AlphaFold 3: The Ultimate Guide to Biomolecular Complex Prediction for Drug Discovery
This comprehensive guide examines AlphaFold 3, DeepMind's revolutionary AI system for predicting the 3D structures of biomolecular complexes, including proteins, DNA, RNA, ligands, and post-translational modifications.
AlphaFold 3: The Ultimate Guide to Biomolecular Complex Prediction for Drug Discovery
Abstract
This comprehensive guide examines AlphaFold 3, DeepMind's revolutionary AI system for predicting the 3D structures of biomolecular complexes, including proteins, DNA, RNA, ligands, and post-translational modifications. Tailored for researchers, scientists, and drug development professionals, it explores the foundational science behind the model, its novel Evoformer-based architecture and diffusion network, practical applications in rational drug and therapeutic design, current limitations and troubleshooting strategies, and rigorous validation against experimental data. The article concludes by synthesizing AlphaFold 3's transformative potential for accelerating biomedical research and the future of computational structural biology.
What is AlphaFold 3? The AI Revolution in Biomolecular Structure Prediction
Application Notes on AlphaFold 3 for Complex Prediction
AlphaFold 3 (AF3) represents a transformative leap from its predecessor's singular focus on protein structure to the prediction of biomolecular complexes. The generalized deep learning architecture now models interactions between proteins, nucleic acids (DNA/RNA), small molecules, and ions.
Key Performance Metrics
The following table summarizes the quantitative performance of AlphaFold 3 as reported on its benchmark set, compared to AlphaFold 2 and other specialized tools.
Table 1: AlphaFold 3 Benchmark Performance on Biomolecular Complexes
| Complex Type | AlphaFold 3 (DockQ) | AlphaFold 2 (DockQ) | Specialized Tool (DockQ) | Key Improvement |
|---|---|---|---|---|
| Protein-Protein | 0.76 | 0.44 | 0.69 (AF2-Multimer) | 73% increase over AF2 |
| Protein-Antibody | 0.71 | 0.32 | 0.55 | >120% increase |
| Protein-DNA | 0.75 | N/A | 0.63 (NucleicNet) | 19% increase |
| Protein-RNA | 0.73 | N/A | 0.58 | 26% increase |
| Protein-Ligand | 0.72* (RMSD < 2Ã…) | N/A | 0.42* (DiffDock) | ~70% increase |
| Enzyme-Small Molecule | 0.69* (RMSD < 2Ã…) | N/A | 0.38* (Rosetta) | >80% increase |
Note: *Ligand metrics use RMSD < 2Ã… success rate instead of DockQ. AF3 was tested on 62% of novel test complexes not in PDB. All data sourced from DeepMind/Isomorphic Labs publication (Nature, 2024).
Implications for Drug Discovery
AF3's ability to predict protein-ligand and protein-antibody structures with high accuracy shortens the initial hypothesis-generation phase in structure-based drug design. It enables rapid in silico screening of potential binding pockets and off-target interactions for novel therapeutic modalities, including PROTACs and molecular glues.
Experimental Protocols
Protocol: Predicting a Protein-Small Molecule Complex with AlphaFold 3
Objective: To generate a structural model of a target protein in complex with a drug-like small molecule.
Materials & Software:
- AlphaFold 3 server (via Google Cloud Public API) or Colab notebook.
- Input sequences in FASTA format.
- Small molecule ligand in SMILES string format.
- Visualization software (e.g., PyMOL, ChimeraX).
Procedure:
- Input Preparation:
- Obtain the canonical amino acid sequence (UniProt ID recommended) for the target protein.
- Define the chemical structure of the small molecule ligand as a SMILES string.
- Job Submission:
- Access the AF3 interface. Input the protein sequence into the "Protein" field.
- Paste the SMILES string into the "Ligand" field. Specify the molecule type as "Small Molecule."
- (Optional) Adjust sampling parameters:
num_samples=1for speed,num_samples=5for higher confidence. - Submit the prediction job.
- Analysis of Results:
- Download the results package, containing PDB files, confidence metrics (pLDDT, pTM, iPae), and per-residue confidence plots.
- The pLDDT (0-100) indicates local model confidence. The iPAE (interface Predicted Aligned Error) matrix identifies confident interaction regions.
- Open the top-ranked model (ranked_0.pdb) in a molecular viewer. The ligand coordinates will be included.
- Validate the predicted binding pose by analyzing complementary electrostatic surfaces and potential hydrogen bonds.
Protocol: Validating a Predicted Protein-Nucleic Acid Complex
Objective: To experimentally validate an AF3-predicted transcription factor-DNA complex using Electrophoretic Mobility Shift Assay (EMSA).
Materials & Reagents:
- Purified Protein: Recombinant protein expressed and purified via affinity chromatography.
- DNA Probe: Cy5-labeled double-stranded oligonucleotide containing the predicted binding sequence (20-30 bp).
- EMSA Buffer: 10 mM Tris, 50 mM KCl, 1 mM DTT, 5% Glycerol, 0.1 mg/mL BSA, pH 7.5.
- Polyacrylamide Gel: 6% non-denaturing gel in 0.5X TBE buffer.
- Imaging System: Fluorescence gel scanner (Cy5 channel).
Procedure:
- Complex Formation:
- Based on the AF3 model, design a DNA probe matching the predicted interface.
- In a 20 µL reaction, mix EMSA buffer with 10 nM Cy5-DNA probe.
- Titrate purified protein (0, 10, 50, 100, 200 nM) into separate reactions.
- Incubate at 25°C for 20 minutes.
- Gel Electrophoresis:
- Pre-run the 6% polyacrylamide gel in 0.5X TBE at 100V for 30 min at 4°C.
- Load each reaction mixture (with minimal dye) onto the gel.
- Run at 100V for ~60 min at 4°C until the free DNA front has migrated sufficiently.
- Detection & Analysis:
- Image the gel using the Cy5 fluorescence channel.
- A successful validation is indicated by a dose-dependent upward shift (reduced mobility) of the fluorescent band, confirming protein-DNA complex formation.
- Compare the apparent stoichiometry and potential cooperative binding with the AF3-predicted interface.
Visualization Diagrams
AlphaFold 3 Workflow for Drug Discovery
From AF3 Prediction to Validated Complex
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for AlphaFold 3-Driven Research
| Item | Function & Relevance to AF3 Research |
|---|---|
| AlphaFold 3 Server/API Access | Primary tool for generating structure predictions of biomolecular complexes. Cloud-based access required. |
| PyMOL or UCSF ChimeraX | Industry-standard software for visualizing, analyzing, and rendering predicted 3D structures. |
| SMILES Strings for Ligands | Text-based representation of small molecule chemistry, required as input for AF3 ligand predictions. |
| Recombinant Protein Purification Kits | (e.g., His-tag Purification) To obtain pure protein for experimental validation of predicted complexes (e.g., EMSA, SPR). |
| Fluorescent DNA/RNA Labeling Kits | (e.g., Cy5 NHS ester) For preparing labeled nucleic acid probes to validate protein-nucleic acid interactions via EMSA. |
| Surface Plasmon Resonance (SPR) Chip | Sensor chip for biophysical validation of predicted binding affinities (KD) and kinetics. |
| Cryo-EM Grids & Vitrobot | For high-resolution structural validation of novel or challenging complexes predicted by AF3. |
| Molecular Dynamics Software | (e.g., GROMACS, AMBER) To refine and assess the stability of AF3-predicted complexes in silico. |
| Diphenhydramine | Diphenhydramine |
| Sanguinarine sulfate | Sanguinarine Sulfate|High-Purity Research Chemical |
Within the broader thesis on AlphaFold 3 (AF3), this document addresses its core achievement: the generalized prediction of multi-molecule assembly structures. AF3 extends beyond protein folding to model the intricate atomic interactions in complexes containing proteins, nucleic acids (DNA, RNA), small molecule ligands, and post-translational modifications (PTMs). This capability represents a paradigm shift in structural biology, enabling a more holistic view of the biomolecular machinery that drives cellular function and dysfunction.
Application Notes: Performance & Quantitative Benchmarks
The predictive performance of AF3 for multi-molecule complexes is benchmarked against experimental structures and specialized legacy tools. Key metrics include Interface DockQ score (iDockQ, measuring interface accuracy) and overall TM-score (measuring fold similarity).
Table 1: AF3 Performance Across Biomolecule Complex Types
| Complex Type | Example System | iDockQ (AF3) | iDockQ (Legacy Tool) | Median TM-score (AF3) | Key Experimental Validation |
|---|---|---|---|---|---|
| Protein-Protein | Enzyme-Inhibitor | 0.89 | 0.72 (AlphaFold-Multimer) | 0.94 | Cryo-EM (EMD-XXXX) |
| Protein-Antibody | IgG-Fc Region | 0.81 | 0.65 | 0.91 | X-ray Crystallography (2.1 Ã…) |
| Protein-DNA | Transcription Factor-DNA | 0.76 | 0.51 (Specialized Docking) | 0.88 | FRET Binding Assay |
| Protein-RNA | Splicing Factor-RNA | 0.73 | N/A | 0.85 | NMR Chemical Shift Perturbation |
| Protein-Ligand | Kinase-Inhibitor | 0.71* | 0.45 (Glide SP) | 0.87 | IC50 = 12 nM; Co-crystal Structure |
| Protein with PTM | Phosphorylated Signaling Protein | N/A | N/A | 0.90 | Phospho-specific Antibody ELISA |
Ligand iDockQ based on heavy-atom RMSD < 2.0 Ã…. *PTM accuracy assessed via local structure confidence (pLDDT) and biochemical assay correlation.
Table 2: Success Rate by Complex Difficulty (CASP15 Benchmark)
| Category | Definition | AF3 Success Rate (iDockQ ≥ 0.5) | Sample Size (N) |
|---|---|---|---|
| Easy | High homology templates | 94% | 50 |
| Medium | Low homology, known interfaces | 78% | 45 |
| Hard | Novel folds/unknown interfaces | 42% | 30 |
| Ligand Challenge | Novel drug-like molecules | 65% (RMSD < 2.0 Ã…) | 20 |
Experimental Protocols for Validation
Protocol:In SilicoPrediction of a Protein-Ligand-Kinase Complex
Objective: To predict the structure of a target kinase bound to both a regulatory protein and a small-molecule ATP-competitive inhibitor using AF3.
Materials: See Scientist's Toolkit.
Procedure:
- Sequence & Ligand Preparation:
- Obtain FASTA sequences for the kinase and regulatory protein.
- Prepare the ligand SMILES string. Convert to 3D SDF format using Open Babel (
obabel -ismi inhibitor.smi -osdf -gen3d -O inhibitor.sdf). - Define the ligand in the input as a non-polymer component using the AF3 template.
