GATK HaplotypeCalleris standard for germline SNVs/indels.GATK Mutect2with a panel-of-normals is used for somatic calls.CNVkitorGATK gCNVare employed for calling copy number alterations from capture data. - Variant Refinement & Annotation: Filter variant calls using
GATK FilterMutectCallsorVariantFiltration. Annotate withVEPorSnpEff.
Diagram Title: Hybridization Capture NGS Pipeline
Computational Demand Comparison
The computational requirements differ significantly, primarily driven by data volume, processing steps, and target region size.
Table 1: Quantitative Comparison of Computational Demands
| Parameter | Amplicon-Based NGS | Hybridization Capture NGS | Notes |
|---|---|---|---|
| Typical Data Yield per Sample | 0.5 - 2 Gb | 5 - 15 Gb | Capture yields more data due to larger targeted region and off-target reads. |
| Primary Storage (FASTQ+BAM) | 2 - 5 GB | 20 - 60 GB | Directly proportional to data yield. |
| CPU Hours (Typical WGS) | 4 - 10 core-hours | 20 - 50 core-hours | Capture requires more time for alignment, BQSR, and complex variant calling. |
| Peak RAM Usage | 8 - 16 GB | 16 - 32 GB | BQSR and some CNV callers in capture pipelines are memory-intensive. |
| Critical Intensive Step | Duplicate Marking | Alignment & BQSR | Amplicon duplicates are a key artifact; Capture requires robust sequence analysis. |
| Pipeline Complexity | Low to Moderate | High | Capture pipelines have more steps and optional analyses (e.g., CNV). |
The Scientist's Toolkit: Key Research Reagent & Software Solutions
Table 2: Essential Materials and Tools for NGS Analysis
| Item | Function | Example Product/Software |
|---|---|---|
| Library Prep Kit | Converts nucleic acid sample into sequencing-ready library. | Illumina DNA Prep, KAPA HyperPlus, Twist Human Core Exome |
| Hybridization Capture Probes | Biotinylated oligonucleotides to enrich specific genomic regions. | IDT xGen Panels, Roche NimbleGen SeqCap, Twist Target Panels |
| Amplicon Panel Primers | PCR primers designed to tile across target regions. | Illumina AmpliSeq, Thermo Fisher Scientific Oncomine |
| Sequence Analysis Suite | Integrated toolkit for NGS data processing. | GATK, DRAGEN (Illumina), BWA, samtools |
| Variant Annotation DB | Provides functional, population, and clinical context for variants. | Ensembl VEP, dbSNP, gnomAD, ClinVar |
| High-Performance Compute (HPC) | Infrastructure for running computationally intensive pipelines. | Local cluster (SLURM), Cloud (AWS, GCP), NVIDIA Parabricks |
| Containerization | Ensures pipeline reproducibility and ease of deployment. | Docker, Singularity, Bioconda |
Application Notes
Cancer Genomics: Hybridization Capture for Tumor-Normal Pair Analysis
Hybridization capture, utilizing panels like the MSK-IMPACT or FoundationOne CDx, is the gold standard for somatic variant detection in cancer. It enables broad genomic profiling from formalin-fixed, paraffin-embedded (FFPE) samples, detecting single nucleotide variants (SNVs), insertions/deletions (Indels), copy number alterations (CNAs), and structural variants (SVs) across hundreds of cancer-related genes. Recent studies (2023-2024) highlight its utility in identifying low-frequency variants in heterogeneous tumors and its critical role in guiding matched targeted therapies.
Key Data from Recent Studies (2023-2024): Table 1: Performance Metrics of Hybridization Capture in Cancer Studies
| Metric | Typical Performance (Large Panels, >300 genes) | Key Challenge |
|---|---|---|
| Sensitivity for SNVs (at 5% VAF) | >99% | Input DNA quality/quantity from FFPE |
| Specificity | >99.9% | Off-target sequencing & background noise |
| Uniformity of Coverage | ~90% of targets within 0.2-5x mean | High GC-rich regions |
| Input DNA Requirement | 50-200 ng | Degraded samples require more input |
| Turnaround Time (Wet-lab to Data) | 5-7 days | Complex bioinformatics for SV/CNV |
Infectious Disease: Amplicon Sequencing for Pathogen Surveillance and Outbreak Investigation
Amplicon-based NGS (e.g., ARTIC Network protocol for SARS-CoV-2) is dominant for pathogen genomic surveillance due to its high sensitivity on low-viral-load samples and resilience to host nucleic acid contamination. It is the method of choice for tracking transmission chains, detecting emerging variants, and diagnosing polymicrobial infections via 16S/ITS rRNA sequencing. Recent applications include rapid characterization of mpox virus outbreaks and antimicrobial resistance gene profiling in bacterial pathogens.
Key Data from Recent Studies (2023-2024): Table 2: Performance Metrics of Amplicon Sequencing in Infectious Disease
| Metric | Typical Performance (Viral/Bacterial Targets) | Key Challenge |
|---|---|---|
| Sensitivity (Ct value <35) | >95% detection rate | Primer mismatches due to new variants |
| Limit of Detection | 10-100 copies per reaction | Background in polymicrobial samples |
| Specificity | High (with optimized primers) | Amplicon crossover contamination |
| Throughput | High (100s-1000s of samples per run) | Barcode assignment errors |
| Turnaround Time (Wet-lab to Data) | 1-3 days | Manual steps in library prep |
Inherited Disorders: Hybridization Capture for Comprehensive Germline Testing
For inherited disorders, hybridization capture using large clinical exome (e.g., Invitae Comprehensive Exome) or whole-genome panels is preferred due to its comprehensive coverage, accurate CNV calling, and ability to detect variants in non-coding regions associated with disease. It is essential for diagnosing heterogeneous conditions like hereditary cancer syndromes, cardiomyopathies, and neurodevelopmental disorders. Recent trends emphasize the integration of RNA-seq capture to assess splicing variants.
Key Data from Recent Studies (2023-2024): Table 3: Performance Metrics in Inherited Disorder Research
| Metric | Hybridization Capture (Clinical Exome) | Amplicon (Focused Panel, <50 genes) |
|---|---|---|
| Diagnostic Yield | 25-40% (broad phenotypes) | 30-50% (specific phenotypes) |
| CNV Detection Accuracy | High (via depth-based analysis) | Limited/Poor |
| Sequence Homology Handling | Good (with careful bait design) | Problematic in pseudogene-rich regions |
| Ability to Add Genes | Flexible (without redesign) | Requires full redesign |
| Cost per Sample | Moderate-High | Low |
Experimental Protocols
Protocol: Hybridization Capture for Somatic Variant Detection in FFPE Tumor Samples
Title: Hybridization Capture-Based Library Preparation from FFPE DNA for Cancer Panel Sequencing.
Key Reagent Solutions:
- FFPE DNA Extraction Kit (e.g., QIAamp DNA FFPE Tissue Kit): Isolates DNA from cross-linked, fragmented tissue.
- Library Prep Kit (e.g., KAPA HyperPrep): Creates sequencing-compatible libraries with unique dual indices (UDIs) to minimize index hopping.
- Hybridization Cocktail & Bait Set (e.g., xGen Hybridization Capture Kit, MSK-IMPACT bait library): Contains biotinylated DNA or RNA baits targeting specific genomic regions.
- Streptavidin Magnetic Beads (e.g., Dynabeads MyOne Streptavidin T1): Binds biotinylated baits to capture target DNA-library hybrids.
- Target Enrichment Bead-Based Cleanup Kit (e.g., AMPure XP Beads): For size selection and purification of captured libraries.
Methodology:
- DNA Shearing & Quality Control: Fragment 50-200 ng of FFPE-derived DNA to ~200bp via sonication. Assess fragment size distribution using a Bioanalyzer/TapeStation (DV200 > 30% recommended).
- Library Construction: Perform end-repair, A-tailing, and ligation of UDI adapters using the library prep kit. Clean up with AMPure XP beads.
- Library Amplification: Perform 6-10 cycles of PCR to amplify the adapter-ligated DNA. Clean up with AMPure XP beads. Quantify by qPCR.
- Hybridization: Pool up to 8 libraries (500 ng total). Denature and hybridize with the biotinylated bait library at 65°C for 16-24 hours in a thermal cycler.
- Capture & Wash: Bind hybridization mix to streptavidin beads. Wash stringently (e.g., 65°C) to remove non-specifically bound DNA.
- Post-Capture Amplification: Elute captured DNA from beads. Perform 12-14 cycles of PCR to generate the final sequencing library. Clean up with AMPure XP beads.
- Sequencing: Quantify by qPCR and sequence on an Illumina platform (e.g., NovaSeq 6000) with paired-end 2x150 bp reads to a mean coverage of >500x for tumor and matched normal.
Protocol: Amplicon Sequencing for Viral Genome Surveillance
Title: Multiplex PCR Amplicon Sequencing for Viral Pathogens (e.g., SARS-CoV-2).
Key Reagent Solutions:
- Viral RNA Extraction Kit (e.g., MagMAX Viral/Pathogen Kit): Purifies RNA from swab/media with magnetic bead technology.
- Reverse Transcription Master Mix (e.g., SuperScript IV): Converts viral RNA to cDNA.
- Multiplex PCR Primer Pools (e.g., ARTIC Network V4.1 primer set): Tiled amplicon primers spanning the viral genome.
- High-Fidelity DNA Polymerase (e.g., Q5 Hot Start): For accurate amplification with minimal errors.
- Library Prep Kit for Amplicons (e.g., Illumina DNA Prep): Attaches sequencing adapters and indices to pooled amplicons.
Methodology:
- RNA to cDNA: Extract RNA. Perform reverse transcription on 5-10 µL of RNA using random hexamers or gene-specific primers.
- Multiplex PCR Set-up: Set up two separate multiplex PCR reactions (primer pools A & B) using 2.5 µL of cDNA, primer pools, and high-fidelity polymerase. Cycle conditions: 98°C for 30s; 35 cycles of (98°C for 15s, 63°C for 5 min); 72°C for 2 min.
- Amplicon Pooling & Cleanup: Combine PCR products from pools A and B. Purify using a 1x ratio of AMPure XP beads to remove primers and non-specific products.
- Library Preparation & Indexing: Quantify purified amplicons. Use 50 ng as input into a tagmentation-based library prep kit (e.g., Illumina DNA Prep) following manufacturer's instructions. Perform a limited-cycle (5-8 cycles) index PCR.
- Library Cleanup & Validation: Clean up libraries with AMPure XP beads (0.9x ratio). Assess library size (~350-450bp) and concentration via TapeStation and qPCR.
- Sequencing: Pool libraries and sequence on an Illumina MiSeq or NextSeq (2x150 bp) to achieve >1000x mean coverage.
Visualization Diagrams
Title: Hybridization Capture Workflow for Cancer Genomics
Title: Amplicon Sequencing Workflow for Pathogen Detection
Title: Case Study Mapping to NGS Method Comparison Thesis
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions
| Item | Function | Example Product |
|---|---|---|
| Biotinylated Capture Baits | Single-stranded DNA/RNA probes that hybridize to target genomic regions for enrichment. | IDT xGen Lockdown Probes |
| High-Fidelity DNA Polymerase | PCR enzyme with low error rate, critical for accurate variant calling. | NEB Q5 Hot Start |
| Unique Dual Index (UDI) Kits | Provides unique combinatorial barcodes for each sample to prevent index hopping. | Illumina IDT for Illumina UDIs |
| Magnetic Beads (SPRI) | Size-selective purification of nucleic acids (e.g., fragment selection, cleanup). | Beckman Coulter AMPure XP |
| FFPE DNA Repair Mix | Enzyme cocktail to fix deamination (C>T artifacts) and fragmentation in FFPE DNA. | NEB FFPE DNA Repair Mix |
| Hybridization Buffer & Enhancers | Optimizes hybridization kinetics and specificity during capture. | Roche NimbleGen SeqCap EZ |
| Multiplex PCR Primer Panels | Pre-designed, tiled primer sets for comprehensive pathogen or gene panel coverage. | ARTIC Network Primers |
| Streptavidin Magnetic Beads | Binds biotinylated DNA-bait complexes for magnetic separation. | Thermo Fisher Dynabeads MyOne Streptavidin T1 |
Optimizing Performance: Troubleshooting Common Pitfalls in Targeted NGS
This application note details critical protocols for mitigating errors inherent to amplicon-based NGS workflows. Within the broader thesis comparing amplicon-based and hybridization capture methods, understanding these artifacts is essential for accurate interpretation of amplicon data. While amplicon sequencing offers high depth and low input requirements, it is uniquely susceptible to PCR-derived errors and biases that can compromise variant calling fidelity, especially in low-frequency and heterozygote detection scenarios relevant to cancer genomics and pathogen detection.