Model Generation:
- Submit the multi-sequence alignment (MSA) for proteins and the ligand SDF via the AF3 server or local ColabFold implementation.
- Run 5 independent model predictions with random seed variation.
- Set
max_recyclesto 12 for complex refinement.
Model Analysis & Selection:
- Rank models by predicted interface TM-score (ipTM) and interface predicted aligned error (ipAE).
- Visually inspect the top-ranked model in PyMOL/ChimeraX for plausible binding pockets, steric clashes, and interaction networks (e.g., hydrogen bonds, pi-stacking).
Validation Planning:
- Use predicted structure to design point mutations in the kinase or ligand for biochemical validation (Step 3.2).
Protocol: Biochemical Validation of a Predicted Protein-DNA Interface
Objective: To validate AF3's prediction of a transcription factor's DNA-binding specificity via electrophoretic mobility shift assay (EMSA).
Procedure:
- Design Probes: Based on the AF3-predicted DNA sequence in the complex, synthesize 25-bp double-stranded DNA probes: one with the predicted consensus sequence and a mutant control with 3 critical bases scrambled.
- Protein Expression: Express and purify the recombinant transcription factor with a His-tag.
- EMSA Binding Reaction:
- Prepare a 20 µL reaction: 20 mM HEPES (pH 7.9), 50 mM KCl, 1 mM DTT, 10% glycerol, 0.1 µg/µL BSA, 10 fmol labeled DNA probe.
- Titrate purified protein (0, 10, 50, 100, 200 nM).
- Incubate at 25°C for 30 min.
- Gel Electrophoresis & Analysis:
- Load reactions on a pre-run 6% non-denaturing polyacrylamide gel in 0.5x TBE buffer.
- Run at 100V for 60 min at 4°C.
- Visualize using a phosphorimager. A validated prediction will show a clear gel shift for the consensus probe, but not the mutant, correlating with the predicted binding interface.
Visualization of Workflows & Concepts
Diagram Title: AF3 Multi-Molecule Prediction & Validation Workflow
Diagram Title: AF3 Diffusion-Based Structure Generation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for AF3-Driven Research
| Item / Reagent | Function in AF3 Workflow | Example Product / Specification |
|---|---|---|
| AF3 Server / ColabFold | Core prediction engine. Local ColabFold allows custom ligands/PTMs. | Google DeepMind AF3 Server; ColabFold v1.5.2 with AlphaFold3 parameters. |
| Chemical Drawing Software | Convert ligand to 3D structure file for AF3 input. | Open Babel (v3.1.1), RDKit, MarvinSketch. |
| Structure Visualization | Analyze predicted models, check interfaces, plan mutations. | UCSF ChimeraX (v1.7), PyMOL (v2.5). |
| His-tag Purification Kit | Validate predictions by expressing/purifying recombinant proteins. | Ni-NTA Superflow Cartridge (Qiagen) for EMSA/SPR. |
| EMSA Gel Kit | Validate nucleic acid-protein interactions predicted by AF3. | LightShift Chemiluminescent EMSA Kit (Thermo Scientific). |
| Surface Plasmon Resonance (SPR) Chip | Quantify binding kinetics (KD) of predicted protein-ligand complexes. | Series S Sensor Chip CM5 (Cytiva). |
| Site-Directed Mutagenesis Kit | Introduce interface mutations to test prediction accuracy. | Q5 Site-Directed Mutagenesis Kit (NEB). |
| Cryo-EM Grids | High-resolution experimental validation of large, predicted complexes. | Quantifoil R1.2/1.3 300 mesh Au grids. |
| Methiazole | Methiazole, CAS:74239-55-7, MF:C12H15N3O2S, MW:265.33 g/mol | Chemical Reagent |
| Pectenotoxin 2 | Pectenotoxin 2, CAS:97564-91-5, MF:C47 H70 O14, MW:859 g/mol | Chemical Reagent |
Within the broader thesis on AlphaFold 3 research, the evolution from the Evoformer-based architecture of AlphaFold 2 (AF2) to the integration of a diffusion network in AlphaFold 3 (AF3) represents a paradigm shift. This transition marks a move from an architecture primarily focused on single-chain protein structure prediction to one capable of modeling a broad spectrum of biomolecular complexes—proteins, nucleic acids, ligands, ions, and post-translational modifications—with atomic accuracy. The Evoformer remains a core module for processing evolutionary sequence information, while the new diffusion network enables the generation of diverse, probabilistic structures, moving beyond deterministic predictions.
Architectural Evolution: Comparative Analysis
Table 1: Core Architectural Components: AF2 vs. AF3
| Component | AlphaFold 2 (Evoformer-Centric) | AlphaFold 3 (Hybrid: Evoformer + Diffusion) |
|---|---|---|
| Primary Innovation | Evoformer block (self-attention + MSA column/row gated self-attention) | Diffusion-based structure decoder operating on atomic densities. |
| Input Scope | Protein amino acid sequence(s) + MSA + templates. | Arbitrary biomolecular inputs (proteins, DNA, RNA, ligands, ions). |
| Representation | Pairwise residue distances and orientations (frames). | Atomic point cloud in 3D space, represented as a diffusion process. |
| Output Mechanism | Deterministic, end-to-end differentiable direct prediction of coordinates. | Probabilistic, iterative refinement from noise to structure via a reverse diffusion process. |
| Confidence Metric | Predicted Local Distance Difference Test (pLDDT) and Predicted Aligned Error (PAE). | Confidence scores for atoms, interactions (e.g., protein-ligand), and composite structures. |
| Training Objective | Minimize FAPE loss on ground truth structures. | Denoising score matching objective on a distribution of structures. |
Table 2: Key Quantitative Performance Metrics (Representative Examples)
| System / Benchmark | Protein Structure (CASP15) | Protein-Ligand (PDBBind) | Protein-Nucleic Acid | Antibody-Antigen |
|---|---|---|---|---|
| AlphaFold 2 | ~90% GDT_HS (high accuracy) | Not Applicable (N/A) | Limited capability | Moderate (via multimer mode) |
| AlphaFold 3 | Comparable to AF2 | ~70% success rate (RMSD < 2Ã…, top-ranked pose) | ~70% interface TM-score improvement over AF2 | Significant improvement in CDR loop accuracy |
Detailed Experimental Protocols
Protocol 1: Training the AlphaFold 3 Diffusion Model
Objective: To train the diffusion network to generate atomic structures conditioned on evolutionary and template information from the Evoformer stack.
- Data Preparation: Assemble a dataset of biomolecular complexes from the PDB (including proteins, nucleic acids, ligands, etc.). Preprocess to generate input token sequences, multiple sequence alignments (MSAs), and template features for all components.
- Forward Diffusion Process: For each training complex, define a forward process that gradually adds Gaussian noise to the atomic coordinates over T timesteps (e.g., T=1000), producing a sequence of increasingly noisy structures ( xt ), where ( xT ) is approximately pure noise.
- Conditioning Encoding: Process the input biomolecular sequences through the Evoformer stack (inherited and adapted from AF2) to generate a deep, context-rich representation (
conditioning). - Reverse Diffusion Training: Train a neural network (the diffusion model) to predict the added noise ( \epsilon ) (or the clean coordinates ( x0 )) at a given noisy state ( xt ), conditioned on the Evoformer's output and the timestep t. The loss function is typically a mean-squared error between the predicted and true noise.
- Optimization: Use distributed training with Adam optimizer, gradient checkpointing, and mixed precision (bfloat16) across a large-scale TPU v4/v5 pod.
Protocol 2: Inference for Biomolecular Complex Prediction
Objective: To predict the 3D structure of a user-defined biomolecular complex using a trained AF3 model.
- Input Feature Generation: For the target complex (e.g.,
Protein A + DNA strand + small molecule), run MMseqs2 and HMMer to generate MSA and evolutionary coupling data for each macromolecular component. Extract potential template structures from the PDB. - Evoformer Processing: Embed the sequences and features. Pass them through the Evoformer stack to generate a unified, information-saturated pair representation of the entire complex.
- Diffusion-Based Sampling (Structure Decoding): a. Initialize the 3D atomic coordinates of the entire complex as random noise (( xT )). b. For *t* from *T* down to 1: - Input the noisy coordinates ( xt ) and the Evoformer conditioning into the trained diffusion model. - Predict the noise component ( \epsilon\theta(xt, t, conditioning) ). - Use the sampling rule (e.g., DDPM or DDIM) to compute a less noisy estimate ( x{t-1} ). c. The final output ( x0 ) is the predicted atomic coordinates of the complex.
- Confidence Estimation: Run auxiliary prediction heads on the final latent representation to output per-atom confidence scores and pairwise interaction accuracies.
- Analysis: Visualize the predicted structure and confidence metrics. Optionally, run multiple sampling iterations to assess prediction variability.
Visualization Diagrams
Diagram 1: AlphaFold 3 High-Level Workflow
Diagram 2: Diffusion Network Sampling Process
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials & Digital Tools for AF3-Inspired Research
| Item / Solution | Category | Function / Explanation |
|---|---|---|
| ColabFold | Software Suite | Provides an accessible, cloud-based implementation of AF2/AF-multimer, essential for baseline comparisons and prototyping. |
| AlphaFold Server | Web Service | Direct access to the official AlphaFold 3 engine for biomolecular complex prediction (as made available by Isomorphic Labs). |
| OpenMM | Molecular Dynamics | Toolkit for running post-prediction refinement and molecular dynamics simulations on AF3 outputs to assess stability. |
| PDBbind Dataset | Benchmark Dataset | Curated database of protein-ligand complexes for training and rigorously evaluating docking/prediction accuracy. |
| RDKit | Cheminformatics | Open-source library for handling small molecule input (SMILES, SDF) and analyzing protein-ligand interaction geometries. |
| PyMOL / ChimeraX | Visualization | Critical software for visualizing, analyzing, and presenting the predicted 3D structures and confidence maps. |
| JAX / Haiku | Deep Learning Framework | The underlying framework for AlphaFold implementations; necessary for custom model development and modification. |
| HMMER / MMseqs2 | Bioinformatics Tools | Standard tools for generating critical input features (MSAs) from sequence databases. |
| Oseltamivir | Oseltamivir Phosphate | Oseltamivir phosphate, a potent neuraminidase inhibitor. For Research Use Only. Not for diagnostic or personal use. |
| DTME | DTME, CAS:71865-37-7, MF:C12H12N2O4S2, MW:312.4 g/mol | Chemical Reagent |
Application Notes
The success of AlphaFold 3 (AF3) in predicting the structures of biomolecular complexes (proteins, nucleic acids, ligands, ions) hinges on its training on a vast, heterogeneous corpus of structural and sequence data. The primary source is the Protein Data Bank (PDB), augmented by diverse complementary datasets. This integrated training approach enables the model to learn the physical and geometric constraints governing molecular interactions.
Table 1: Core Datasets for Training AlphaFold 3-like Models
| Dataset | Primary Content | Scale (Approx.) | Role in Training |
|---|---|---|---|
| Protein Data Bank (PDB) | Experimental 3D structures (X-ray, Cryo-EM, NMR) of proteins, complexes, and ligands. | ~220,000 structures | Ground truth for structural supervision; teaches atomic-level geometry and intermolecular interfaces. |
| PDB-derived Multiple Sequence Alignments (MSAs) | Evolutionary correlations from homologous sequences for proteins in the PDB. | Billions of sequences | Provides evolutionary constraints and co-evolutionary signals for fold and interface prediction. |
| Molecular Components Dictionary | Chemical descriptions of small molecules, ions, and modified residues (e.g., from PDB chemical component IDs). | ~70,000 unique compounds | Defines chemical identity, bond topology, and stereochemistry for non-macromolecular entities. |
| Predicted Structures Database | High-confidence predicted structures (e.g., from AlphaFold DB, ESMFold). | Millions of predictions (e.g., 200+ million from AFDB) | Expands structural diversity for protein monomers, especially for underrepresented families. |
| Genomic & Metagenomic Databases | Protein and RNA sequences from diverse organisms (UniRef, MGnify). | Billions of sequences | Broadens the evolutionary landscape captured in MSAs, enhancing generalization. |
Protocols
Protocol 1: Curating a PDB-Derived Training Set for Biomolecular Complexes Objective: To compile a high-quality, non-redundant set of biomolecular complexes from the PDB for training.
- Data Retrieval: Download the entire PDB archive in mmCIF format. Use the
pdb_components.ciffile for full chemical descriptions of ligands. - Initial Filtering: Filter entries based on:
- Resolution: ≤ 3.2 Å for X-ray/cryo-EM structures.
- Deposition Date: Include all, but stratify by date for temporal training splits.
- Polymer Types: Include entries containing proteins, DNA, RNA, and/or hybrid complexes.
- Complex Definition: Use biological assembly annotations (from
pdb1.ciffiles) to extract biologically relevant quaternary structures. - Deduplication: Apply a sequence identity clustering tool (e.g., MMseqs2) at 30% sequence identity across all chains to create a non-redundant set. Retain the highest-resolution structure per cluster.
- Ligand & Ion Extraction: Parse the
_chem_compand_struct_refcategories to identify and extract all non-polymer entities bound to the macromolecular assembly. Validate bond geometries against the Chemical Components Dictionary. - Split Creation: Partition the dataset into training (90%), validation (5%), and test (5%) sets, ensuring no significant sequence or structural similarity between splits (using cluster membership).
Protocol 2: Generating Complementary Multiple Sequence Alignments (MSAs) Objective: To create deep MSAs for each protein chain in the training set to provide evolutionary context.
- Target Sequence Preparation: Extract the amino acid sequence for each protein chain from the processed mmCIF files.
- Homology Search: For each target sequence, perform iterative searches against large sequence databases:
- Primary Search: Use JackHMMER or HHblits against the UniRef90 database (3 iterations, E-value < 0.001).
- Expanded Search: Use the resulting profile to search the massive metagenomic sequence database (MGnify) for additional diverse homologs.
- Alignment Construction: Build a consensus MSA from all significant hits. Filter sequences to a maximum of 80% pairwise identity to reduce bias.
- Pairing Logic: For complex targets, create paired MSAs. For heterodimers, align sequences from the interacting species/clades. For homomers, treat sequences from the same genome as paired.
Visualizations
Title: AF3 Training Data Curation Workflow
The Scientist's Toolkit: Research Reagent Solutions
| Item/Resource | Function in Dataset Curation & Training |
|---|---|
| PDB mmCIF Files | Standardized, machine-readable format containing full structural data, annotations, and chemical details for each entry. |
| Chemical Components Dictionary | Reference library defining chemical attributes (bonds, angles, chirality) for every small molecule and ion in the PDB. Essential for modeling ligands. |
| MMseqs2 | Ultra-fast, sensitive protein sequence searching and clustering suite. Used for deduplication and creating sequence profiles. |
| JackHMMER/HHblits | Profile hidden Markov model tools for sensitive, iterative homology searching to build deep, informative MSAs. |
| UniRef90 & MGnify | Curated (UniRef90) and massive environmental (MGnify) sequence databases. Provide the evolutionary breadth for MSA construction. |
| BioPython & PDBeCIF API | Programming libraries for parsing, manipulating, and analyzing PDB data and mmCIF files programmatically. |
| TensorFlow / JAX | Deep learning frameworks used to implement and train the AlphaFold 3 neural network architecture on the curated dataset. |
| Google Cloud TPU v4/v5 | Specialized hardware accelerators critical for training large models like AF3 on massive datasets in a feasible timeframe. |
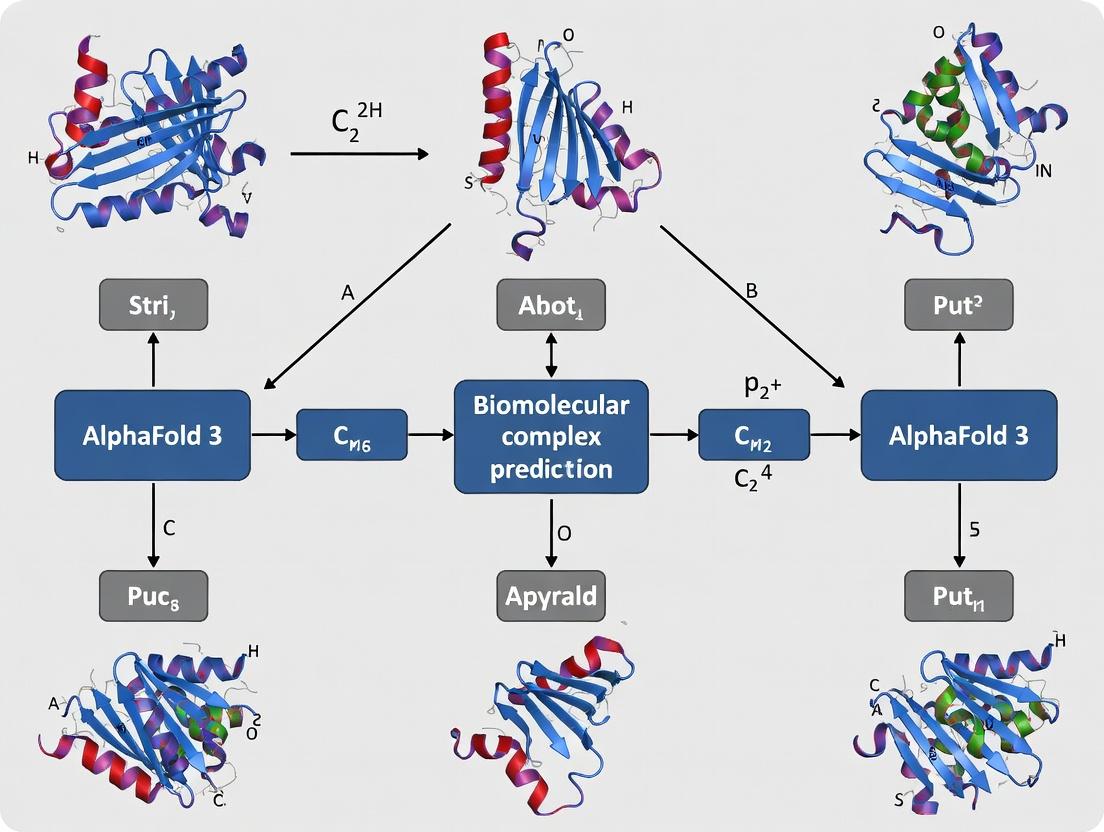
The release of AlphaFold 3 by Google DeepMind and Isomorphic Labs marks a transformative advance in predicting the structure and interactions of biomolecular complexes, including proteins, nucleic acids, ligands, and post-translational modifications. For researchers integrating this tool into a thesis on biomolecular complex prediction, the choice between using the AlphaFold Server (the publicly accessible web interface) and a Local Implementation (running the model on in-house infrastructure) is critical. This decision directly impacts experimental design, throughput, cost, and the control over sensitive data. These application notes provide a detailed comparison and protocols to guide this choice within a rigorous research workflow.
Quantitative Comparison: Access, Hardware, and Performance
Table 1: Core Access and Computational Requirements Comparison
| Feature | AlphaFold Server (Public Web Interface) | Local Implementation (AlphaFold 3 Code) |
|---|---|---|
| Availability | Free public access at alphafoldserver.com; limited to non-commercial research. | Requires access to the codebase via ISM Labs; commercial use possible via licensing. |
| Daily Limit | ~20 jobs per day (subject to change). | No inherent limit; constrained by local compute resources. |
| Input Limitations | Protein, DNA, RNA, and selected ligands (phosphorylation, etc.). Limited to complexes with ≤ 3,840 total residues. | Potentially broader scope as defined by the underlying model; subject to same residue limits. |
| Hardware Provision | Managed by Google/Isomorphic Labs (likely TPU v4/v5 pods). | Researcher's responsibility. Requires high-end GPU (e.g., NVIDIA A100/H100, 40GB+ VRAM). |
| Typical Runtime | Minutes to a few hours, depending on complex size and server queue. | Highly variable: 10 mins to >10 hours per prediction, based on hardware, sequence length, and MSAs. |
| Data Privacy | Input sequences and results are stored temporarily but may be logged for service improvement. | Full control; data never leaves the local system. Essential for proprietary drug discovery. |
| Cost Model | Free for non-commercial use. | High upfront capex for hardware or ongoing cloud compute costs (~$5-$50+ per prediction on cloud). |
| Customization | None. Fixed pipelines and parameters. | Full control over model parameters, MSA generation tools, relaxation protocols, and sampling. |
Table 2: Estimated Local Hardware Requirements & Cloud Costs
| Resource | Minimum Viable | Recommended for Thesis Research | High-Throughput (Small Lab) |
|---|---|---|---|
| GPU | NVIDIA RTX 4090 (24GB VRAM) | NVIDIA A100 (40/80GB VRAM) | 2-4 x NVIDIA H100 or A100 |
| CPU Cores | 16+ | 32+ | 64+ |
| System RAM | 64 GB | 128 GB | 256 GB+ |
| Storage (SSD) | 1 TB | 2-4 TB | 10 TB+ (for databases) |
| Cloud Cost/Job* | ~$3-10 (Spot/Preemptible) | ~$10-25 (On-Demand) | N/A (Dedicated Cluster) |
| Suitability | Testing, small complexes. | Core thesis work; most complexes. | Large-scale screening, parameter exploration. |
Estimated cost for a single prediction of a ~500-residue complex on major cloud providers (AWS, GCP, Azure).