Table 1: Common Amplicon-Specific Errors and Their Estimated Frequencies
| Error Type | Primary Cause | Typical Frequency Range | Impact on Variant Calling |
|---|---|---|---|
| Polymerase Misincorporation | Taq polymerase errors during early cycles | 10^-5 to 10^-4 per base per cycle | False positive SNVs, especially at low allele frequency |
| Chimeric Reads (PCR Recombination) | Incomplete extension generating template-switching artifacts | 0.5% to 2.0% of total reads | False structural variants, false haplotype associations |
| PCR Duplicates | Amplification of identical template molecules | Highly variable (10-90%+ of reads) | Inflates coverage metrics, obscures true library complexity |
| Allele Dropout (ADO) | Primer-template mismatch, poor primer design, low input | 1% to 20%+ at heterozygous loci | False homozygosity, loss of heterozygosity (LOH) artifacts |
| Amplification Bias | GC content, secondary structure, primer efficiency | Several-fold coverage difference | Uneven coverage, regions with insufficient depth for calling |
Table 2: Efficacy of Mitigation Strategies on Error Reduction
| Mitigation Strategy | Target Error | Key Metric | Typical Reduction Achieved* |
|---|---|---|---|
| High-Fidelity Polymerase | Misincorporation | Error rate per base | 3- to 10-fold vs. standard Taq |
| Unique Molecular Identifiers (UMIs) | PCR Duplicates & Some Late Errors | Duplicate read fraction | >95% of PCR duplicates removed |
| Limited PCR Cycles | All PCR-derived errors | Final Cycle Number | Linear reduction with fewer cycles |
| Optimized Primer Design | Allele Dropout, Bias | On-target rate, Uniformity | ADO can be reduced to <2% |
| Duplicate Consensus Calling (with UMIs) | Polymerase Errors | Final SNV FDR | Can reduce error rate to ~10^-7 |
*Reduction is highly dependent on specific protocol and sample quality.
Experimental Protocols
Protocol 2.1: UMI-Based Error Correction and Duplicate Removal
Objective: To generate accurate, duplicate-corrected sequencing data from amplicon libraries.
Materials:
- Genomic DNA sample
- UMI-tagged gene-specific primers (Integrated DNA Technologies)
- Q5 Hot Start High-Fidelity 2X Master Mix (New England Biolabs)
- AMPure XP beads (Beckman Coulter)
- Next-generation sequencer (Illumina recommended)
Methodology:
- Primer Design: Design primers with a degenerate UMI (8-12 random bases) at the 5' end, followed by a fixed handle and then the target-specific sequence.
- First PCR (Limited Cycles):
- Set up reaction: 50 ng gDNA, 0.5 µM UMI-primers, 1X Q5 Master Mix.
- Cycling: 98°C 30s; 8-12 cycles of (98°C 10s, 65°C 30s, 72°C 30s); 72°C 2 min.
- Purification: Clean amplicons with 0.8X AMPure XP beads. Elute in 25 µL EB buffer.
- Indexing PCR (Add Illumina Adapters):
- Use 5 µL of purified first PCR product as template.
- Use standard Illumina indexing primers and polymerase.
- Run for 6-8 cycles.
- Purification & Sequencing: Clean final library with 0.9X AMPure XP beads. Quantify, pool, and sequence on an Illumina platform with paired-end reads.
- Bioinformatic Processing (Key Steps):
- UMI Extraction: Identify UMI sequence from read header or first bases.
- Read Grouping: Group reads originating from the same original molecule by mapping position and UMI sequence, allowing for 1-2 errors in the UMI.
- Consensus Building: For each read group, generate a consensus sequence by majority rule or quality-weighted alignment.
- Duplicate Removal: Deduplicate based on UMI-group identity.
Protocol 2.2: Optimized Multiplex PCR to Minimize ADO and Bias
Objective: To achieve uniform coverage and minimize allele dropout in a multi-gene panel.
Materials:
- HS Probes Master Mix (Roche) or similar multiplex-ready enzyme
- Pre-designed, empirically validated primer pools (e.g., from Illumina or Twist Bioscience)
- Qubit dsDNA HS Assay Kit (Thermo Fisher)
Methodology:
- Primer Pool Balancing: Use primer concentrations pre-optimized for balanced amplification. If designing custom panels, use software (e.g., Primer3, Multiplex Manager) and validate empirically.
- Reaction Setup:
- Use 20-50 ng of high-quality gDNA.
- Utilize a polymerase master mix specifically formulated for high multiplexing.
- Keep total primer concentration within vendor specification (typically 0.1-1 µM aggregate).
- Thermocycling with a Touchdown/Ramp Protocol:
- 95°C 5 min.
- Touchdown: 10 cycles of (95°C 30s, 65-57°C (-0.8°C/cycle) 30s, 72°C 1 min).
- Standard: 25 cycles of (95°C 30s, 57°C 30s, 72°C 1 min).
- 72°C 5 min.
- Use a slow ramp rate (e.g., 1°C/sec) between annealing and extension steps.
- Post-PCR Cleanup and QC: Purify with AMPure XP beads (0.8X). Quantify yield with Qubit and assess size distribution via capillary electrophoresis (e.g., Bioanalyzer). Uniform smear indicates balanced amplification.
Visualizations
UMI Error Correction Workflow
Causes and Mitigation of ADO and Bias
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Error-Mitigated Amplicon Sequencing
| Reagent / Material | Vendor Examples | Critical Function in Error Mitigation |
|---|---|---|
| High-Fidelity DNA Polymerase | NEB Q5, Roche High Fidelity, KAPA HiFi | Reduces polymerase misincorporation errors by 3-10x due to 3’→5’ exonuclease proofreading. |
| Unique Molecular Identifier (UMI) Adapters/Primers | IDT Duplex Seq adapters, Swift Biosciences Accel-NGS | Tags each original DNA molecule with a unique barcode to enable bioinformatic error correction and true duplicate removal. |
| Multiplex PCR Optimization Master Mix | Roche Multiplex PCR Kit, Qiagen Multiplex PCR Plus | Specially formulated buffers and enzymes to minimize primer-dimer and imbalance in complex primer pools, reducing ADO. |
| PCR Enhancers (DMSO, Betaine) | Sigma-Aldrich, Thermo Fisher | Reduce amplification bias from GC-rich regions or secondary structure by lowering DNA melting temperature. |
| Solid Phase Reversible Immobilization (SPRI) Beads | Beckman Coulter AMPure XP, MagBio HighPrep PCR | Size-selective cleanup to remove primer dimers and non-specific products that consume sequencing capacity and introduce noise. |
| Liquid Handling Robotics | Hamilton STAR, Beckman Coulter Biomek | Enables precise, reproducible low-volume pipetting essential for consistent multiplex PCR and UMI library prep. |
| Digital PCR System | Bio-Rad QX200, Thermo Fisher QuantStudio | Absolute quantification of input DNA and amplicons to optimize input and cycle number, minimizing over-cycling. |
Application Notes
Within the broader thesis comparing amplicon-based and hybridization-capture NGS methods, this document addresses three persistent challenges in hybridization-capture (HybCap) workflows: off-target binding, uneven coverage, and inefficient capture of high-GC regions. While HybCap excels in variant discovery across large, custom genomic regions, these technical hurdles can compromise data quality, increase sequencing costs, and necessitate careful protocol optimization.
Key Challenges & Quantitative Summary: The following table summarizes core challenges and representative quantitative impacts from recent literature and internal validation.
Table 1: Quantitative Impact of Key Hybrid-Capture Challenges
| Challenge | Typical Metric Impact | Observed Range | Consequence |
|---|---|---|---|
| Off-Target Binding | Off-target rate (fraction of reads) | 20-50% | Reduced on-target efficiency, increased sequencing cost for desired coverage. |
| Coverage Uniformity | Fold-80 penalty (top 20% mean / bottom 20% mean) | 1.5 - 3.5x | Increased sequencing depth required to cover low-coverage regions; risk of missing variants. |
| High GC Content | Relative capture yield (vs. GC-neutral region) | 40-70% drop for >70% GC | Gaps or severe under-coverage in promoters, first exons, and other GC-rich functional elements. |
Protocols
Protocol 1: Optimized Hybridization for Uniformity and Specificity Objective: To maximize on-target specificity and improve coverage uniformity by optimizing hybridization conditions and using custom blocking reagents. Materials: See "Research Reagent Solutions" (Table 2). Procedure:
- Fragmented DNA Preparation: Shear 100-200 ng of genomic DNA to a peak of 150-200 bp using a focused-ultrasonicator. Repair ends, adenylate, and ligate with unique dual-indexed adapters.
- Library Amplification: Amplify library with 6-8 PCR cycles using a high-fidelity polymerase. Clean up with 1.0x SPRSelect beads.
- Hybridization Cocktail Setup (on ice):
- Library DNA: 100-200 ng.
- Custom Blockers (e.g., Cot-1, xGen Universal Blockers): 2-5 µL.
- GC Enhancer Solution (e.g., xGen Hybridization Buffer): 7.5 µL.
- Custom Additive (e.g., PEG 8000): 1.5 µL (final conc. ~6%).
- Biotinylated Capture Probes: 1 µL of relevant panel.
- Nuclease-free water to 15 µL total.
- Denaturation & Hybridization: Denature at 95°C for 10 minutes. Immediately incubate at 58°C for 16-24 hours in a thermal cycler with heated lid (105°C).
- Post-Hybridization Wash & Capture:
- Pre-warm Streptavidin Beads and wash twice with Bead Wash Buffer.
- Add hybridization mix to beads. Incubate at 58°C for 45 min with agitation.
- Perform three stringent washes at 58°C with pre-warmed Stringent Wash Buffer for 5 minutes each.
- Perform two room-temperature washes with Bead Wash Buffer.
- Post-Capture Amplification: Elute captured DNA in low-EDTA TE. Amplify with 12-14 PCR cycles. Clean up with 0.9x SPRSelect beads. Quantify via qPCR.
Protocol 2: Targeted Enrichment of High-GC Regions Objective: To specifically improve the capture efficiency of genomic regions with >70% GC content. Materials: As in Protocol 1, with specific high-GC panel. Procedure:
- Follow Protocol 1, steps 1-2 for library prep.
- Modified Hybridization Setup: Increase the proportion of GC Enhancer Solution to 50% of the total hybridization volume. Maintain the 6% PEG 8000 additive.
- Optimized Thermal Cycling: Use a two-stage hybridization:
- Stage 1: Denature at 98°C for 5 min.
- Stage 2: Hybridize at 65°C for 48 hours. This longer, higher-temperature incubation promotes probe binding to high-GC targets.
- Reduced Stringency Washes: To retain bound high-GC fragments, perform only one stringent wash at 65°C for 10 minutes, followed by two standard room-temperature washes.
- Follow Protocol 1, step 6 for post-capture processing.
Visualizations
Diagram 1: Optimized Hybrid-Capture Workflow (100 chars)
Diagram 2: Challenge Mechanisms & Solutions (94 chars)
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions
| Reagent / Material | Function & Rationale |
|---|---|
| xGen Universal Blockers (IDT) or Cot-1 DNA | Blocks hybridization of probes to repetitive genomic elements, significantly reducing off-target capture. |
| xGen Hybridization Buffer (IDT) or Rapid Hyb Buffer (Cytiva) | Contains chemical agents (e.g., dextran sulfate) that increase probe effective concentration and improve kinetics, especially for high-GC targets. |
| Polyethylene Glycol (PEG) 8000 | Molecular crowding agent that accelerates hybridization, improving overall efficiency and uniformity. |
| Dimethyl Sulfoxide (DMSO) or Betaine | Additives that destabilize DNA secondary structures, facilitating probe access to high-GC regions. |
| Locked Nucleic Acid (LNA) or Super-GT Probes | Modified nucleotide probes with increased melting temperature (Tm), enhancing binding to challenging, high-GC targets. |
| Streptavidin Magnetic Beads (MyOne C1) | High-binding-capacity beads for efficient capture of biotinylated probe-target complexes. |
| High-Fidelity PCR Master Mix (e.g., KAPA HiFi) | Essential for minimal-bias amplification of pre- and post-capture libraries, preserving representation. |
| SPRSelect / AMPure XP Beads | For consistent size selection and cleanup, critical for removing adapter dimers and excess primers. |
This application note details wet-lab optimizations for next-generation sequencing (NGS) library preparation, framed within a broader thesis comparing amplicon-based and hybridization capture methods. The primary focus is on protocol modifications that enhance sensitivity (detection of true positives) and specificity (reduction of false positives) for both methodologies, crucial for applications in oncology, infectious disease, and inherited genetic disorder research.
Table 1: Impact of Protocol Modifications on Performance Metrics
| Optimization Parameter | Amplicon-Based Method (Typical Improvement) | Hybridization Capture (Typical Improvement) | Key Modification |
|---|---|---|---|
| Input DNA/RNA Quantity | Sensitivity ↓ below 10 ng; Specificity ↓ due to duplicates | Sensitivity ↓ below 50 ng; Specificity stable | Use of duplicate molecular tags (UMIs) and PCR/preamplification cycles adjustment. |
| Enzymatic Master Mix | Sensitivity: +5-15%; Specificity: +3-10% (Hot-start polymers) | Sensitivity: +2-8%; Specificity: marginal | Switch to high-fidelity, hot-start polymerase for amplicon; optimized ligase for capture. |
| Hybridization Temperature & Time | Not Applicable | Sensitivity: +10-25%; Specificity: +5-15% | Incremental temperature optimization (+/- 5°C from standard) and time (4-24 hr). |
| Post-Capture Wash Stringency | Not Applicable | Sensitivity: -5% (if too high); Specificity: +20% | Increase wash temperature (e.g., +2-5°C) or add formamide (e.g., 10%). |
| PCR Cycle Number | Sensitivity: +; Specificity: - (if >20 cycles, duplicates ↑) | Sensitivity: +; Specificity: - (if >12 cycles) | Minimize cycles: Amplicon: 14-18; Capture post-enrichment: 8-12. |
| UMI Incorporation & Deduplication | Sensitivity: Neutral; Specificity: +15-30% | Sensitivity: Neutral; Specificity: +10-25% | Integration of UMIs in initial PCR/ligation and bioinformatic collapsing. |
| Blocking Reagent Optimization | Sensitivity: +5% (reduce primer-dimer); Specificity: +8% | Sensitivity: +20% (reduce off-target); Specificity: +25% | Use of cot DNA, specific blockers (e.g., IDT xGen), RNAse A for rRNA. |
Table 2: Comparative Performance Post-Optimization
| Metric | Optimized Amplicon-Based | Optimized Hybridization Capture | Assay Context |
|---|---|---|---|
| Sensitivity (at 0.5% VAF) | 95-99% | 97-99.5% | Somatic SNV detection. |
| Specificity | 99.8% | 99.9% | Somatic SNV detection. |
| Uniformity of Coverage | >98% (targeted) | 90-95% (across megabase panels) | On-target reads. |
| GC-Bias | Low (short amplicons) | Moderate-High (requires optimization) | Coverage in GC-rich regions. |
| Input DNA Flexibility | 1-50 ng (highly flexible) | 50-200 ng (optimal) | FFPE and low-input applications. |
| Hands-on Time | Low (single-tube PCR) | High (multiple steps) | Protocol workflow. |
Detailed Experimental Protocols
Protocol 1: Optimized Amplicon-Based NGS for Low-Input FFPE DNA
Objective: Maximize sensitivity and specificity for a 50-gene hotspot panel from 10 ng of FFPE DNA. Materials: See "Research Reagent Solutions" table. Procedure:
- DNA Repair: Treat 10 ng of FFPE DNA with 1X DNA Damage Repair Mix (e.g., NEB FFPE Repair Mix) for 20 minutes at 20°C. Purify using 1.8X SPRI beads.
- Initial Tagmentation (Optional): For some panels, use a controlled tagmentation step (e.g., Illumina Nextera) to fragment DNA, followed by purification.
- UID/UMI Labeling & Target Amplification:
- Prepare PCR mix: 1X High-Fidelity Hot-Start Polymerase Master Mix, 500 nM forward and reverse UID-containing panel-specific primers, 2 µL of repaired DNA.
- Thermocycler: 98°C for 2 min; 14 cycles of [98°C for 30 sec, 60°C for 30 sec, 72°C for 1 min]; 72°C for 5 min.
- Indexing PCR: Clean up PCR 1 product with 0.9X SPRI beads. Perform a second, limited-cycle (6-8 cycles) PCR to add full Illumina adapter indices and sequencing primers.
- Final Clean-up & QC: Purify with 0.9X SPRI beads. Quantify by qPCR (e.g., Kapa Library Quant) and check fragment size (e.g., Bioanalyzer). Pool and sequence.
Protocol 2: Optimized Hybridization Capture for a 2 Mb Comprehensive Cancer Panel
Objective: Achieve high uniformity and specificity for a large panel from 100 ng of genomic DNA. Materials: See "Research Reagent Solutions" table. Procedure:
- Library Preparation: Fragment 100 ng gDNA (e.g., Covaris shearing to ~250 bp). Perform end-repair, A-tailing, and ligation of UMI adapters (e.g., IDT Duplex Sequencing adapters) using a high-efficiency ligase. Clean up with 0.9X SPRI beads.
- Pre-Capture PCR: Amplify library with 6-8 cycles using a high-fidelity polymerase. Clean up with 0.9X SPRI beads. Quantify accurately.
- Hybridization:
- Mix 250 ng of pre-capture library with 5 µg of specific blocking oligos (e.g., IDT xGen Universal Blockers), 1 µg of Cot-1 DNA, and 500 ng of biotinylated capture probes (panel-specific) in 1X hybridization buffer.
- Denature at 95°C for 10 min, then hybridize at 65°C for 16-20 hours in a thermocycler with heated lid.
- Post-Capture Washing & Amplification:
- Bind to streptavidin beads (e.g., MyOne C1). Wash sequentially with: a) High Stringency Wash Buffer I at room temp, b) Pre-warmed High Stringency Wash Buffer II at 67°C (key step for specificity), twice.
- Perform on-bead post-capture PCR (10 cycles) to amplify enriched libraries.
- Final Clean-up & QC: Purify, quantify by qPCR, and assess size distribution. Pool at equimolar ratios for sequencing.
Visualizations
Diagram 1: Key Decision Points for Method Selection
Title: Workflow for NGS Method Selection
Diagram 2: Shared vs. Distinct Optimization Levers
Title: Optimization Levers for NGS Methods
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Protocol Optimization
| Item | Function & Role in Optimization | Example Product(s) |
|---|---|---|
| High-Fidelity Hot-Start Polymerase | Reduces PCR errors and primer-dimer formation, improving specificity. Essential for both methods. | NEB Q5 Ultra II, Kapa HiFi HotStart, Takara PrimeStar GXL. |
| Duplex UMI Adapters | Enables accurate error correction and removal of PCR/sequencing duplicates by tagging original DNA molecules, drastically improving specificity. | IDT Duplex Sequencing Adapters, Twist UMI Adapters. |
| Hybridization Blockers | Block repetitive sequences (e.g., Alu, LINE) and adapter-adapter interactions during capture, increasing on-target rate and specificity. | IDT xGen Universal Blockers, Roche NimbleGen SeqCap HE Universal Oligo. |
| Biotinylated Capture Probes | Target-specific oligonucleotides to enrich genomic regions of interest. Pool design and tiling density impact sensitivity/coverage uniformity. | IDT xGen Lockdown Probes, Twist Target Enrichment Probes. |
| Solid-Phase Reversible Immobilization (SPRI) Beads | For size selection and clean-up. Ratios (e.g., 0.9X vs 1.8X) are critical for removing primer dimers and optimizing library size distribution. | Beckman Coulter AMPure XP, Kapa Pure Beads. |
| Strand-Displacing Polymerase for RCA | Used in some amplicon approaches (e.g., AmpliSeq) to improve uniformity from low-input samples. | Phi29 Polymerase. |
| Formamide or SSC-Based Wash Buffers | Increase stringency of post-capture washes, removing non-specifically bound fragments to improve specificity. | Included in Agilent SureSelect, Roche NimbleGen kits. |
| DNA/RNA Damage Repair Mix | Repairs nicks, deamination (FFPE artifacts), and breaks in degraded samples, recovering sensitivity. | NEB FFPE DNA Repair Mix, NEBNext FFPE RNA Repair Mix. |
Bioinformatic Filtering Strategies to Remove Technical Noise and Artifacts
Within the broader context of comparing amplicon-based and hybridization-capture Next-Generation Sequencing (NGS) methods, the mitigation of technical noise and artifacts is paramount. Both approaches are susceptible to distinct, methodology-specific artifacts alongside common sequencing errors. Effective bioinformatic filtering is essential to ensure the accuracy of variant calling, taxonomic assignment, and all downstream analyses. This document provides application notes and protocols for noise/artifact identification and removal, tailored to the strengths and weaknesses of each NGS library preparation technique.
The optimal filtering strategy is informed by the underlying technology.
Table 1: Primary Technical Artifacts by NGS Method
| Artifact/Source | Amplicon-Based Sequencing | Hybridization Capture Sequencing |
|---|---|---|
| PCR Errors | High. Duplicate reads from PCR amplification dominate. Early-cycle errors are propagated. | Moderate. Duplicates occur but are less frequent relative to unique fragments. |
| Cross-Contamination/Index Hopping | High risk due to similar amplicon sizes. Barcode swapping generates false positives. | Moderate risk. Heterogeneous fragment sizes reduce swap impact. |
| Sequencing Errors | Present in all technologies (Illumina, Ion Torrent, etc.). | Present in all technologies. |
| Mapping/Alignment Bias | Lower complexity; easier alignment but prone to primer-dimers mapping. | High complexity; challenging alignment in repetitive regions, leading to false calls. |
| Method-Specific Noise | Chimeras, primer-specific bias, heterogeneous amplification efficiency. | Off-target capture, low on-target efficiency, non-uniform coverage. |
Core Filtering Strategies & Protocols
Protocol 1: Removal of PCR Duplicates
Objective: To eliminate reads originating from the same PCR template molecule, preserving only unique starting fragments.
Materials & Workflow:
- Input: Aligned sequencing reads (BAM/SAM file).
- Tool Selection: Use
picard MarkDuplicates(broadly applicable) orsamtools rmdup(faster, for paired-end). - Execution (Picard):
- Critical Parameter for Capture Data: Set
BARCODE_TAGoption for duplex Unique Molecular Identifier (UMI)-based protocols to accurately identify pre-PCR molecules. - Output: BAM file with duplicate reads removed and associated metrics.
Note for Amplicon Data: Standard duplicate marking is often ineffective for amplicons due to identical start/end positions. Use UMIs incorporated during reverse transcription or initial PCR is essential.
Protocol 2: Strand Bias and Low-Frequency Variant Filtering
Objective: To remove false positive variants resulting from sequencing artifacts or mapping errors, common in both methods but with different profiles.
Materials & Workflow:
- Input: Raw variant calls (VCF file) from callers like GATK Mutect2, VarScan2, or FreeBayes.
- Tool: GATK FilterMutectCalls or bcftools filter.
- Execution (GATK):
- Key Filters to Apply (Customize in bcftools):
- Depth (
DP): Minimum total depth (e.g., DP > 10). - Strand Bias (
FSor SP): Fisher’s Exact Test for strand bias (e.g., FS < 20). - Allele Frequency (
AF): Minimum alternate allele fraction (e.g., AF > 0.01 for capture; may be higher for amplicon). - Mapping Quality (
MQ): Minimum median mapping quality (e.g., MQ > 40).
- Depth (
Table 2: Suggested Initial Filtering Thresholds by Method
| Filter | Amplicon-Based (e.g., Tumor) | Hybridization Capture (e.g., cfDNA) |
|---|---|---|
| Minimum Depth (DP) | > 100 | > 50 |
| Alternate Allele Count | ≥ 3 | ≥ 5 |
| Strand Bias (FS) | < 40 | < 30 |
| Allele Frequency | > 0.005 | > 0.002 |
Protocol 3: Contamination and Cross-Contamination Management
Objective: To identify and filter out reads arising from sample-to-sample contamination (index hopping) or environmental sources.
Materials & Workflow:
- Prevention: Use unique dual indices (UDIs) and enzymatic solutions to repair index hopping.
- Detection (Microbiome/Taxonomic):
- Tool:
decontam(R package) orKraken2with bracken. - Input: ASV/OTU table and negative control samples.
- Method: Apply the "prevalence" method to identify taxa more abundant in negative controls than true samples.