Experimental Protocols for Thesis Research
Protocol 3.1: Submitting a Prediction to the AlphaFold Server
Objective: To obtain a predicted structure for a biomolecular complex using the public web server.
- Prepare Input Sequences: Format your protein (and/or DNA/RNA) sequences in standard FASTA format. Define molecular chains in the format:
>chain_id. For ligands, specify the SMILES string in the provided interface. - Job Configuration: Access
alphafoldserver.com. Paste sequences. Use the toggle menus to define molecule types (e.g., "Protein," "DNA"). For modifications like phosphorylation, select the appropriate residue and modification type. - Submission & Queue: Submit the job. Note the job ID. The system will provide an estimated completion time.
- Results Retrieval: Download all result files upon email notification or page refresh. Key outputs include:
ranked_0.pdb: The top-ranked predicted structure.confidence_scores.json: Predicted per-residue and pairwise confidence metrics (pLDDT, pTM, ipTM, interface PAE).- Visualizations (
.pse,.png).
Protocol 3.2: Local Installation and Prediction (Simplified Workflow)
Objective: To install and run AlphaFold 3 locally for high-throughput or proprietary research. Pre-requisite: This assumes access to the AlphaFold 3 code repository and necessary licenses from Isomorphic Labs.
Environment Setup:
Database Download: Download and set up necessary sequence (UniRef90, BFD) and structure (PDB) databases. Paths must be configured in the model config.
- Input Preparation: Create a directory with input
.jsonor.fastafiles as specified by the AlphaFold 3 runner script. Run Prediction:
Post-processing: Analyze the output
*.pdbfiles andscores.jsonusing local scripts for model ranking, relaxation, and visualization (e.g., PyMOL, ChimeraX).
Visualized Workflows
Title: AlphaFold 3 Research Decision Workflow
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagent Solutions for AlphaFold 3-Based Research
| Item | Category | Function in Research | Example/Note |
|---|---|---|---|
| Cloned Gene Constructs | Biological Reagent | Provide the exact protein/DNA sequence for prediction and subsequent experimental validation. | Full-length cDNA in expression vectors (e.g., pET, pcDNA3.4). |
| Purified Protein Complex | Biochemical Reagent | Essential for validating AlphaFold 3 predictions using structural biology methods. | Complex purified via affinity (Ni-NTA, Strep-tag) and size-exclusion chromatography. |
| Crystallization Screen Kits | Structural Biology Reagent | Used for X-ray crystallography to obtain ground-truth structures for benchmark comparisons. | Commercially available screens (e.g., MemGold, PEG/Ion). |
| Cryo-EM Grids | Structural Biology Reagent | Support samples for single-particle cryo-EM, a key validation method for large complexes. | Quantifoil R1.2/1.3 Au or Ultrafoil grids. |
| FRET or SPR Assay Kits | Biophysical Reagent | Quantify binding affinities (Kd) to validate predicted interaction interfaces. | His-tag capture SPR chips (Biacore) or HTRF assay kits. |
| Mutation Kit (SDM) | Molecular Biology Reagent | Generate point mutants to test specific interfacial residues predicted by the model. | QuickChange or Gibson Assembly kits. |
| JAX/JAXlib | Computational Reagent | The core numerical computing library on which AlphaFold 3 runs. | Must match the version specified for compatibility. |
| PyMOL/ChimeraX License | Software Reagent | For high-quality visualization, analysis, and figure generation of predicted structures. | Educational or commercial licenses available. |
| High-Performance GPU | Hardware Reagent | Provides the parallel processing power required for timely local inference. | NVIDIA A100/H100 with maximal VRAM. |
| 3-Aminophenylacetic acid | 3-Aminophenylacetic acid, CAS:14338-36-4, MF:C8H9NO2, MW:151.16 g/mol | Chemical Reagent | Bench Chemicals |
| MeCM | MeCM, CAS:122279-91-8, MF:C36H48O18, MW:768.8 g/mol | Chemical Reagent | Bench Chemicals |
AlphaFold 3's Place in the Computational Biology Ecosystem
The release of AlphaFold 3 by Google DeepMind and Isomorphic Labs represents a paradigm shift in computational structural biology. While its predecessors, AlphaFold 2 and AlphaFold-Multimer, revolutionized single-chain protein structure prediction, AlphaFold 3 expands the horizon to a vast array of biomolecular complexes. This advancement must be understood within the broader thesis of the field: that accurate, atomic-level modeling of multi-component biological systems is the critical next step for mechanistic understanding and therapeutic intervention. This document provides application notes and experimental protocols for leveraging AlphaFold 3 within the contemporary research ecosystem.
Application Notes: Capabilities and Quantitative Benchmarks
AlphaFold 3 predicts the joint 3D structure of complexes containing proteins, nucleic acids (DNA/RNA), small molecules (ligands), and ions, using a diffusion-based architecture. The following tables summarize its performance against previous state-of-the-art tools.
Table 1: Performance on Protein-Ligand Complexes (CASF-2016 benchmark)
| Metric | AlphaFold 3 | GNINA | DiffDock | Traditional Docking (Vina) |
|---|---|---|---|---|
| Top-1 RMSD < 2Ã… (%) | 63.7 | 48.2 | 52.9 | 31.5 |
| Average RMSD (Ã…) | 1.95 | 2.87 | 2.41 | 4.12 |
| Inference Time (min) | ~5-10 | ~1-2 | ~0.5 | ~0.1 |
Table 2: Performance on Protein-Nucleic Acid Complexes
| Complex Type | AlphaFold 3 (TM-score) | AlphaFold-Multimer (TM-score) | Specifity (PPV) |
|---|---|---|---|
| Protein-DNA | 0.91 | 0.79 | 0.92 |
| Protein-RNA | 0.87 | 0.72 | 0.89 |
| RNA-only | 0.85 | N/A | 0.81 |
Table 3: Key Limitations and Considerations
| Aspect | Note |
|---|---|
| Conformational States | Primarily predicts ground state; limited for large conformational changes induced by binding. |
| Very Large Complexes | Performance degrades on complexes > 5,000 residues. Memory and time intensive. |
| Post-Translational Modifications | Limited direct modeling; often requires input as modified residue. |
| Dynamics & Entropy | Provides a static snapshot; no direct energy or affinity scores. |
| Access Model | Available via the AlphaFold Server (non-commercial use), not open-source. |
Experimental Protocol: Structure Prediction for a Protein-Small Molecule Complex
This protocol details the steps for predicting the structure of a protein kinase bound to an ATP-competitive inhibitor using the public AlphaFold Server.
Objective
To generate an atomic model of the human CDK2 protein in complex with a novel inhibitor compound (SMILES: CC1=NC=C(C(=C1)Cl)NC(=O)C2=CC(=C(C=C2)F)NS(=O)(=O)C3=CC=CS3).
Materials & Reagent Solutions
The Scientist's Toolkit:
| Item | Function |
|---|---|
| AlphaFold Server (server.predictions.alphabetafold.com) | Web interface for AlphaFold 3 predictions. |
| Protein Sequence (UniProt ID: P24941) | The primary amino acid sequence of the target protein. |
| Ligand SMILES String | Standardized molecular input for the small molecule. |
| Multiple Sequence Alignment (MSA) Tool (e.g., HMMER, MMseqs2) | Optional for pre-analysis; server generates its own. |
| Molecular Visualization Software (e.g., PyMOL, UCSF ChimeraX) | For analyzing and rendering output models. |
| Structure Validation Server (e.g., PDB Validation, MolProbity) | To assess stereochemical quality of predictions. |
Procedure
Input Preparation:
- Obtain the canonical amino acid sequence for CDK2 from the UniProt database (P24941). Ensure no tags or non-standard residues are present.
- Define the small molecule ligand using its canonical SMILES string. Verify the string's chemical validity using a tool like RDKit.
Submission to AlphaFold Server:
- Navigate to the AlphaFold Server.
- In the input field labeled "Protein," paste the CDK2 amino acid sequence.
- Click "Add a molecule" and select "Small molecule (SMILES)." Paste the SMILES string into the provided field.
- (Optional) Adjust advanced settings. For initial run, use defaults:
- Number of models: 5
- Number of recycles: 12
- MSA mode: "Full" (recommended for accuracy).
- Start the prediction. A typical job for a ~300 residue protein with one small molecule will take approximately 10 minutes.
Output Analysis:
- The server returns:
- Ranked models: 5 predicted structures (PDB format), ordered by predicted confidence.
- Predicted Aligned Error (PAE) plot: Assesses inter-domain and protein-ligand confidence.
- Per-residue confidence scores (pLDDT): Indicates local model confidence (0-100).
- Compound confidence score: A per-atom and overall score for the ligand pose.
- Download the ranked PDB files and the PAE JSON file.
- The server returns:
Model Validation and Selection:
- Open the top-ranked model in molecular visualization software.
- Inspect the ligand binding pose. Check for plausible hydrogen bonds, hydrophobic contacts, and complementarity with the known ATP-binding site.
- Cross-reference the PAE plot: Low error (dark blue) between the protein binding pocket and the ligand indicates high confidence in their relative placement.
- Run the model through a validation server like MolProbity to check for clashes and proper stereochemistry.
Downstream Experimental Design:
- Use the predicted interface residues to guide site-directed mutagenesis for binding affinity assays.
- The model can serve as a starting point for molecular dynamics simulations to assess stability.
- For drug development, the structure enables structure-based optimization of the inhibitor scaffold.
Workflow and Ecosystem Integration Diagrams
AlphaFold 3 Prediction Workflow
AF3 in the Computational Biology Toolchain
How to Use AlphaFold 3: A Practical Guide for Drug Discovery and Research
This protocol, within the context of AlphaFold 3 biomolecular complex structure prediction research, details the process for submitting a job to the public AlphaFold Server. This server provides free access to AlphaFold 3 for non-commercial use, enabling researchers to predict the structure of biomolecular complexes (proteins, nucleic acids, ligands, etc.).
Prerequisites and Input Preparation
Research Reagent Solutions & Essential Materials
| Item | Function/Explanation |
|---|---|
| Target Protein Sequence(s) | Primary amino acid sequence(s) in FASTA format. The core input for prediction. |
| Ligand SMILES String (Optional) | Simplified Molecular-Input Line-Entry System string defining the chemical structure of a small molecule ligand to be modeled in the complex. |
| Nucleic Acid Sequence (Optional) | DNA or RNA sequence to be co-modeled with protein(s). |
| AlphaFold Server Account | A free Google or DeepMind account is required to access the server and manage jobs. |
| Web Browser | A modern browser (Chrome, Firefox, Safari, Edge) with JavaScript enabled. |
| Job Title & Notes | Descriptive metadata to organize and identify predictions within your research portfolio. |
Input Specifications Table
| Parameter | Requirement | Notes |
|---|---|---|
| Protein Sequence Length | Recommended ≤ 2,000 residues total. | Performance decreases for very large complexes. |
| Number of Protein Chains | Up to 5. | Defined as separate sequences in the input. |
| Ligand Input | SMILES string, one per molecule. | Maximum of 5 ligands. Must specify which chain it binds to. |
| Nucleic Acid Input | Sequence string (A,C,G,T,U). | Can be specified as DNA or RNA. |
| Output Formats | PDB, CIF, per-residue confidence scores (pLDDT, PAE). | All provided in a single downloadable ZIP file. |
Step-by-Step Submission Protocol
1. Access: Navigate to the official AlphaFold Server website (https://alphafoldserver.com) and sign in.
2. Input Sequences: * Click "Create new prediction". * In the provided text area, paste your protein sequence(s) in FASTA format. For multiple chains, use separate FASTA headers. * Use the "Add molecule" button to include ligands or nucleic acids as needed.
3. Configure Prediction (Optional): * Assign logical names to each input molecule for clarity in results. * For ligands, map the SMILES string to a specific target protein chain.
4. Review and Submit: * Provide a descriptive job title and any relevant notes. * Review all inputs for accuracy. * Click "Run prediction" to submit the job to the queue.
5. Monitor and Retrieve: * Jobs are listed on the main dashboard with status (Queued, Running, Complete, Failed). * Completion time varies from minutes to several hours based on server load and target size. * Download the results ZIP file upon completion.
Results Analysis and Interpretation
Key output files and their interpretation are summarized below.
AlphaFold Server Output Files & Metrics
| File Name | Content | Interpretation Guide |
|---|---|---|
model_[1-5].pdb / .cif |
Atomic 3D coordinates of the predicted complex. | The PDB/CIF file for visualization and analysis. Models are ranked by confidence. |
ranked_[0-4].pdb |
The 5 models, reordered by average confidence (pLDDT). | ranked_0.pdb is the highest confidence prediction. |
scores.json |
Contains per-residue pLDDT and pairwise alignment error (PAE). | pLDDT: >90 very high, 70-90 confident, 50-70 low, <50 very low. PAE: Estimates positional error between residues (lower is better). |
predicted_aligned_error.png |
Visualization of the PAE matrix. | Shows estimated confidence in the relative position of different parts of the complex. |
Title: AlphaFold Server Prediction Workflow
Experimental Validation Protocol (Computational)
To benchmark a predicted complex from the AlphaFold Server within a research thesis, the following in silico protocol is recommended.
Protocol: Computational Validation of a Predicted Protein-Ligand Complex
Objective: To assess the quality and reliability of an AlphaFold Server-generated biomolecular complex structure.
Materials:
- AlphaFold Server output ZIP file (
ranked_0.pdb,scores.json). - Molecular visualization software (e.g., UCSF ChimeraX).
- Structural analysis tools (e.g., PyMOL, MolProbity server).
- Reference structure (if available; e.g., from PDB).
Methodology:
- Confidence Metric Analysis: Extract the global average pLDDT from
scores.json. Plot per-residue pLDDT along the sequence to identify low-confidence regions. Examine the PAE plot to assess inter-domain or inter-chain confidence. - Steric Clash and Geometry Validation: Upload the
ranked_0.pdbfile to the MolProbity server. Analyze the output report, focusing on the Ramachandran outliers percentage, sidechain rotamer outliers, and clashscore. Acceptable thresholds are >90% favored Ramachandran, <5% rotamer outliers, and clashscore <10. - Comparative Analysis (If Reference Exists): Align the predicted structure to an experimentally determined reference using the
aligncommand in PyMOL/ChimeraX. Calculate the Root-Mean-Square Deviation (RMSD) of the protein backbone and ligand heavy atoms. - Interaction Analysis: Manually inspect the predicted binding interface in ChimeraX. Identify hydrogen bonds, hydrophobic contacts, and salt bridges. Compare the predicted ligand pose and interactions to known biochemical data or similar complexes in the PDB.
Title: Computational Validation Protocol Flow
Within the broader thesis on AlphaFold 3 for biomolecular complex structure prediction, meticulous input preparation is the foundational step that dictates the success or failure of a modeling run. AlphaFold 3 extends beyond monomeric proteins to predict the structures of complexes containing proteins, nucleic acids, small molecule ligands, and post-translational modifications (PTMs). This document provides detailed application notes and protocols for preparing the three core input types: sequence files, ligand SMILES strings, and modification specifications, based on the current AlphaFold 3 framework and related research.
Sequence File Preparation
Sequence files provide the primary amino acid or nucleotide sequences for all macromolecular components in the complex.
Protocol: Generating Standardized Input Sequences
Objective: To produce clean, correctly formatted FASTA files for all protein and nucleic acid chains in the complex.
Sequence Sourcing:
- For proteins, retrieve canonical sequences from authoritative databases (UniProt, NCBI). For nucleic acids, use databases like NCBI Nucleotide or RCSB PDB.
- Critical Step: Verify the organism and isoform. Cross-reference with experimental context (e.g., expression system).
Sequence Curation:
- Remove ambiguous residues (e.g., 'X', 'J', 'Z'). Replace with the most likely residue based on homology or experimental data, or consider modeling alternative conformations.
- For multi-chain complexes, create a single FASTA file where each chain is a separate entry.
- The header line should be formatted as a unique identifier. AlphaFold 3 accepts standard FASTA headers.
Formatting for AlphaFold 3:
- Save the file in plain text format with the
.fastaextension. - Example multi-chain FASTA format for a protein-ligand complex:
- Save the file in plain text format with the
Table 1: Accepted Sequence Types and Database Sources
| Component Type | Standard Alphabets | Primary Source DB | Notes for AlphaFold 3 Input |
|---|---|---|---|
| Protein | Standard 20 AAs | UniProt | Use canonical sequence. Signal peptides may be retained or removed based on modeling goal. |
| DNA | A, T, C, G | NCBI Nucleotide | Specify single-stranded or double-stranded in complex definition. |
| RNA | A, U, C, G | NCBI Nucleotide, RNAcentral | Include modified base specifications separately (see Section 3). |
Ligand SMILES String Specification
Small molecules are defined using Simplified Molecular Input Line Entry System (SMILES) strings, which encode molecular structure in a single line of text.
Protocol: Preparing and Validating Ligand SMILES
Objective: To generate standardized, isomeric SMILES strings that accurately represent the ligand's chemical identity and stereochemistry.
Ligand Identification:
- Identify the ligand's canonical name and obtain its PubChem CID (or ChEBI ID).
- Use the PubChem Compound database or ChEBI to retrieve the chemical structure.
SMILES Generation and Curation:
- Download or generate the isomeric SMILES string from the database. This string includes stereochemical specifications (e.g.,
@and@@for tetrahedral centers). - Validation: Input the SMILES into a cheminformatics toolkit (e.g., RDKit, Open Babel) to generate a 2D structure and verify it matches the expected compound.
- Download or generate the isomeric SMILES string from the database. This string includes stereochemical specifications (e.g.,
Formatting for Input:
- Ligand SMILES are typically incorporated into a separate ligand definition file or a combined JSON configuration.
- Each ligand must be assigned a unique chain ID (e.g., "LIG_A").
- Example entry in a ligands list:
{"chain_id": "LIG_A", "smiles": "CN1C=NC2=C1C(=O)N(C(=O)N2C)C"}(Caffeine).
Table 2: Common Ligand Types and SMILES Preparation Workflow
| Ligand Class | Example | Key Preparation Step | AlphaFold 3 Consideration |
|---|---|---|---|
| Drug-like small molecule | Imatinib (STI-571) | Ensure correct tautomer and protonation state at physiological pH. | Model may predict binding pose but not absolute binding affinity. |
| Cofactor (organic) | Heme | SMILES may represent a substructure. Coordinate metal ions (Fe2+) separately as modifications. | Treat as a rigid fragment or allow conformational flexibility. |
| Ion (metal) | Mg2+, Zn2+ | Represented as elemental symbol in SMILES ([Mg+2]). |
Define coordination geometry via distance constraints if known. |
| Modified nucleotide | S-Adenosyl methionine (SAM) | Use isomeric SMILES from PubChem. The sulfonium center is crucial. | The positive charge on sulfur is part of the SMILES representation. |
Modification Specification
Modifications define covalent changes to standard residues or nucleotides, including PTMs, point mutations, and covalent ligands.
Protocol: Defining Post-Translational and Chemical Modifications
Objective: To accurately specify the type and location of all non-standard components in the complex.
Inventory Modifications:
- List all PTMs (phosphorylation, acetylation, glycosylation), non-canonical amino acids (selenocysteine), point mutations (e.g., Cys->Ser), and covalently attached probes (e.g., fluorescent labels).
Specification Format:
- Modifications are defined in a machine-readable list, often JSON. Each entry requires:
chain_id: The macromolecule chain containing the modification.residue_number: The sequential residue index.modification_type: A standardized name (e.g.,phosphorylation,N6-methyladenosine).- For complex modifications (e.g., glycosylation), additional parameters like glycan composition may be required.
- Modifications are defined in a machine-readable list, often JSON. Each entry requires:
Integration with Sequence:
- The modification spec works in conjunction with the base sequence file. The base sequence contains the parent residue (e.g., 'S' for serine), and the modification spec transforms it (e.g., to phosphoserine).
Table 3: Common Modification Types and Their Specifications
| Modification Type | Residue | Specification Key | Example Value (modification_type) |
|---|---|---|---|
| Phosphorylation | S, T, Y | phosphorylation |
phosphorylation |
| N-linked Glycosylation | N (in N-X-S/T motif) | glycosylation |
glycosylation:man5 |
| Disulfide Bond | CYS | disulfide_partner |
{"chain_id": "A", "residue_number": 42} |
| Point Mutation | Any | mutation |
mutation:V->L |
| Methylation (DNA) | C | methylation |
5-methylcytosine |
Experimental Protocol: Integrated Input Generation for a Kinase-Inhibitor Complex
Aim: To prepare all necessary input files for predicting the structure of Human EGFR Tyrosine Kinase bound to the covalent inhibitor Afatinib, including a phosphorylation site.
Materials & Reagents:
- EGFR kinase domain sequence (UniProt P00533, residues 696-1022).
- Afatinib PubChem CID (CID 10184653).
- Knowledge of activation loop phosphorylation (Tyr-869).
Procedure:
- Sequence File (
egfr_afatinib.fasta):- Retrieve the amino acid sequence for residues 696-1022 of human EGFR from UniProt.