- Tool:
- Detection (Human Genomics):
- Tool:
VerifyBamID2orContamination.py(GATK). - Method: Estimates cross-sample contamination by comparing allele frequencies to a known population dataset.
- Tool:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents & Tools for Artifact Mitigation
| Item | Function/Benefit | Method Relevance |
|---|---|---|
| Unique Dual Indices (UDIs) | Minimizes index hopping by ensuring each sample pair is unique. Critical for multiplexed amplicon panels. | Both (Critical for Amplicon) |
| UMI Adapters (Duplex) | Allows bioinformatic consensus calling to remove PCR and sequencing errors. Gold standard for low-frequency variant detection. | Both (Critical for Capture liquid biopsy) |
| Hybridization Capture Blockers (e.g., Cot-1 DNA, xGen) | Suppresses off-target capture of repetitive elements, improving on-target efficiency and uniformity. | Hybridization Capture Only |
| High-Fidelity DNA Polymerase (e.g., Q5, KAPA HiFi) | Reduces PCR-induced nucleotide substitution errors during library amplification. | Both |
| PCR Clean-up Beads (SPRI) | Removes primer dimers and size-selects fragments, crucial for clean amplicon libraries. | Both (Critical for Amplicon) |
Visualization of Filtering Workflows
Filtering Workflow for NGS Methods
Key Sources of Technical Noise
Achieving Optimal Sequencing Depth and Coverage for Variant Calling in Each Method
Within a comparative thesis on amplicon-based versus hybridization capture Next-Generation Sequencing (NGS) methods, achieving optimal sequencing depth and coverage is paramount for accurate and reliable variant calling. This protocol details the experimental and bioinformatic strategies necessary to determine and achieve these critical parameters for each method, ensuring robust detection of single nucleotide variants (SNVs), insertions, and deletions (Indels).
Key Definitions and Targets
Sequencing Depth (Coverage): The average number of reads that align to a specific genomic region. Coverage Uniformity: The evenness of read distribution across targeted regions. Variant Calling Sensitivity: The probability of detecting a true variant.
Optimal targets differ by method and application:
Table 1: Recommended Sequencing Parameters for Variant Calling
| Parameter | Amplicon-Based (Germline) | Amplicon-Based (Low-Frequency Variant) | Hybridization Capture (Germline) | Hybridization Capture (Somatic) |
|---|---|---|---|---|
| Minimum Mean Depth | 100x | 500 - 5,000x | 100x | 200-300x |
| Target Depth for >95% Sensitivity | 150x | 1,000x (for 1% allele frequency) | 150x | 300x |
| Acceptable Coverage Uniformity | >90% bases at >0.2x mean depth | >90% bases at >0.5x mean depth | >80% bases at >0.2x mean depth | >80% bases at >0.2x mean depth |
| Typical Duplicate Rate | High (PCR-derived) | Very High | Moderate (can be reduced with UMIs) | Moderate (UMIs recommended) |
Experimental Protocols
Protocol 1: Determining Optimal Depth via Downsampling Experiment
Objective: Empirically determine the depth required for variant calling saturation in your specific assay.
Materials:
- High-depth sequencing data from a well-characterized control sample (e.g., Genome in a Bottle GIAB, Horizon Multiplex I cfDNA Reference Standard).
- Bioinformatics workstation with SAMtools, BEDTools, GATK, or other variant caller.
Procedure:
- Generate High-Depth Data: Sequence your control sample to a very high depth (>500x for capture, >5,000x for amplicon) using your standard library prep method.
- Downsampling: Use
samtools view -sor Picard'sDownsampleSamto create subsets of your aligned BAM file at incremental depths (e.g., 50x, 100x, 150x, 200x, 300x, 500x). - Variant Calling: Call variants on each downsampled BAM using your standard pipeline (e.g., GATK HaplotypeCaller for germline, Mutect2 for somatic).
- Sensitivity Analysis: Compare variants called at each downsampled depth to the "truth set" from the high-depth data or known reference standard. Plot sensitivity (True Positives / (True Positives + False Negatives)) against depth.
- Determine Saturation Point: Identify the depth at which the increase in sensitivity plateaus (<2% increase per 50x depth increment). This is your optimal depth for that sample and method.
Protocol 2: Assessing and Optimizing Coverage Uniformity for Hybridization Capture
Objective: Evaluate and improve the evenness of coverage across the target bed file.
Materials:
- Hybridization capture kit (e.g., IDT xGen, Roche NimbleGen, Agilent SureSelect).
- Appropriate blocking agents (e.g., Cot Human DNA, IDT xGen Universal Blockers).
- Bioanalyzer/TapeStation for QC.
Procedure:
- Library Preparation: Prepare sequencing library following manufacturer's protocol with sheared gDNA (150-300bp insert size).
- Capture Reaction:
- Use 500-1000ng of pre-amplified library.
- Critical Step: Include recommended amounts of vendor-specific universal and/or index-specific blocking oligos to prevent adapter capture.
- For difficult/gene-rich regions, consider adding a custom booster probe pool.
- Perform hybridization at 65°C for 16-24 hours.
- Post-Capture PCR: Use minimal PCR cycles (typically 8-12) to amplify captured libraries. Over-amplification increases duplicates and skews uniformity.
- Data Analysis:
- Align reads to reference genome (e.g., using BWA-MEM).
- Calculate coverage metrics using
picard CalculateHsMetricsormosdepth. - Key metric:
PCT_TARGET_BASES_20X(percentage of target bases covered at ≥20x). Aim for >90%.
- Troubleshooting Poor Uniformity:
- If uniformity is low, titrate probe-to-input library ratio. Excess probe can increase off-target binding.
- Increase hybridization time to 24 hours for more equilibrium binding.
- Re-design probes for consistently low-coverage regions.
Protocol 3: Minimizing PCR Artifacts and Duplicates in Amplicon Sequencing
Objective: Generate highly uniform amplicon data with minimal false positives from PCR errors.
Materials:
- High-fidelity, low-bias DNA polymerase (e.g., Q5, KAPA HiFi).
- Unique Molecular Identifiers (UMI) adapters.
- Two-step amplification protocol: target amplification + index addition.
Procedure:
- Primer Design: Design amplicons to be 150-250bp. Use software (e.g., Primer3) to check for secondary structures and ensure uniform Tm (±1°C).
- Multiplex PCR Optimization:
- Perform a primer concentration titration (50nM-500nM) to balance amplicon yield.
- Use a touchdown PCR program (e.g., start annealing at 65°C, decrease by 0.5°C/cycle for 10 cycles, then 15 cycles at constant 60°C) to improve specificity.
- Limit total PCR cycles to ≤25 whenever possible.
- UMI Integration (Critical for Low-Frequency Variants):
- In the first PCR, use primers containing a random UMI sequence (8-12bp).
- Purify amplicons and perform a second, limited-cycle (4-8 cycles) PCR to add full sequencing adapters and indices.
- Bioinformatic Duplicate Collapsing:
- Use tools like
fgbioorUMI-toolsto group reads originating from the same original molecule by their UMI and alignment position. - Generate a consensus read for each group to eliminate PCR errors and define a single, accurate count for each molecule.
- Use tools like
- Variant Calling: Call variants from the UMI-collapsed BAM file using a caller sensitive to low-frequency variants (e.g.,
VarScan2,LoFreq). Set minimum supporting reads based on UMI counts, not raw reads.
Visualization of Workflows
Title: NGS Variant Calling Workflow Comparison
Title: Optimal Depth Determination Experiment
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions
| Item | Function | Example Brands/Products |
|---|---|---|
| High-Fidelity DNA Polymerase | Reduces PCR errors during library amplification, critical for accurate variant calling. | NEB Q5, KAPA HiFi, Takara PrimeSTAR GXL |
| Unique Molecular Identifier (UMI) Adapters | Enables bioinformatic distinction of PCR duplicates from original molecules, essential for low-frequency variant detection. | IDT Duplex Seq, Twist UMI, Swift Biosciences Accel-NGS |
| Hybridization Capture Probes | Biotinylated oligos to enrich genomic regions of interest from a fragmented library. | IDT xGen, Roche NimbleGen SeqCap, Agilent SureSelect |
| Universal Blocking Oligos | Block adapter sequences during capture to prevent off-target enrichment and improve uniformity. | IDT xGen Universal Blockers |
| Methylated or Non-Methylated Cot DNA | Blocks repetitive genomic sequences during hybridization to improve on-target specificity. | Thermo Fisher, Invitrogen |
| Quantitative QC Kits | Accurately measure library concentration and size distribution pre-sequencing. | Agilent Bioanalyzer/TapeStation, KAPA Library Quantification Kits |
| Benchmark Reference Standards | Provide known variant positions to validate assay sensitivity, specificity, and limit of detection. | Genome in a Bottle (GIAB), Horizon Discovery, Seracare |
| Bioinformatics Pipelines | Integrated toolkits for alignment, duplicate handling, variant calling, and filtering. | GATK, Sentieon, DRAGEN, BCFtools |
Application Notes
Within a comprehensive thesis comparing amplicon-based and hybridization capture Next-Generation Sequencing (NGS) methods, optimizing the cost-benefit equation is paramount. This analysis moves beyond simple per-sample reagent costs to include hands-on time (a critical labor cost) and sequencing efficiency (data yield and quality). The optimal choice is context-dependent, varying with project scale, sample number, genomic target size, and available laboratory automation.
Key Trade-offs:
- Amplicon-Based (e.g., Multiplex PCR): Lower reagent costs and minimal hands-on time for small targets (< 500 kb). However, amplification bias and high duplication rates reduce sequencing efficiency for larger targets or high-throughput screens.
- Hybridization Capture (e.g., Probe-based): Higher upfront reagent costs and longer, more complex hands-on protocols. Superior for sequencing efficiency across large, contiguous targets (e.g., whole exomes, large gene panels > 500 kb), providing more uniform coverage and higher on-target rates.
Quantitative Comparison Summary:
Table 1: Comparative Metrics for Amplicon vs. Hybridization Capture (Typical Range for 100-200 Target Genes)
| Metric | Amplicon-Based (Multiplex PCR) | Hybridization Capture | Notes |
|---|---|---|---|
| Reagent Cost per Sample | $20 - $80 | $80 - $250 | Highly dependent on vendor, panel size, and sample multiplexing. |
| Total Hands-On Time | 4 - 8 hours | 12 - 24 hours (over 2-3 days) | Capture includes library prep and hybridization/wash steps. |
| Typical On-Target Rate | > 90% | 50% - 80% | Amplicon inherently targeted; capture efficiency depends on probe design. |
| Coverage Uniformity | Low (Prone to Dropouts) | High | Amplification bias vs. uniform probe hybridization. |
| Input DNA Requirement | Low (1-10 ng) | Moderate to High (50-200 ng) | Capture is less efficient with low-input samples. |
| Best Suited For | Small panels, pathogen detection, low-input samples, high-sample-count screens. | Large panels, whole exome, contiguous genomic regions, requiring uniform coverage. |
Table 2: Cost-Benefit Decision Matrix
| Project Parameter | Recommended Method | Primary Rationale |
|---|---|---|
| Target Size < 500 kb, Sample # > 1000 | Amplicon | Lower per-sample cost & hands-on time dominate; sequencing efficiency less critical. |
| Target Size > 500 kb, Sample # < 100 | Hybridization Capture | Higher sequencing efficiency and coverage uniformity justify upfront cost. |
| Limited Budget, Moderate Target Size | Amplicon | Minimizes reagent expenditure. |
| Limited Lab Personnel Time | Amplicon | Significantly lower hands-on time. |
| Requirement for High Coverage Uniformity | Hybridization Capture | Avoids amplification bias and dropouts. |
| Low-Quality/FFPE or Low-Input DNA | Amplicon (with specific kits) | More robust to degraded/fragmented DNA. |
Experimental Protocols
Protocol 1: High-Throughput Amplicon-Based NGS Library Preparation Objective: Generate indexed NGS libraries from 96 genomic DNA samples for a 50-gene panel. Materials: See The Scientist's Toolkit below. Procedure:
- DNA Normalization: Dilute all gDNA samples to 5 ng/µL in a 96-well plate.
- Multiplex PCR Amplification:
- Prepare a master mix containing: 1X PCR buffer, 3.5 mM MgCl₂, 200 µM dNTPs, 0.5 µM pooled primer mix, 0.05 U/µL DNA polymerase.
- Aliquot 9 µL of master mix into each well of a 96-well PCR plate.
- Add 1 µL (5 ng) of normalized gDNA per well.