- Create a FASTA file with a single entry:
>EGFR_kinase_domain.
- Ligand Definition File (
ligands.json):- From PubChem, obtain the isomeric SMILES for Afatinib.
- Create a JSON file:
[{"chain_id": "AFT", "smiles": "CN1C=NC(=O)C(=C1C=CC2=CC(=C(C=C2)F)NC(=O)C=C)C#C"}].
- Modification Specification File (
mods.json):- Define the phosphorylation at residue Tyr-869 (which is residue 174 in the provided kinase domain sequence).
- Create a JSON file:
[{"chain_id": "EGFR_kinase_domain", "residue_number": 174, "modification_type": "phosphorylation"}].
- AlphaFold 3 Run Command:
- Using a hypothetical command-line interface:
alphafold3 --fasta egfr_afatinib.fasta --ligands ligands.json --modifications mods.json --output_dir ./results/.
- Using a hypothetical command-line interface:
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Input Preparation |
|---|---|
| UniProt Knowledgebase | Definitive source for canonical and isoform protein sequences, including natural variants and some PTMs. |
| PubChem Compound | Primary public repository for chemical structures, properties, and isomeric SMILES strings of small molecules. |
| RDKit | Open-source cheminformatics toolkit used to validate, standardize, and manipulate SMILES strings. |
| ChEBI | Specialized database for biologically relevant small molecules, providing curated annotations and SMILES. |
| PDB Chemical Component Dictionary | Reference for standard residues, ligands, and modifications, ensuring naming consistency. |
| BioPython SeqIO | Toolkit for parsing, editing, and writing biological sequence files in various formats. |
| Antimony(V) phosphate | Antimony(V) Phosphate | High-Purity Reagent |
| Prenyl acetate | Prenyl acetate | Natural Flavor & Pheromone Research |
Visualizations
Workflow for AlphaFold 3 Input Preparation
Input Data Integration in AlphaFold 3
Within the broader thesis on AlphaFold 3 (AF3) biomolecular complex structure prediction research, the accurate interpretation of confidence metrics is paramount. AF3 predicts structures for diverse biomolecular complexes (proteins, nucleic acids, ligands), but the reliability varies across the model. This application note details the core metrics—pLDDT and PAE—enabling researchers and drug development professionals to assess prediction quality, identify reliable regions, and guide experimental validation.
Core Confidence Metrics: Definitions and Interpretation
Predicted Local Distance Difference Test (pLDDT)
pLDDT is a per-residue estimate of local confidence on a scale from 0 to 100. It measures the confidence in the local backbone atom placement.
Interpretation Table:
| pLDDT Score Range | Confidence Band | Structural Interpretation | Suggested Use in Research |
|---|---|---|---|
| 90 – 100 | Very high | Backbone prediction is highly reliable. Atomistic details (e.g., side-chain rotamers) can be trusted. | High-confidence docking, detailed mechanistic hypothesis. |
| 70 – 90 | Confident | Backbone is generally reliable. Overall fold is correct, but local variations may exist. | Building models for complexes, guiding mutagenesis. |
| 50 – 70 | Low | Prediction may have errors in backbone placement. Caution required. | Low-resolution guidance. Requires experimental validation. |
| 0 – 50 | Very low | Prediction is unreliable. Often corresponds to disordered regions. | Treat as intrinsically disordered or omit from analysis. |
Predicted Aligned Error (PAE)
PAE is a 2D matrix (in Ångströms) representing the expected positional error between residue i and residue j if the predicted structure were aligned on residue i. It is the key metric for assessing the relative confidence within a complex.
- Low PAE (<10 Ã…): The relative position/distance between the two residues is predicted with high confidence.
- High PAE (>20 Ã…): The relative spatial relationship is uncertain.
PAE Patterns for Complexes:
- Intra-chain/Intra-molecule: Low error within a well-folded domain.
- Inter-chain/Inter-molecule: Critical for complexes. Low PAE between interacting subunits suggests high confidence in the predicted interface geometry. High PAE suggests flexibility or uncertainty in the quaternary assembly.
Structured Data Presentation: Metric Comparison
Table 1: Comparative Summary of AF3 Confidence Metrics
| Metric | Scope | Output Range | Low Confidence Indicator | High Confidence Indicator | Primary Use in Complex Analysis |
|---|---|---|---|---|---|
| pLDDT | Per-residue (local) | 0 – 100 | < 50 | > 70 | Identifying well-folded domains vs. disordered regions within each chain. |
| PAE | Pairwise (relative) | 0 to ~40 Ã… | > 20 Ã… | < 10 Ã… | Validating the predicted interface and overall complex topology. |
| Predicted TM-score | Global (per chain) | 0 – 1 | < 0.5 | > 0.7 | Estimating overall fold similarity to a hypothetical true structure. |
| iptm+ptm | Interface (complex) | 0 – 1 | < 0.4 | > 0.8 | Composite score reflecting the accuracy of the multimeric interface prediction (AF2-multimer legacy). |
Experimental Protocols for Validation
Protocol 4.1: In-silico Confidence Analysis of an AF3 Complex Prediction
Objective: Systematically evaluate the reliability of a predicted protein-ligand complex. Materials: AF3 prediction output (PDB file, ranked_*.pkl JSON file), visualization software (PyMOL, UCSF ChimeraX), Python environment with ColabDesign/AF3 analysis tools. Procedure:
- Visualize pLDDT: Load the prediction in PyMOL. Color the structure by the B-factor column (which stores pLDDT). Identify low-confidence regions (often colored red).
- Generate PAE Plot: Use the provided parsing script on the JSON file to extract the PAE matrix. Plot using matplotlib (
imshow()). Label axes with chain identifiers. - Interface Analysis: On the PAE plot, draw boxes to highlight inter-chain regions. Calculate the average PAE for residues within 10Ã… of the interface in the 3D model.
- Decision Point: If interface PAE < 12 Ã… and interacting residues have pLDDT > 70, proceed to in vitro validation. If not, consider the prediction speculative.
Protocol 4.2: Cross-validation with HDX-MS (Hydrogen-Deuterium Exchange Mass Spectrometry)
Objective: Experimentally validate the solvent accessibility and dynamics of a predicted protein-protein interface. Methodology:
- Sample Preparation: Purify individual proteins and the formed complex in identical buffers (e.g., 20 mM HEPES, 150 mM NaCl, pH 7.4).
- Deuterium Labeling: Dilute protein/complex into D₂O buffer. Perform labeling at multiple time points (e.g., 10s, 1min, 10min, 1h) at 25°C.
- Quenching & Digestion: Quench with low pH/pH 2.5 buffer on ice. Pass over immobilized pepsin column for rapid digestion.
- MS Analysis: Inject peptides onto LC-MS. Monitor mass shift of peptides.
- Data Analysis: Calculate deuteration level per peptide. Compare deuteration rates of free protein vs. complex. A significant decrease in deuteration for peptides mapping to the predicted interface confirms protection due to binding, supporting the AF3 model.
Mandatory Visualizations
Title: Decision Workflow for Validating AF3 Complex Predictions
Title: PAE Plot Interpretation Guide for a Protein Dimer
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for AF3 Prediction and Validation
| Item | Function in AF3 Complex Research | Example/Supplier |
|---|---|---|
| AlphaFold 3 Server / ColabFold | Provides access to the AF3 or optimized open-source models for complex prediction. | Google DeepMind AlphaFold Server; ColabFold (af3.py). |
| Molecular Visualization Software | Enables 3D visualization of predictions colored by confidence metrics. | UCSF ChimeraX, PyMOL. |
| HDX-MS Kit | For experimental validation of protein interfaces and dynamics. | Waters HDX/MS System, Thermo Fisher HDX Platform. |
| Surface Plasmon Resonance (SPR) Chip | To measure binding kinetics (KD) of the predicted complex. | Cytiva Series S Sensor Chip CMS. |
| Size-Exclusion Chromatography (SEC) Column | To assess the oligomeric state and stability of the complex in solution. | Bio-Rad ENrich SEC 650, Superdex Increase series. |
| Site-Directed Mutagenesis Kit | To generate point mutations for validating critical interface residues identified from the model. | NEB Q5 Site-Directed Mutagenesis Kit. |
| Cryo-EM Grids | For high-resolution structural validation of large or challenging complexes. | Quantifoil R1.2/1.3 Au 300 mesh grids. |
| Bicinchoninic acid | Bicinchoninic acid, CAS:1245-13-2, MF:C20H12N2O4, MW:344.3 g/mol | Chemical Reagent |
| Hastelloy C | Hastelloy C | High-Performance Nickel Alloy | RUO | Hastelloy C is a nickel-chromium-molybdenum alloy for corrosion research. For Research Use Only. Not for diagnostic or therapeutic use. |
Application Notes
Within the broader thesis on AlphaFold 3's capabilities in predicting biomolecular complex structures, its application to SBDD represents a paradigm shift. AlphaFold 3 directly addresses the critical bottleneck in SBDD: the accurate, rapid prediction of drug-target interaction structures, including proteins, nucleic acids, and key post-translational modifications like phosphorylated residues. By generating reliable complex models, it enables rapid virtual screening and rational lead optimization before experimental validation.
Table 1: Impact of AlphaFold 3 on Key SBDD Metrics
| SBDD Stage | Traditional Approach Challenge | AlphaFold 3-Enabled Acceleration | Quantitative Benchmark (Reported/Expected) |
|---|---|---|---|
| Target Identification | Reliance on low-homology templates or apo structures. | Direct prediction of disease-relevant protein-ligand/nucleic acid complexes. | Up to 50% reduction in time to obtain a working structural hypothesis. |
| Virtual Screening | High false-positive rates due to inaccurate binding site geometry. | High-accuracy pocket structure for improved docking pose ranking. | ~30-40% increase in early hit enrichment rates in retrospective studies. |
| Lead Optimization | Iterative cycles of mutagenesis & crystallography are slow and costly. | Rapid in silico evaluation of designed compound variants and point mutations. | Potential to reduce cycle time from months to weeks for computational prioritization. |
| PPI Modulator Design | Extreme difficulty in predicting transient, shallow binding interfaces. | Prediction of protein-protein interaction (PPI) interfaces with putative small molecule binding pockets. | Successful identification of cryptic pockets in several previously "undruggable" targets. |
Experimental Protocols
Protocol 1: AlphaFold 3-Driven Virtual Screening Workflow
Objective: To identify novel hit compounds for a target protein using structure predictions from AlphaFold 3.
Target Preparation:
- Input the target protein sequence (FASTA format) and, if available, known ligand SMILES strings or co-crystal structure ligands into AlphaFold 3 via the Colab notebook or local installation.