- Thermal cycle: 95°C for 5 min; [95°C for 30 sec, 60°C for 30 sec, 72°C for 60 sec] x 35 cycles; 72°C for 5 min. Hold at 4°C.
- PCR Clean-up: Using a magnetic bead-based clean-up system, purify the amplicons. Elute in 20 µL of nuclease-free water.
- Indexing PCR (Attach Indices and Full Adapters):
- Prepare a master mix containing: 1X PCR buffer, 200 µM dNTPs, 0.5 µM unique dual index primers, 0.05 U/µL DNA polymerase.
- Combine 5 µL of purified amplicon with 15 µL of indexing master mix.
- Thermal cycle: 95°C for 3 min; [95°C for 30 sec, 55°C for 30 sec, 72°C for 60 sec] x 8 cycles; 72°C for 5 min. Hold at 4°C.
- Final Library Clean-up: Perform a double-sided magnetic bead size selection to remove primer dimers and excess primers. Elute in 30 µL of buffer.
- QC and Pooling: Quantify each library by fluorometry. Normalize concentrations and pool equimolar amounts of all 96 libraries.
- Sequencing: Denature and dilute the final pool for loading onto the sequencer (e.g., Illumina MiSeq/NextSeq).
Protocol 2: Hybridization Capture for Exome Sequencing Objective: Prepare indexed libraries from 24 samples and enrich for the human exome. Materials: See The Scientist's Toolkit below. Procedure:
- Shearing & Library Prep: Using a mechanical shearing system (e.g., Covaris), fragment 100 ng of each gDNA sample to a target size of 200-250 bp. Perform end-repair, A-tailing, and adapter ligation using a commercial library preparation kit. Include unique dual indices.
- Post-Ligation PCR Amplification: Amplify libraries with 6-8 cycles of PCR. Purify using magnetic beads.
- Library QC and Normalization: Quantify libraries by fluorometry. Check size distribution by capillary electrophoresis. Pool 200-500 ng of each library equimolarly into a single tube. Dry the pool in a vacuum concentrator.
- Hybridization: Resuspend the dried pool in hybridization buffer containing blocking oligonucleotides (to suppress adapter-adapter interactions). Add the biotinylated exome probe library. Denature at 95°C for 5-10 minutes and then incubate at 65°C for 16-24 hours.
- Capture with Streptavidin Beads:
- Pre-wash streptavidin-coated magnetic beads.
- Add the bead slurry to the hybridization reaction and incubate at 65°C for 45 minutes with gentle mixing.
- Place the tube on a magnet, discard supernatant.
- Stringency Washes: Perform a series of washes at 65°C (with agitation) using increasingly stringent buffers (e.g., low salt to high salt/SDS buffers) to remove non-specifically bound DNA.
- Elution and Post-Capture PCR: Resuspend beads in nuclease-free water. Elute captured DNA by heating to 95°C for 10 minutes. Immediately transfer the supernatant to a new tube. Amplify the captured library with 10-14 cycles of PCR using universal primers.
- Final Clean-up: Purify the final PCR product with magnetic beads. Quantify and assess size distribution.
- Sequencing: Denature, dilute, and load the final enriched pool onto a sequencer (e.g., Illumina NovaSeq).
Visualizations
Title: Amplicon vs. Capture Method Decision Flow (93 chars)
Title: Hybridization Capture Experimental Workflow (58 chars)
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for NGS Target Enrichment
| Category | Item | Function & Relevance |
|---|---|---|
| Nucleic Acid Handling | Magnetic Beads (SPRI) | Universal clean-up, size selection, and concentration of DNA libraries. Critical for both methods. |
| Low EDTA TE Buffer | Elution and storage buffer; EDTA can inhibit enzymatic steps if too concentrated. | |
| Amplicon-Specific | High-Fidelity, Hot-Start DNA Polymerase | Reduces PCR errors and primer-dimer formation during multiplex amplification. |
| Pooled Primer Panels | Target-specific primers for multiplex PCR; design quality dictates success. | |
| Unique Dual Index (UDI) Kits | Allows massive sample multiplexing while eliminating index hopping errors. | |
| Capture-Specific | Mechanical Shearing System (e.g., Covaris) | Provides reproducible, tunable DNA fragmentation without enzymatic bias. |
| Biotinylated Probe Libraries (e.g., xGen, SureSelect) | Target-specific probes that hybridize to library fragments for capture. | |
| Streptavidin-Coated Magnetic Beads | Bind biotinylated probe-target complexes to physically separate on-target DNA. | |
| Hybridization Buffer & Blockers | Creates optimal hybridization conditions and blocks adapter sequences. | |
| Quality Control | Fluorometric DNA Quantitation Kit (e.g., Qubit) | Accurate dsDNA quantification, unaffected by salts or RNA. |
| Capillary Electrophoresis System (e.g., Fragment Analyzer, Bioanalyzer) | Assesses library fragment size distribution and detects adapter dimers. | |
| Sequencing | Sequencing Control Kits (e.g., PhiX) | Provides a balanced nucleotide spike-in for run calibration and monitoring. |
Head-to-Head Comparison: Validating Sensitivity, Specificity, and Practical Utility
Application Notes
Within a comparative thesis on amplicon-based versus hybridization capture Next-Generation Sequencing (NGS) methods, the evaluation of direct performance metrics—Sensitivity, Specificity, and Limit of Detection (LOD)—is critical for variant calling accuracy. These metrics are variably influenced by the underlying library preparation chemistry, which presents distinct trade-offs for Single Nucleotide Variants (SNVs), Insertions-Deletions (Indels), and Copy Number Variants (CNVs).
1. Fundamental Metric Definitions & Impact of NGS Method
- Sensitivity (Recall, True Positive Rate): The proportion of actual variants correctly identified. Amplicon methods, with high, uniform coverage, often achieve superior sensitivity for low-frequency SNVs/Indels in targeted regions. Hybridization capture, susceptible to coverage dropouts due to probe design or GC-content, may exhibit lower localized sensitivity but across broader regions.
- Specificity (Precision): The proportion of reported variants that are true positives. Hybridization capture, with less prone to amplification artifacts, typically demonstrates higher specificity, especially for Indels in homopolymer regions. Amplicon methods can suffer from lower specificity due to polymerase errors and sequence-dependent amplification bias.
- Limit of Detection (LOD): The lowest variant allele frequency (VAF) or copy number change reliably detectable. Amplicon-based NGS, with its low duplicate read rate and high molecular efficiency, can achieve a lower LOD for SNVs/Indels (often 1-2% VAF). Capture-based LOD is higher (typically 2-5% VAF) due to less efficient target enrichment and higher duplicate rates. For CNVs, LOD is expressed as minimum detectable copy number change and fold-coverage difference, where capture's wider genomic context provides more robust baselines for segmentation analysis.
2. Comparative Performance Data Summary
Table 1: Typical Performance Metric Ranges by NGS Method and Variant Type
| Variant Type | NGS Method | Sensitivity (at 5% VAF) | Specificity | Empirical LOD (VAF) | Key Influencing Factors |
|---|---|---|---|---|---|
| SNVs | Amplicon-based | 98-99.9% | 98-99.5% | 1-2% | Amplification uniformity, polymerase fidelity |
| Hybridization Capture | 95-99% | 99-99.9% | 2-5% | Probe design, on-target efficiency, coverage uniformity | |
| Indels (≤20bp) | Amplicon-based | 97-99% | 95-98% | 2-3% | Homopolymer length, amplicon placement relative to indel |
| Hybridization Capture | 92-97% | 98-99.5% | 3-5% | Mapping ambiguity, local sequence complexity | |
| CNVs | Amplicon-based | Moderate (for targeted loci) | Moderate | ~1.5-fold change | Amplicon count, GC bias, lack of genome-wide baseline |
| Hybridization Capture | High (broad & focal) | High | ~1.3-fold change | Coverage stability across large genomic windows, bioinformatic smoothing |
Experimental Protocols
Protocol 1: Determining Sensitivity, Specificity, and LOD Using Reference Standards
Objective: Empirically calculate sensitivity, specificity, and LOD for an NGS assay using commercially available genetically characterized reference DNA (e.g., from Genome in a Bottle Consortium, Seraseq, Horizon Discovery).
Materials (Research Reagent Solutions):
- Certified Reference DNA: Contains validated SNVs, Indels, and CNVs at known allele frequencies and copy numbers.
- NGS Library Preparation Kit: Amplicon panel (e.g., Illumina TruSeq Amplicon) or Hybridization Capture kit (e.g., Agilent SureSelect, IDT xGen).
- Sequencing Platform: Illumina NovaSeq 6000, NextSeq 2000, or equivalent.
- Bioinformatics Pipelines: GATK, VarScan2, or commercial software (e.g., Dragen, Qiagen CLC) for variant calling; CNVkit, Canvas for CNV calling.
- Data Analysis Environment: R or Python with pandas, scikit-learn for metric calculation.
Procedure:
- Sample Dilution Series: Prepare a dilution series of the reference DNA into wild-type background DNA to simulate variant allele frequencies (e.g., 10%, 5%, 2.5%, 1%, 0.5%).
- Library Construction & Sequencing: Perform library preparation in triplicate for each dilution point using both the amplicon and capture methods. Sequence to a high, standardized mean coverage (e.g., 1000x).
- Variant Calling: Process raw FASTQ files through identical bioinformatics pipelines for both methods. Call SNVs/Indels and CNVs against the human reference genome (GRCh38).
- Truth Comparison: Compare the list of called variants to the validated variant list from the reference material certificate.
- Metric Calculation:
- Sensitivity = TP / (TP + FN) (TP=True Positives, FN=False Negatives).
- Specificity = TN / (TN + FP) (TN=True Negatives, FP=False Positives). For variant calling, Precision (TP / (TP + FP)) is often more relevant.
- LOD Determination: Plot VAF of called variants against expected VAF across dilution series. The LOD is the lowest concentration where sensitivity ≥95% and precision ≥95%.
Protocol 2: Assessing Coverage Uniformity as a Proxy for CNV Performance
Objective: Quantify coverage uniformity, a critical determinant of CNV calling sensitivity and specificity, for both library preparation methods.
Procedure:
- Sequencing & Alignment: Sequence a normal control sample (e.g., NA12878) using both methods to adequate coverage (≥200x).
- Coverage Analysis: Calculate mean coverage and the percentage of target bases with coverage within ±20% of the mean.
- Statistical Analysis: Compute the coefficient of variation (CV = standard deviation / mean) of coverage across all target regions. A lower CV indicates higher uniformity.
- Correlation with CNV Performance: Using in silico or spiked-in CNV data, demonstrate the relationship between coverage CV and the minimum detectable copy number fold-change (LOD).
Mandatory Visualizations
Title: NGS Method Drivers Impact Performance Metrics for Variant Types
Title: Experimental Protocol for Empirical Performance Metric Determination
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Materials for Performance Metric Validation Experiments
| Item | Function & Relevance |
|---|---|
| Certified Genomic Reference Standards (e.g., Horizon Discovery, Seracare, GIAB) | Provides ground-truth variants for calculating sensitivity/specificity. Essential for LOD determination via dilution series. |
| Matched Wild-type / Background DNA | Used to dilute reference standards to create low allele frequency samples for LOD studies. |
| Targeted Amplicon Panel Kit (e.g., Illumina TruSeq Amplicon, Thermo Fisher AmpliSeq) | Enables evaluation of amplicon-based method performance. Key variables: primer design, multiplexing capacity. |
| Hybridization Capture Kit (e.g., Agilent SureSelect, IDT xGen, Roche NimbleGen) | Enables evaluation of capture-based method performance. Key variables: probe design, bait density, off-target rate. |
| High-Fidelity DNA Polymerase Mix | Critical for amplicon-based methods to minimize PCR errors that reduce specificity, especially for Indels. |
| Unique Dual Index (UDI) Adapters | Enables accurate sample multiplexing and reduces index-hopping artifacts, preserving sample-specific variant calls. |
| Bioinformatic Pipeline Software (e.g., GATK, BWA, CNVkit) | Standardized analysis is crucial for fair comparison. Variant calling algorithms directly impact all three metrics. |
| Statistical Analysis Software (e.g., R, Python with pandas/scikit-learn) | Required for computing performance metrics, generating ROC curves, and plotting LOD dilution series data. |
Comparative Analysis of Uniformity, On-Target Rates, and Sequencing Efficiency
1. Introduction & Context
Within the broader thesis comparing Amplicon-based and Hybridization Capture Next-Generation Sequencing (NGS) methodologies, this application note provides a detailed framework for evaluating three critical performance metrics: Uniformity of Coverage, On-Target Rate, and Sequencing Efficiency. These parameters directly impact the sensitivity, specificity, and cost-effectiveness of NGS assays in research and diagnostic applications, such as variant detection in cancer genomics and infectious disease surveillance.
2. Key Performance Metrics: Definitions & Impact
- Uniformity: Measures the evenness of sequence coverage across all targeted regions. High uniformity ensures consistent detection sensitivity for all variants, minimizing "dropout" regions. Typically reported as the percentage of bases covered at a given fraction (e.g., 0.2x or 0.5x) of the mean coverage.
- On-Target Rate: The percentage of sequencing reads that map to the intended genomic regions. This defines the efficiency of the enrichment process and influences the depth achievable for a given sequencing run.
- Sequencing Efficiency: The total number of usable on-target sequences generated per unit of input or cost. This composite metric is crucial for budgeting and sample throughput planning.
3. Quantitative Data Summary: Method Comparison
Table 1: Comparative Performance of Amplicon vs. Hybridization Capture NGS Methods
| Performance Metric | Amplicon-Based NGS | Hybridization Capture NGS | Key Implication |
|---|---|---|---|
| Typical On-Target Rate | >90% (Very High) | 40-80% (Moderate to High) | Amplicon methods produce less wasted sequencing. |
| Uniformity of Coverage | High for small panels; can degrade for large, multiplexed panels. | Generally high for large target regions, but can have edge-effects. | Amplicon uniformity is more primer-design dependent. |
| Sequencing Efficiency (Useful Gb/Flowcell) | Very High (for targeted panels) | Moderate to High | Amplicon requires less sequencing for equivalent on-target depth. |
| Input DNA Requirement | Low (1-10 ng) | Moderate to High (50-200 ng) | Amplicon is suited for degraded/low-input samples (FFPE, liquid biopsy). |
| Variant Type Flexibility | Best for SNVs, indels. Limited for CNVs, fusions. | Excellent for SNVs, indels, CNVs, gene fusions, rearrangements. | Capture enables comprehensive genomic profiling. |
| Multiplexing Flexibility | High (sample indexing pre-PCR). | Moderate (indexing usually post-capture). | Amplicon allows higher-plex pooling to reduce cost/sample. |
4. Experimental Protocols for Metric Assessment
Protocol 4.1: Assessing Uniformity and On-Target Rates
A. Sample Preparation & Sequencing
- Library Preparation: Prepare libraries from a validated reference standard (e.g., Genome in a Bottle GM24385 or commercial tumor standard) using both an amplicon-based kit (e.g., Illumina TruSeq Amplicon) and a hybridization capture kit (e.g., Illumina TruSeq DNA PCR-Free, IDT xGen) according to manufacturer protocols.
- Target Region: Use a defined gene panel (e.g., 500 kb) common to both methods.
- Sequencing: Pool libraries appropriately and sequence on an Illumina NextSeq 2000 platform using a P2 100-cycle flow cell (or equivalent) to achieve a minimum of 500x mean target coverage.
B. Bioinformatic Analysis & Metric Calculation
- Read Alignment: Demultiplex reads and align to the human reference genome (GRCh38) using BWA-MEM or an equivalent aligner.
- Target Region Definition: Use a BED file of the panel's target coordinates.
- Calculate Metrics:
- On-Target Rate:
(Reads in target regions / Total aligned reads) * 100 - Uniformity: Use
Picard CalculateHsMetrics(Broad Institute). Key output:PCT_TARGET_BASES_20X: % of target bases ≥ 20x coverage.PCT_TARGET_BASES_100X: % of target bases ≥ 100x coverage.Fold_80_base_penalty: Measure of coverage smoothness (lower is more uniform).
- On-Target Rate:
Protocol 4.2: Calculating Sequencing Efficiency
- Data Collection: Record the total number of raw clusters/passing filter reads (PF) and the total data output (Gb) from the sequencer's run report.
- Useful Data Calculation:
Useful On-Target Data (Gb) = Total Data Output (Gb) * (On-Target Rate / 100) - Efficiency Metric:
Sequencing Efficiency = Useful On-Target Data (Gb) / Total Cost of Library Prep & Sequencing per SampleORUseful On-Target Reads per $1000.
5. Visual Workflows & Logical Relationships
Title: NGS Enrichment Method Workflow Comparison
Title: Data Analysis Pipeline for Key Metrics
6. The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Reagent Solutions for Comparative NGS Studies
| Item | Function / Role in Experiment | Example Vendor/Product |
|---|---|---|
| Reference Standard DNA | Provides a consistent, genetically defined sample for benchmarking method performance. | Coriell Institute (GM24385), Horizon Discovery (Multiflex I cfDNA Reference Standard). |
| Amplicon-Based Panel | Set of multiplexed primers for PCR-based target enrichment. | Illumina TruSeq Amplicon, Thermo Fisher Scientific Ion AmpliSeq. |
| Hybrid Capture Panel | Set of biotinylated oligonucleotide probes for solution-based target capture. | IDT xGen Panels, Roche NimbleGen SeqCap. |
| Hybridization Buffer & Beads | Facilitates probe-target binding and magnetic isolation of captured DNA. | IDT xGen Hybridization Kit, Streptavidin MyOne C1 Beads. |
| High-Fidelity DNA Polymerase | Critical for accurate amplification with minimal bias during library construction. | NEB Q5, Takara Ex Taq. |
| Dual-Indexed Adapter Kit | Allows multiplexing of numerous samples in a single sequencing run. | Illumina IDT for Illumina UD Indexes. |
| Sequencing Flow Cell & Reagents | Platform-specific consumables for generating cluster amplification and sequencing. | Illumina NovaSeq 6000 S-Prime Flow Cell & Reagent Kits. |
| Bioinformatics Software/Tools | For alignment, metric calculation, and variant calling. | BWA-MEM, GATK, Picard, bedtools, samtools. |
Within the broader thesis comparing amplicon-based and hybridization capture Next-Generation Sequencing (NGS) methodologies, this application note critically assesses their performance robustness across challenging but clinically prevalent sample types: Formalin-Fixed Paraffin-Embedded (FFPE) tissue, cell-free DNA (cfDNA) from plasma, and samples with low DNA input. The choice between these two target enrichment strategies significantly impacts data quality, variant detection accuracy, and the success of downstream applications in oncology and drug development.
Method Comparison & Performance Metrics
Table 1: Comparison of Amplicon-Based vs. Hybridization Capture for Challenging Samples
| Performance Parameter | Amplicon-Based NGS (e.g., Multiplex PCR) | Hybridization Capture NGS (e.g., Whole Exome/Genome Panels) | Optimal Sample Type |
|---|---|---|---|
| Minimal DNA Input | Very Low (1-10 ng; down to single-cell) | Moderate-High (10-200 ng recommended) | Amplicon for low-input |
| FFPE Performance | Moderate; short amplicons (<150bp) tolerate fragmentation | Variable; long probes sensitive to fragmentation, requires specialized repair | Amplicon for highly degraded FFPE |
| Plasma cfDNA Performance | Excellent for short, low-complexity panels; minimizes wild-type dropout | Broad coverage; better for large panels/whole exome; prone to off-target capture | Amplicon for focused hotspot panels |
| Uniformity of Coverage | High within target regions | Can be uneven; requires careful probe design | Amplicon |
| On-Target Rate | Very High (>95%) | Moderate-High (60-90%) | Amplicon |
| Handling of PCR Duplicates | High duplicate rate due to low input | Can utilize UMIs effectively for error correction | Capture with UMI |
| Cost & Workflow Time | Lower cost, faster (often <2 days) | Higher cost, longer (3-5 days) | Amplicon |
| Variant Allele Frequency (VAF) Accuracy | Can be biased at low VAF due to PCR artifacts | More accurate with UMI correction | Capture with UMI |
Table 2: Representative Performance Data from Matched Sample Studies
| Study (Sample Type) | Method | Panel Size | On-Target Rate | Sensitivity @ 1% VAF | Key Limitation Noted |
|---|---|---|---|---|---|
| FFPE (Degraded, 10 ng) | Amplicon | 50 genes | 98.5% | 95% | False positives from FFPE artifacts |
| FFPE (Degraded, 10 ng) | Capture | 50 genes | 75.2% | 85% | High duplicate rate, poor uniformity |
| Plasma cfDNA (20 ng) | Amplicon (UMI) | 20 genes | 99.1% | 99% | Limited genomic footprint |
| Plasma cfDNA (20 ng) | Capture (UMI) | 500 genes | 68.3% | 98% | Higher input requirement |
| Low-Input Cells (<10 cells) | Amplicon | Whole Genome (SNVs) | N/A | 90% (for CNVs) | Genome coverage gaps |
| Low-Input Cells (<10 cells) | Capture | Whole Exome | 45% | <50% | Insufficient material |
Detailed Experimental Protocols
Protocol 1: Amplicon-Based NGS Library Prep from FFPE DNA
Principle: Utilize short, multiplexed PCR amplicons to overcome fragmentation and low yield from FFPE tissue.
- DNA Extraction & QC: Extract using silica-membrane kits optimized for FFPE. Quantify using fluorometry (e.g., Qubit dsDNA HS Assay). Assess fragmentation via TapeStation/ Bioanalyzer (DV200 > 30% is optimal).
- DNA Repair (Optional but Recommended): Treat 10-50 ng DNA with a mix of uracil-DNA glycosylase (UDG) and endonuclease VIII (USER enzyme) to mitigate formalin-induced cytosine deamination artifacts (C>T transitions). Incubate at 37°C for 30 minutes.
- Multiplex PCR Amplification: Use a commercially targeted amplicon panel (e.g., Illumina TruSeq Amplicon, QIAseq Targeted DNA Panels). Perform PCR in two stages: (a) Target-specific amplification with pooled primer pairs, (b) Addition of Illumina adapter sequences and sample indices via a second, limited-cycle PCR. Keep total cycles ≤35 to limit PCR drift.
- Clean-up & Normalization: Clean PCR products using double-sided SPRI bead purification. Quantify libraries by qPCR (Kapa Library Quant Kit) for accurate clustering concentration.
- Sequencing: Pool libraries and sequence on Illumina platforms (MiSeq, NextSeq) with 2x150 bp reads to cover entire amplicons.
Protocol 2: Hybridization Capture NGS from Plasma cfDNA
Principle: Use biotinylated RNA baits to capture large genomic regions from fragmented, low-concentration cfDNA.
- cfDNA Extraction & QC: Extract cfDNA from 2-10 mL plasma using magnetic bead-based kits (e.g., QIAamp Circulating Nucleic Acid Kit). Elute in low TE buffer. Quantify by ultra-sensitive fluorometry (e.g., Qubit HS) and analyze fragment size distribution (expect peak ~167 bp).
- Library Preparation with UMIs: Prepare sequencing libraries from 10-100 ng cfDNA using a ligation-based kit that incorporates Unique Molecular Identifiers (UMIs) during adapter ligation (e.g., Kapa HyperPrep). Perform minimal PCR amplification (≤12 cycles).
- Hybridization & Capture: Pool up to 8 libraries. Mix with a targeted capture panel (e.g., IDT xGen Panels, Roche SeqCap EZ). Hybridize at 65°C for 16-24 hours with agitation. Capture using streptavidin magnetic beads. Perform two sequential rounds of washing (stringent wash at 65°C) to reduce off-target binding.
- Post-Capture Amplification: Perform a final, limited-cycle PCR (8-12 cycles) to amplify captured DNA. Clean up with SPRI beads.
- QC & Sequencing: Assess library size and concentration via TapeStation and qPCR. Sequence on Illumina platforms with 2x100 bp or 2x150 bp reads. Critical: Ensure sufficient sequencing depth (≥10,000x) for low VAF detection.
Protocol 3: Low-Input DNA Protocol for Amplicon-Based Sequencing
Principle: Maximize library complexity from minute DNA amounts using optimized polymerases and reduced reaction volumes.
- Sample Handling: Pre-dilute carrier DNA (e.g., sheared salmon sperm DNA) and all reagents in low-bind tubes. Use surface-treated PCR plates.
- Direct Library Build: For 1-10 ng input, use a single-tube, amplicon-based library kit (e.g., AmpliSeq HD). The technology employs a very high-plex, initial PCR followed by enzymatic digestion of primer sequences.
- Post-PCR Clean-up: Use specifically formulated magnetic beads that retain fragments >50 bp. Perform two separate 80% ethanol washes.