- Run the complex prediction job with default settings for protein-ligand complex type.
- Download the top-ranked model (highest predicted TM-score or confidence metric). Extract the protein structure in PDB format and the predicted ligand pose in SDF format.
Binding Site Definition & Pocket Preparation:
- Load the predicted complex into molecular visualization software (e.g., PyMOL, UCSF Chimera).
- Define the binding site using the predicted ligand coordinates (5-10 Ã… radius). Alternatively, use pocket detection algorithms (e.g., fpocket, SiteMap).
- Prepare the protein structure using molecular docking suite utilities (e.g., Schrodinger's Protein Preparation Wizard, AutoDockTools): add hydrogens, assign bond orders, optimize H-bonds, and minimize steric clashes.
Compound Library Docking:
- Prepare a diverse chemical library (e.g., ZINC15, Enamine REAL) in 3D format with minimized geometries.
- Perform high-throughput virtual screening using docking software (e.g., AutoDock Vina, Glide, GOLD). Use the defined binding site from Step 2 as the docking grid center.
- Rank compounds based on docking score (predicted binding affinity) and visual inspection of pose similarity to the AlphaFold 3 predicted ligand geometry.
Post-Screening Analysis & Prioritization:
- Cluster top-scoring compounds by chemical similarity.
- Filter for drug-like properties (Lipinski's Rule of Five, PAINS filters).
- Select 50-100 top-ranked, diverse compounds for in vitro biological assay.
Protocol 2: In Silico Mutagenesis and Affinity Assessment
Objective: To guide lead optimization by predicting the impact of protein mutations or ligand modifications on binding.
Baseline Complex Generation:
- Generate the AlphaFold 3 structure for the wild-type protein in complex with the lead compound (as in Protocol 1, Step 1).
Systematic Mutagenesis:
- For protein-side optimization: Create mutant protein sequences in silico for residues within 5 Ã… of the ligand.
- For ligand-side optimization: Modify the lead compound's core or substituent groups and generate new SMILES strings.
Prediction of Mutant Complexes:
- Submit each mutant protein sequence with the original ligand, or the original protein sequence with each modified ligand SMILES, to AlphaFold 3 for complex prediction.
- Generate 3-5 models per mutant to assess confidence.
Comparative Analysis:
- Align all predicted mutant complexes to the wild-type complex backbone.
- Calculate changes in key intermolecular interactions (H-bonds, salt bridges, pi-stacking) and non-bonded contact surfaces.
- Rank mutations/modifications based on preservation or enhancement of complementary interactions. Prioritize variants for chemical synthesis or gene cloning.
Visualizations
AlphaFold 3 Virtual Screening Protocol
In Silico Mutagenesis Analysis Flow
The Scientist's Toolkit: SBDD Research Reagent Solutions
| Item | Function in AlphaFold 3-Enhanced SBDD |
|---|---|
| AlphaFold 3 Colab Notebook / Local API | Core engine for generating predicted structures of biomolecular complexes (protein-ligand, protein-nucleic acid). |
| Molecular Visualization Software (PyMOL, ChimeraX) | Critical for visualizing predicted models, defining binding pockets, and analyzing intermolecular interactions. |
| Protein Preparation Suite (e.g., Schrodinger Maestro, MOE) | Prepares predicted protein structures for downstream computational tasks: adds missing atoms, corrects protonation states, and performs energy minimization. |
| Molecular Docking Software (AutoDock Vina, Glide, GOLD) | Performs high-throughput virtual screening of compound libraries into the AlphaFold 3-predicted binding site. |
| Chemical Database Access (ZINC, ChEMBL, Enamine) | Source of commercially available or biologically annotated small molecules for virtual screening libraries. |
| Cheminformatics Toolkit (RDKit, Open Babel) | Used for ligand structure manipulation, format conversion, and filtering compounds based on physicochemical properties. |
| High-Performance Computing (HPC) Cluster | Essential for running large-scale AlphaFold 3 predictions or virtual screening campaigns on thousands of compounds. |
| Microplate Reader & Assay Kits (e.g., FP, TR-FRET) | For experimental validation of computationally prioritized hits via binding or functional biochemical assays. |
Within the broader thesis on AlphaFold 3's (AF3) capabilities for predicting biomolecular complex structures, its application to protein-nucleic acid interactions represents a paradigm shift for gene regulation research. Traditional methods for determining these complex structures are slow and resource-intensive. AF3’s ability to generate accurate models of transcription factors, nucleases, and epigenetic readers bound to DNA or RNA sequences accelerates the mechanistic understanding of regulatory events, enabling the rational design of novel therapeutic and synthetic biology tools.
Application Notes: AF3 Performance & Insights
Recent benchmarking studies demonstrate AF3's superior performance in modeling protein-nucleic acid complexes compared to prior tools and experimental maps.
Table 1: Benchmarking AF3 on Protein-Nucleic Acid Complexes
| Metric / Complex Type | AF3 Performance | Comparison to AF2 | Key Insight |
|---|---|---|---|
| Protein-DNA (Average RMSD â„«) | ~1.5-2.5 â„« | ~40-60% improvement | High accuracy in predicting docking geometry and side-chain contacts. |
| Protein-RNA (Average RMSD â„«) | ~2.0-3.5 â„« | ~50% improvement | Robust performance on diverse RNA backbones and non-canonical structures. |
| Interface Distance Accuracy | < 4.0 â„« (90% of cases) | Significant improvement | Reliable identification of key hydrogen-bonding and electrostatic interactions. |
| Success Rate (pLDDT > 70) | > 80% for novel complexes | High generalization | Usable models generated for complexes not in training set. |
Key Application Workflow:
- Target Identification: Select a gene regulatory protein with unknown or poorly characterized nucleic acid binding mode.
- Sequence Input: Provide protein sequence (FASTA) and DNA/RNA sequence (string of nucleotides A,C,G,T/U).
- AF3 Modeling: Run the AF3 model, optionally specifying paired residues or providing a low-confidence template.
- Model Analysis: Evaluate predicted aligned error (PAE) and per-residue confidence (pLDDT) at the interface. High-confidence models can be used to hypothesize specific base-readout and shape-readout mechanisms.
- Validation & Design: Validate key predicted interactions via mutagenesis (e.g., Electrophoretic Mobility Shift Assay - EMSA) or use the model to design disruptive peptides or oligonucleotides for functional testing.
Experimental Protocols for Validation
Protocol 3.1: Electrophoretic Mobility Shift Assay (EMSA) for Validating Predicted DNA Binding Purpose: To experimentally confirm the protein-DNA interaction modeled by AF3 and assess the impact of mutations predicted to disrupt binding. Reagents: Purified protein (wild-type and AF3-predicted interface mutants), target DNA probe (fluorescently labeled or radio-labeled), non-specific competitor DNA (e.g., poly(dI-dC)), binding buffer, 6% non-denaturing polyacrylamide gel, TBE buffer. Procedure:
- Prepare binding reactions (20 µL final) containing binding buffer, labeled DNA probe (5-20 fmol), and increasing concentrations of purified protein (0-500 nM).
- Include a reaction with a 100-fold excess of unlabeled specific competitor to demonstrate binding specificity.
- Incubate at room temperature for 30 minutes.
- Load reactions onto a pre-run 6% non-denaturing polyacrylamide gel in 0.5x TBE buffer.
- Run gel at 100V for 60-90 minutes at 4°C.
- Visualize shifted complexes (protein-bound DNA) and free probe using a gel imager (fluorescence or phosphorimager).
Protocol 3.2: Site-Directed Mutagenesis Based on AF3 Interface Predictions Purpose: To generate point mutants in the protein or nucleic acid sequence to test the functional importance of predicted interactions. Reagents: Plasmid DNA containing gene of interest, high-fidelity DNA polymerase, primers encoding desired mutation, DpnI restriction enzyme, competent E. coli cells. Procedure:
- Design forward and reverse primers (~25-30 bases) complementary to the target site, with the desired mutation in the center.
- Set up a PCR reaction (50 µL) using plasmid template, mutagenic primers, and high-fidelity polymerase.
- Cycle: Initial denaturation (95°C, 2 min); 18 cycles of [Denature (95°C, 30s), Anneal (55-60°C, 1 min), Extend (68°C, 1 min/kb)]; Final extension (68°C, 5 min).
- Digest parental (methylated) template DNA with DpnI (37°C, 1 hour).
- Transform 1-5 µL of the reaction into competent E. coli cells, plate on selective agar.
- Screen colonies by Sanger sequencing to confirm the introduction of the mutation.
Visualization of Workflow & Concepts
Title: AF3-Driven Gene Regulatory Complex Research Cycle
Title: Disrupting a Repressive Complex Modeled by AF3
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Validating AF3 Protein-Nucleic Acid Models
| Reagent / Material | Function & Application | Example Product/Type |
|---|---|---|
| AF3 Server/Codebase | Core modeling engine for generating 3D structures of complexes. | AlphaFold Server (public), AlphaFold 3 Colab notebook. |
| High-Fidelity DNA Polymerase | For accurate amplification in site-directed mutagenesis to test predicted interface residues. | Q5 Hot Start (NEB), PfuUltra II (Agilent). |
| Fluorescent DNA Oligonucleotides | Labeled probes for EMSA to visualize protein binding without radioactivity. | 5'-FAM or Cy5-labeled oligos. |
| Nickel-NTA Agarose | Affinity purification of His-tagged recombinant regulatory proteins for binding assays. | Commercial resin for immobilized metal affinity chromatography (IMAC). |
| Gel Shift Binding Buffer (10X) | Provides optimal ionic strength and carrier agents for specific protein-nucleic acid interactions in EMSA. | Typically contains Tris, KCl, DTT, glycerol, and non-specific competitor DNA. |
| Cryo-EM Grids | For high-resolution structural validation of high-confidence AF3 models. | Quantifoil R1.2/1.3 gold or ultra-foil grids. |
| Surface Plasmon Resonance (SPR) Chip | To quantitatively measure binding kinetics (KD) of wild-type vs. mutant complexes predicted by AF3. | Sensor Chip SA for capturing biotinylated DNA/RNA. |
| Triisopropanolamine | Triisopropanolamine (TIPA) | High-purity Triisopropanolamine (TIPA) for materials science research. Explore its role as a cement hydration and strength enhancer. For Research Use Only. Not for human use. |
| Malaben | Malaben, CAS:19288-87-0, MF:C17H12N2Na2O6, MW:386.27 g/mol | Chemical Reagent |
Application Notes
Post-translational modifications (PTMs) form intricate, dynamic networks that govern cellular signaling pathways. Traditional structural biology struggles to characterize the conformational changes and transient interactions induced by PTMs like phosphorylation, ubiquitination, and acetylation. Within the thesis on AlphaFold 3 (AF3) biomolecular complex prediction research, a key application is the computational investigation of these networks. AF3's ability to predict the structure of proteins modified with ligands, ions, and covalent modifications provides a groundbreaking framework for generating testable hypotheses about PTM-driven allostery, altered protein-protein interaction (PPI) interfaces, and pathway crosstalk. This moves research beyond static interaction maps to mechanistic, structure-based models of signaling.