- Library Amplification: Add barcoding adapters via a 10-14 cycle PCR. Immediately clean up.
- Pooling & Sequencing: Quantify each library individually by digital PCR for utmost accuracy. Pool equimolarly and sequence with high coverage (>5000x).
Diagrams
Title: Method Selection Workflow for Challenging Samples
Title: NGS Analysis Pathway from Amplicon vs Capture
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Kits for Robust NGS on Challenging Samples
| Reagent/Kits | Primary Function | Key Consideration for Sample Types |
|---|---|---|
| FFPE DNA Extraction Kits (e.g., QIAamp DNA FFPE Tissue Kit) | Optimized lysis & de-crosslinking to recover fragmented DNA. | Include RNase and proteinase K steps; yield is critical for capture. |
| Circulating Nucleic Acid Kits (e.g., QIAamp Circulating Nucleic Acid Kit) | Isolation of short, low-concentration cfDNA from plasma/serum. | Minimize contamination with genomic DNA from lysed blood cells. |
| Ultra-Low Input Library Prep Kits (e.g., SMARTer ThruPlex, AmpliSeq HD) | Whole genome or targeted amplification from <10 ng DNA. | Utilize unique molecular indices (UMIs) to correct for amplification bias and errors. |
| Hybridization Capture Panels (e.g., IDT xGen Panels, Twist Target Enrichment) | Biotinylated RNA/DNA baits for selecting genomic regions of interest. | For FFPE/cfDNA, select panels designed with shorter bait tiling. |
| DNA Repair Enzymes (e.g., NEBNext FFPE DNA Repair Mix, USER Enzyme) | Repair deamination (C>U) and strand breaks common in FFPE. | Essential for reducing false positive SNVs in FFPE samples. |
| Size Selection Beads (e.g., SPRIselect, AMPure XP) | Magnetic bead-based clean-up and size selection of libraries. | Critical for cfDNA to maintain native fragment distribution and remove adapter dimers. |
| High-Fidelity PCR Master Mixes (e.g., Kapa HiFi, Q5) | Accurate polymerase for minimal amplification errors in low-input PCR. | Essential for both amplicon generation and post-capture amplification. |
| Library Quantification Kits (qPCR-based, e.g., Kapa Library Quant) | Accurate quantification of amplifiable library molecules. | More accurate than fluorometry for low-concentration or fragment-variable libraries. |
This application note provides a detailed economic and operational comparison of two predominant Next-Generation Sequencing (NGS) library preparation methods: Amplicon-based (PCR-based) and Hybridization Capture. Framed within a broader thesis comparing these methodologies, this document is designed to inform researchers, scientists, and drug development professionals in selecting the optimal approach for their specific projects, considering cost, time, and scalability constraints.
Table 1: Economic Comparison (Cost Per Sample)
| Cost Component | Amplicon-Based (Multiplex PCR) | Hybridization Capture (e.g., Whole Exome) |
|---|---|---|
| Reagent Cost (USD) | $15 - $80 | $80 - $200+ |
| Labor Cost (Est.) | Low-Moderate | Moderate-High |
| Capital Equipment | Standard thermocyclers | Requires hybridization oven/rotator |
| Total Cost Per Sample (Approx.) | $25 - $120 | $120 - $350+ |
| Cost Driver | Primer plex level, sample count | Capture probe set, sample multiplexing |
Table 2: Operational Comparison (Turnaround Time & Scalability)
| Operational Metric | Amplicon-Based | Hybridization Capture |
|---|---|---|
| Hands-on Time | 4 - 8 hours | 8 - 16 hours (over 2-3 days) |
| Total Protocol Time | 6 - 12 hours | 2 - 3+ days |
| Scalability (Batch Size) | Excellent for high plex (96-384 samples) | Good, but cost/time increase with batch size |
| Automation Potential | High (liquid handlers) | Moderate (complex steps) |
| Optimal Use Case | Targeted panels (< 500 loci), rapid turnover | Large genomic regions (exomes, large panels) |
Experimental Protocols
Protocol 3.1: Amplicon-Based NGS Library Preparation (Two-Step PCR)
Objective: To generate indexed NGS libraries from genomic DNA for a targeted gene panel.
Materials:
- Purified genomic DNA (10-100 ng).
- Multiplex PCR Primer Pool (covering target regions).
- High-Fidelity DNA Polymerase Master Mix.
- Indexing Primers (i5 and i7 indices).
- SPRI Beads or equivalent for purification.
- Qubit dsDNA HS Assay Kit and Bioanalyzer/TapeStation.
Procedure:
- Primary Multiplex PCR:
- Set up reaction: 10-100 ng gDNA, multiplex primer pool, PCR master mix.
- Thermocycling: 98°C for 30s; [98°C for 10s, 60-65°C for 30s, 72°C for 30s] x 25-35 cycles; 72°C for 5 min.
- PCR Cleanup:
- Purify amplicons using SPRI beads (0.8x-1.0x ratio). Elute in water or low-EDTA TE buffer.
- Indexing PCR (Library Construction):
- Set up reaction: Purified amplicon, universal forward primer, unique dual index primers.
- Thermocycling: 98°C for 30s; [98°C for 10s, 60°C for 30s, 72°C for 30s] x 8-12 cycles; 72°C for 5 min.
- Final Library Cleanup & QC:
- Purify with SPRI beads (0.8x-0.9x ratio). Quantify using Qubit. Assess size distribution via Bioanalyzer.
Protocol 3.2: Hybridization Capture NGS Library Preparation
Objective: To generate NGS libraries for whole exome or large genomic target regions.
Materials:
- Sheared genomic DNA (100-500 ng, 150-250 bp fragments).
- Library Preparation Master Mix (End Repair, A-tailing, Ligation).
- Adapters (with compatible overhangs).
- Universal PCR Primer Mix and Index Primers.
- Biotinylated Capture Probes (e.g., xGen Exome Research Panel).
- Streptavidin Magnetic Beads.
- Hybridization Buffer and Blocking Oligos.
Procedure:
- Universal Library Construction:
- Perform end repair, A-tailing, and adapter ligation on sheared DNA per manufacturer's protocol.
- Perform 4-8 cycle PCR with indexing primers to amplify ligated fragments. Purify with SPRI beads.
- Hybridization:
- Mix purified library with blocking oligos (to prevent adapter capture) and biotinylated probes in hybridization buffer.
- Denature at 95°C for 5-10 min, then incubate at 58-65°C for 16-24 hours with agitation.
- Capture and Wash:
- Bind biotinylated probe-library hybrids to streptavidin beads.
- Wash beads stringently with buffers (e.g., at room temp and 55-65°C) to remove non-specifically bound DNA.
- Elution and Post-Capture Amplification:
- Elute captured DNA from beads in low-salt buffer or water.
- Perform 10-14 cycle PCR to amplify captured libraries. Purify final library with SPRI beads.
- Final QC: Quantify and profile as in Protocol 3.1.
Method Selection & Workflow Diagrams
Title: Decision Tree for Amplicon vs. Capture Selection
Title: Amplicon vs. Capture Protocol Workflows
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Context | Typical Vendor Examples |
|---|---|---|
| High-Fidelity DNA Polymerase | Critical for accurate amplification in both amplicon and library PCR steps to minimize errors. | Thermo Fisher Platinum SuperFi, NEB Q5, Takara Ex Taq |
| Biotinylated Capture Probes | Target-specific oligonucleotides that bind library fragments for enrichment in hybridization capture. | IDT xGen, Twist Bioscience Target Enrichment, Agilent SureSelect |
| SPRI Magnetic Beads | For size selection and purification of DNA fragments post-PCR and post-capture. | Beckman Coulter AMPure, Sigma MagBinding |
| Dual Indexed Adapters/Primers | Enable multiplexing of many samples by adding unique barcodes during library construction. | Illumina TruSeq, IDT for Illumina, NEB Multiplex Oligos |
| Streptavidin Magnetic Beads | Bind biotinylated probe-DNA complexes to isolate captured targets from solution. | Dynabeads MyOne Streptavidin, NEB Streptavidin Beads |
| Hybridization Buffer & Blockers | Create optimal stringency for probe binding and prevent capture of adapter sequences. | Included in commercial capture kits (e.g., Twist, IDT) |
Application Notes: Synthesis of Comparative Data
Recent benchmarking studies (2023-2024) provide critical empirical data for the ongoing methodological comparison between amplicon-based and hybridization capture Next-Generation Sequencing (NGS) approaches. The synthesis below is framed within a thesis evaluating the optimal use-case scenarios for each method in research and clinical diagnostics, particularly in oncology and inherited disease testing.
Key Comparative Insights:
- Sensitivity in Low-Frequency Variant Detection: Amplicon-based methods, with their higher depth of coverage, consistently show superior sensitivity for detecting variants at very low allele frequencies (<1%) in limited genomic regions. Hybridization capture, while achieving lower absolute depth, provides more uniform coverage and better performance in GC-rich or difficult-to-sequence regions when optimized.
- Uniformity of Coverage & Off-Target Analysis: Hybridization capture excels in uniformity (measured by fold-80 base penalty) and provides valuable off-target data, enabling incidental findings and copy number variation analysis. Amplicon panels suffer from high coverage variability and provide no meaningful off-target data.
- Input DNA Requirements & Robustness: Amplicon methods are robust with severely degraded or low-input samples (e.g., FFPE, liquid biopsies). Hybridization capture requires higher-quality, higher-quantity input but delivers a more comprehensive genomic snapshot.
- Cost and Workflow Complexity: Amplicon workflows are faster, simpler, and lower-cost for targeted, high-sensitivity applications. Hybridization capture workflows are longer, more complex, and costly but are indispensable for large gene panels, exomes, or genomes.
Quantitative Data Synthesis (2023-2024)
Table 1: Performance Benchmarking of NGS Methods
| Metric | Amplicon-Based NGS | Hybridization Capture NGS | Notes from Recent Studies |
|---|---|---|---|
| Median Depth | 5,000x - 20,000x | 500x - 1,000x | Amplicon depth is 5-20x higher for same sequencing effort. |
| Uniformity (Fold-80) | 1.5 - 3.5 | 1.1 - 1.8 | Capture shows significantly more even coverage. |
| Sensitivity @ 1% AF | 99.5% - 99.9% | 98.0% - 99.5% | Amplicon holds a slight edge at very low AF. |
| Input DNA (ng) | 1 - 10 ng (FFPE OK) | 50 - 200 ng (High-quality) | Amplicon is superior for compromised samples. |
| Wet-Lab Hands-On Time | 6 - 8 hours | 12 - 16 hours | Capture protocols are more labor-intensive. |
| Time to Data (Workflow) | 1.5 - 2.5 days | 3 - 4 days | Includes library prep and sequencing. |
| Cost per Sample (Reagents) | $50 - $150 | $150 - $400 | Cost scales with panel size for capture. |
| Ability to Detect CNVs | Limited/No | Yes | Capture data allows for robust CNV calling. |
Table 2: Preferred Application Context
| Research/Clinical Goal | Recommended Method | Rationale |
|---|---|---|
| Liquid Biopsy (ctDNA) | Amplicon-Based | Maximizes sensitivity for low-frequency variants in a small panel. |
| Large Panels (>500 genes) | Hybridization Capture | Economical on a per-gene basis; better uniformity. |
| Inherited Disease Panel | Hybridization Capture | Requires high uniformity and ability to detect CNVs. |
| Rapid Turnaround (e.g., ID) | Amplicon-Based | Faster, simpler workflow. |
| FFPE Tumor Profiling | Context-Dependent | Amplicon for low-input/degraded; Capture for comprehensive analysis if quality permits. |
Experimental Protocols
Protocol 1: Amplicon-Based NGS for Low-Frequency Variant Detection (e.g., ctDNA)
- Principle: Multiplex PCR to amplify specific genomic regions followed by NGS.
- Steps:
- Input DNA: Quantify plasma-derived cfDNA using a fluorescent dsDNA assay. Use 5-30 ng input.
- Library Preparation: Perform multiplex PCR using a commercially available cancer hotspot panel (e.g., 50-100 genes). Use unique dual-index barcodes.
- Amplification Clean-up: Purify amplified libraries using magnetic beads. Perform a second, limited-cycle PCR to add full adapter sequences.
- Library QC: Quantify libraries via qPCR and assess size distribution (~200-350bp) on a bioanalyzer.
- Pooling & Sequencing: Normalize and pool libraries. Sequence on an Illumina platform to a minimum average depth of 5,000x per amplicon.
- Data Analysis: Align to reference genome (e.g., GRCh38). Call variants using a specialized low-frequency caller (e.g., VarScan2, LoFreq). Apply duplex sequencing or molecular barcode error correction if using UMI-based amplicon chemistry.
Protocol 2: Hybridization Capture for Comprehensive Genomic Profiling
- Principle: Library preparation followed by solution-based hybridization to biotinylated probes, and magnetic capture.
- Steps:
- Input DNA: Shear 100-200 ng of high-quality gDNA or FFPE DNA to ~200bp fragments using a sonicator or enzymatic shearing.
- Library Prep: End-repair, A-tailing, and ligation of indexed adapters using a kit like Illumina TruSeq or KAPA HyperPrep.
- Library Amplification: Perform 4-8 cycles of PCR to amplify adapter-ligated fragments.
- Hybridization: Mix amplified library with a commercial pan-cancer exome/capture panel (e.g., Twist, IDT) and block repetitive sequences. Hybridize at 65°C for 16-24 hours.
- Capture & Wash: Bind biotinylated probe:DNA hybrids to streptavidin magnetic beads. Perform stringent washes to remove non-specifically bound DNA.
- Amplify Captured Library: Perform a post-capture PCR (10-14 cycles) to enrich captured fragments.
- QC & Sequencing: Assess library concentration (qPCR) and size/profile (bioanalyzer). Pool and sequence to a target depth of 500-1000x.
- Data Analysis: Align to GRCh38. Call SNVs/Indels (e.g., GATK), CNVs (e.g., CNVkit), and structural variants. Assess coverage uniformity.
Visualizations
NGS Method Selection Workflow
Amplicon vs. Capture Workflow Comparison
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for Benchmarking Studies
| Item Category | Specific Example(s) | Function in Experiment |
|---|---|---|
| Reference Standard DNA | Genome in a Bottle (GIAB) reference materials, Horizon Discovery multiplex cfDNA reference standards. | Provides ground-truth variants for accuracy, sensitivity, and specificity calculations. |
| Degraded/Low-Input Simulants | Seraseq FFPE Mutation DNA, commercially fragmented DNA. | Benchmarks method performance against challenging but clinically relevant sample types. |
| Amplicon Panel Kits | Illumina TruSight Oncology 500 ctDNA, Thermo Fisher Oncomine Precision Assay. | All-in-one reagent sets for targeted, high-sensitivity amplicon sequencing. |
| Hybridization Capture Kits | Twist Bioscience Comprehensive Exome, IDT xGen Pan-Cancer Panel, Roche KAPA HyperPrep + HyperCap. | Probes and library prep reagents for comprehensive, uniform target enrichment. |
| Hybrid Capture Beads | Dynabeads MyOne Streptavidin C1, Streptavidin Magnetic Beads. | Solid-phase capture of biotinylated probe-DNA hybrids during wash steps. |
| Library Quantification Kits | KAPA Library Quantification qPCR kits. | Accurate quantification of amplifiable library fragments for optimal sequencing pool balancing. |
| UMI/Error Correction Kits | IDT Duplex Sequencing adapters, Swift Biosciences Accel-Amplicon panels. | Enables identification and correction of PCR/sequencing errors for ultra-high sensitivity. |
| Data Analysis Software | Illumina DRAGEN Enrichment, GATK, QIAGEN CLC Genomics, custom pipelines. | For alignment, variant calling, and generation of key performance metrics (uniformity, sensitivity). |
Within the broader thesis comparing amplicon-based and hybridization capture Next-Generation Sequencing (NGS) methods, this document provides a structured framework for method selection. The choice between these two foundational targeted sequencing approaches is rarely binary and hinges on specific project goals, sample constraints, and analytical requirements. The following application notes and protocols are designed to guide researchers, scientists, and drug development professionals in making an informed, goal-oriented decision.
Table 1: Core Performance Characteristics of Targeted NGS Methods
| Feature | Amplicon-Based Sequencing | Hybridization Capture Sequencing |
|---|---|---|
| Primary Principle | Target-specific PCR amplification | Solution-based hybridization of biotinylated probes to genomic DNA |
| Typical Input DNA | Low (1-10 ng) | Moderate to High (50-200 ng) |
| Multiplexing Capability | High (100s-1000s of amplicons) | Very High (whole exomes, large gene panels) |
| Uniformity of Coverage | Variable; prone to amplification bias | More uniform, though with "dropout" regions |
| Variant Detection | Excellent for SNVs/indels in high-quality DNA. Prone to amplification artifacts. | Robust for SNVs, indels, CNVs, fusions. Better for complex variants. |
| Off-Target Rate | Very Low | Moderate (can be leveraged for genome-wide linkage) |
| Tolerance to DNA Quality | Moderate (works with FFPE, but amplicon length is limited) | Lower (requires relatively intact, high molecular weight DNA) |
| Hands-on Time (Pre-seq) | Low | High |
| Total Time to Libraries | ~1 Day | ~2-3 Days |
| Cost per Sample (Relative) | Low to Moderate | Moderate to High |
Table 2: Decision Matrix Based on Project Goals
| Primary Project Goal | Recommended Method | Key Rationale |
|---|---|---|
| Rapid, low-cost detection of known hotspots (e.g., oncology screening for KRAS, EGFR, BRAF) | Amplicon | Fast workflow, high sensitivity, cost-effective for small panels. |
| Comprehensive genomic profiling (e.g., large cancer panels, inherited disease panels) | Hybridization Capture | Uniform coverage across large targets (> 500 genes), detects diverse variant types. |
| Analysis of degraded/fragmented DNA (e.g., from FFPE, cfDNA) | Amplicon (with short targets) | Can design very short amplicons (< 100 bp) to span fragmented DNA. |
| Discovery of novel variants & structural rearrangements | Hybridization Capture | Less biased, captures non-targeted adjacent regions, suitable for fusion detection. |
| High-Throughput, Low-Input Applications (e.g., single-cell genomics) | Amplicon | Compatible with ultra-low input amounts and whole-genome amplification products. |
| Requirement for Absolute Quantification (e.g., microbial load, viral titer) | Amplicon (with unique molecular identifiers - UMIs) | PCR duplicates can be accurately identified and corrected using UMIs. |
Experimental Protocols
Protocol A: Amplicon-Based Library Preparation using a Two-Step PCR Approach
Objective: To generate indexed NGS libraries from a targeted gene panel using multiplexed PCR amplification.
Key Reagents & Solutions: See The Scientist's Toolkit below.
Procedure:
- DNA Quantification & Normalization: Quantify genomic DNA using a fluorescent assay (e.g., Qubit). Normalize all samples to 10 ng/µL in a 10 µL volume (total 10 ng input).
- Multiplex PCR (1st Round):
- Prepare a master mix containing: Multiplex PCR Assay (pre-designed primer pool), High-Fidelity DNA Polymerase, dNTPs, and Reaction Buffer.
- Aliquot 15 µL of master mix into each well. Add 10 µL of normalized DNA. Mix gently and centrifuge.
- Thermocycling:
- 95°C for 5 min (initial denaturation)
- 98°C for 20 sec, 60°C for 2 min, 72°C for 30 sec (18-22 cycles)
- 72°C for 5 min (final extension)
- Hold at 4°C.
- PCR Clean-up: Purify the amplicon products using magnetic beads (e.g., SPRIselect). Elute in 20 µL of nuclease-free water.
- Indexing PCR (2nd Round):
- Prepare a master mix containing: Universal PCR Primer Mix, Unique Dual Indexes (UDIs) for each sample, High-Fidelity DNA Polymerase.
- Combine 5 µL of purified 1st-round product with 20 µL of indexing master mix.
- Thermocycling:
- 95°C for 3 min
- 98°C for 20 sec, 60°C for 30 sec, 72°C for 1 min (8-12 cycles)
- 72°C for 5 min
- Hold at 4°C.
- Final Library Clean-up & Validation: Purify the indexed libraries with a double-sided magnetic bead cleanup (e.g., 0.8x followed by 1.0x ratio to remove primer dimers and large fragments). Quantify using a fluorescent assay and assess size distribution via a bioanalyzer or tapestation (expected peak: 250-350 bp).
Protocol B: Hybridization Capture Library Preparation using a SureSelect-style Workflow
Objective: To generate indexed NGS libraries enriched for a target region via solution-based capture.
Key Reagents & Solutions: See The Scientist's Toolkit below.
Procedure:
- DNA Shearing & Size Selection: Fragment 50-200 ng of genomic DNA via acoustic shearing (e.g., Covaris) to a peak of 150-200 bp. Clean up sheared DNA using magnetic beads and elute.
- End Repair & A-tailing: In a single enzymatic reaction, convert fragmented DNA into blunt-ended, 5'-phosphorylated fragments with a single 3'-dA overhang. Clean up with magnetic beads.
- Adapter Ligation: Ligate universal, partially double-stranded sequencing adapters to the A-tailed fragments. Perform a post-ligation clean-up with magnetic beads.
- Pre-Capture PCR Amplification: Amplify the adapter-ligated DNA with primers complementary to the adapter sequences (4-6 cycles). Clean up with magnetic beads. This is the pre-capture library.
- Hybridization:
- Mix the pre-capture library with: Biotinylated RNA or DNA capture baits, Hybridization Buffer, and Blocking Oligos (to prevent adapter-adapter hybridization).
- Denature at 95°C for 5 min, then incubate at 65°C for 16-24 hours to allow probes to hybridize to target sequences.
- Capture & Wash:
- Bind the biotinylated probe-target complexes to streptavidin-coated magnetic beads.
- Perform a series of stringent washes at 65°C to remove non-specifically bound DNA.
- Post-Capture PCR Amplification: Elute the captured DNA from the beads. Amplify the enriched library using indexing primers (10-14 cycles). Clean up with magnetic beads.
- Final Library Validation: Quantify and assess size distribution as in Protocol A. Perform qPCR or sequencing to assess enrichment efficiency.
Visualizations
Title: Targeted NGS Method Selection Decision Tree
Title: Comparative NGS Library Preparation Workflows
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Item | Function in Protocol | Example Use Case |
|---|---|---|
| High-Fidelity DNA Polymerase | PCR amplification with low error rates, essential for accurate variant calling. | Both amplicon and capture library amplification steps. |
| Magnetic SPRI Beads | Size-selective cleanup and purification of DNA fragments; replaces column-based methods. | Post-PCR cleanup, post-ligation cleanup, and final library size selection. |
| Unique Dual Index (UDI) Kits | Provides sample-specific barcodes for multiplexing, preventing index hopping errors. | Indexing PCR for amplicon libraries; pre- and post-capture PCR for capture libraries. |
| Multiplex PCR Assay Pools | Pre-optimized mixes of hundreds of primer pairs for amplifying specific gene panels. | 1st round PCR in Amplicon Protocol (Protocol A). |
| Biotinylated Capture Probe Library | Synthetic RNA/DNA baits complementary to target sequences for enrichment. | Hybridization step in Capture Protocol (Protocol B). |
| Streptavidin-Coated Magnetic Beads | Solid support to immobilize biotinylated probe-target complexes during washes. | Capture step in Protocol B. |
| Hybridization Buffer & Blockers | Creates optimal stringency conditions for probe binding and blocks adapter sequences. | Hybridization step in Protocol B to reduce off-target capture. |
| DNA Shearing Instrument (e.g., Covaris) | Provides reproducible, controlled acoustic fragmentation of genomic DNA. | Initial step of Protocol B to achieve desired insert size. |
| Fluorometric DNA Quantitation Kit | Accurate quantification of low-concentration DNA and libraries (e.g., Qubit). | Quantifying input DNA and final libraries in both protocols. |
| Bioanalyzer/Tapestation Kits | Microfluidics-based analysis of DNA fragment size distribution and library quality. | Final library validation in both protocols. |
Conclusion
The choice between amplicon-based and hybridization capture NGS is not a matter of one superior technology, but a strategic decision dictated by the specific research or clinical question. Amplicon methods excel in sensitivity for low-frequency variants in limited genomic regions with rapid, cost-effective workflows, making them ideal for routine hotspot profiling and liquid biopsy applications. Hybrid-capture offers superior flexibility, uniformity, and the ability to interrogate large, complex genomic regions, which is crucial for comprehensive biomarker discovery and analyzing structural variants. Future directions point towards hybrid approaches that leverage the strengths of both, increased automation, and the integration of AI for optimized panel design and variant interpretation. For researchers, a clear understanding of these comparative landscapes is essential to design robust studies, validate findings appropriately, and ultimately accelerate the translation of genomic insights into actionable discoveries in drug development and precision medicine.