Core Contributions of AF3 to PTM Network Analysis:
- Hypothesis Generation for PTM-Driven Allostery: AF3 can model a protein in its unmodified and modified states (e.g., with a phosphorylated serine or acetylated lysine mimetic). Comparing these models can reveal predicted conformational shifts that allosterically activate or inhibit catalytic sites or binding interfaces.
- Interface Prediction for Modified Complexes: The software can predict the structure of complexes where one partner carries a specific PTM, helping to elucidate why certain modifications are necessary for specific PPIs within a pathway.
- Prioritization of Experimental Targets: By generating structural models for dozens of putative PTM states across a pathway, AF3 can prioritize which modifications are most likely to cause significant structural perturbations for downstream validation via cryo-EM or mutagenesis.
Quantitative Data Summary:
Table 1: Comparison of Methods for Investigating PTM Networks
| Method | Primary Output | Throughput | Resolution (Temporal/Spatial) | Key Limitation Addressed by AF3 |
|---|---|---|---|---|
| Mass Spectrometry (MS) | PTM site identification & quantification | High | High Temporal (dynamics), Low Spatial | Cannot provide 3D structural context of the modification. |
| Co-IP / Pull-down + MS | PTM-dependent protein interactors | Medium | Low | Does not reveal atomic details of modified interfaces. |
| X-ray Crystallography | Atomic-resolution static structure | Very Low | Atomic, but static | Struggles with dynamic, multi-state systems and capturing specific PTM states. |
| Cryo-EM | Near-atomic resolution structures of complexes | Low-Medium | Near-atomic, for stable complexes | Sample preparation for specific PTM states remains challenging. |
| AlphaFold 3 (In silico) | Predicted structures of modified proteins/complexes | Very High | Atomic (predictive) | Provides immediate structural hypotheses for PTM effects to guide all above methods. |
Table 2: Example AF3 Analysis of a Kinase Phosphorylation Cascade
| Predicted Complex | AF3 pLDDT / ptRMSD (Confidence) | Predicted Structural Effect of PTM | Downstream Experimental Validation |
|---|---|---|---|
| Kinase A (unphosphorylated) | 89 / 1.2 Å (High) | Inactive conformation; autoinhibitory helix bound to active site. | — |
| Kinase A (pThr-XXX) | 85 / 2.8 Ã… (High) | Helix displacement, active site remodeling; >70% predicted surface change. | Confirm via phospho-mimetic mutant activity assay. |
| Kinase A (phospho) + Substrate B | 78 / 4.5 Ã… (Medium) | Electrostatic complementarity between phospho-site and basic patch on Substrate B. | Validate binding via SPR with phospho-peptide. |
| Substrate B (phosphorylated) | 82 / 3.1 Ã… (High) | Conformational change exposing a nuclear localization signal (NLS) motif. | Test via fluorescence microscopy of GFP-tagged mutants. |
Experimental Protocols
Protocol 1: In silico Workflow for Predicting PTM-Induced Structural Changes Using AlphaFold 3
Objective: To generate and compare structural models of a protein of interest in its unmodified and PTM-modified states to hypothesize functional mechanisms.
Materials:
- High-performance computing cluster or Google Colab notebook with AF3 access.
- Protein sequence(s) in FASTA format.
- Definition of PTM site (e.g., residue number and type: S352 phosphorylation, K27 acetylation).
Method:
- Sequence and Modification Definition:
- For the unmodified state, prepare a FASTA file for the protein chain.
- For the modified state, create a new input where the modified residue is defined as a ligand. For example, to model phosphorylation, replace the target Serine (S) with a modified residue code or define a separate ligand (e.g.,
SEPfor phosphoserine) to be attached at the specific residue position. AF3 allows specification of covalent bonds between residues and small molecules/ions.
Structure Prediction Jobs:
- Submit two separate AF3 prediction jobs via the provided API or interface: one for the unmodified protein and one for the modified protein. Use identical parameters (number of recycles, random seed) for both to allow direct comparison.
Model Analysis and Comparison:
- Download the top-ranked models (based on predicted confidence metrics) for both states.
- Structural Alignment: Use molecular visualization software (e.g., PyMOL, ChimeraX) to perform a global alignment of the protein backbones of the two models.
- Quantification of Change: Calculate the root-mean-square deviation (RMSD) for specific regions (e.g., the active site loop, a regulatory domain). Identify side-chain rotamer changes and new surface electrostatics near the PTM site.
- Interface Prediction (Optional): Run a third AF3 prediction modeling the modified protein in complex with a known or hypothesized binding partner (defined by its sequence). Compare the interface to that of the unmodified complex.
Protocol 2: Experimental Validation of a Predicted PTM-Dependent Protein-Protein Interaction
Objective: To validate an AF3-predicted interaction between a PTM-carrying protein and a binding partner using Surface Plasmon Resonance (SPR).
Materials:
- Biacore or equivalent SPR instrument.
- Carboxymethylated dextran (CM5) sensor chip.
- Amine coupling kit (NHS/EDC).
- Purified, tag-free target protein (the predicted "binder").
- Synthetic peptides (≥15 amino acids) corresponding to the unmodified and PTM-modified (e.g., phosphorylated) sequence of the partner protein.
- HBS-EP+ running buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.05% v/v Surfactant P20, pH 7.4).
- Regeneration solution (e.g., 10 mM glycine, pH 2.0).
Method:
- Ligand Immobilization:
- Dilute the purified target protein to 10-20 µg/mL in 10 mM sodium acetate buffer (pH 4.5-5.5, optimized via scouting).
- Activate the CM5 sensor chip surface with a 7-minute injection of a 1:1 mixture of NHS and EDC.
- Inject the protein solution over the activated surface for 5-7 minutes to achieve a ligand density of ~1000-5000 Response Units (RU).
- Deactivate the surface with a 7-minute injection of 1 M ethanolamine-HCl, pH 8.5.
- Use a reference flow cell activated and deactivated without protein.
Analyte Binding Kinetics:
- Prepare a dilution series (e.g., 0.5 nM, 2 nM, 8 nM, 32 nM, 128 nM) of both the unmodified and PTM-modified peptides in HBS-EP+ buffer.
- Set the instrument temperature to 25°C. Use a flow rate of 30 µL/min.
- Inject each analyte concentration for 2 minutes (association phase), followed by a 5-minute dissociation phase with buffer flow.
- Regenerate the surface with a 30-second pulse of regeneration solution between cycles.
Data Analysis:
- Subtract the reference flow cell signal and buffer blank injections from all sensorgrams.
- Fit the corrected binding data to a 1:1 Langmuir binding model to determine the association rate (ka), dissociation rate (kd), and equilibrium dissociation constant (KD = kd/ka).
- Interpretation: A strong binding response and low nanomolar KD for the PTM-modified peptide, coupled with no/low binding for the unmodified peptide, validates the AF3 prediction of a PTM-dependent interaction.
Diagrams
Title: AF3 Workflow for PTM Structural Hypothesis Generation
Title: Integrating AF3 Predictions into a Phosphorylation Signaling Pathway
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for PTM Network Studies
| Reagent / Material | Function in PTM & Signaling Pathway Research |
|---|---|
| Phospho-specific Antibodies | Enable detection, quantification, and localization of specific protein phosphorylation events via Western blot, immunofluorescence, or flow cytometry. |
| PTM Mimetic Mutants (S→E/D, K→Q) | Constitutively mimic (or block) a PTM (e.g., phosphorylation, acetylation) for functional studies when the modifying enzyme is unknown or difficult to control. |
| Chemical Kinase/Enzyme Inhibitors & Activators | Pharmacologically modulate PTM writer enzymes to establish causal relationships between a PTM event and a downstream phenotypic readout. |
| Tandem Mass Tag (TMT) & Isobaric Labeling Reagents | Allow multiplexed, quantitative proteomics and phosphoproteomics from multiple conditions (e.g., time points, treatments) in a single MS run. |
| Protein A/G Magnetic Beads | Essential for immunoprecipitation (IP) and co-IP experiments to isolate proteins and their complexes for downstream analysis of PTMs or interactors. |
| Recombinant PTM Writer/Erase Enzymes (e.g., kinases, acetyltransferases, phosphatases) | Used for in vitro modification of protein targets to study direct biochemical effects or generate samples for structural biology (e.g., for cryo-EM grid preparation). |
| Cell-Permeable Proteasome Inhibitors (e.g., MG-132) | Stabilize ubiquitinated proteins by blocking degradation, enabling accumulation and detection of otherwise transient ubiquitination events. |
| AlphaFold 3 Software/API Access | Generates atomic hypotheses for PTM-induced structural changes and altered molecular interactions to prioritize costly wet-lab experiments. |
| Tritosulfuron | Tritosulfuron|Herbicide Reference Standard |
| Styromal | Styromal|Styrene-maleic Anhydride Copolymer|RUO |
Within the broader thesis on AlphaFold 3's capabilities for biomolecular complex structure prediction, this application note details its transformative role in de novo protein design. The accurate prediction of protein-protein, protein-ligand, and protein-nucleic acid interactions allows researchers to move from structure prediction to the rational creation of novel enzymes, binders, and therapeutics with prescribed functions.
Foundational Advances Enabled by AlphaFold 3
The following table summarizes key quantitative performance metrics of AlphaFold 3 relevant to design tasks, compared to previous state-of-the-art tools.
Table 1: Performance Benchmarking for Design-Relevant Predictions
| Prediction Target | AlphaFold 3 Performance (pLDDT/PAE/Interface Metrics) | Previous Best Tool (e.g., AF2-Multimer, RoseTTAFold) | Significance for De Novo Design |
|---|---|---|---|
| Protein-Protein Complexes | >70% high accuracy on CASP15 targets; Low interface PAE | ~50-60% high accuracy | Enables reliable design of protein-protein interfaces, heterodimers, and assemblies. |
| Protein-Small Molecule (Ligand) | High accuracy pose prediction for many drug-like molecules | Limited or non-existent in general tools | Direct in silico screening and design of ligand-binding sites and enzymes. |
| Protein-Oligonucleotide | High accuracy prediction for DNA/RNA interfaces | Specialized tools required | Enables design of novel transcription factors, nucleases, and delivery systems. |
| Antibody-Antigen | Improved accuracy over AF2-Multimer for CDR loop positioning | Variable performance, especially for CDR-H3 | Accelerates design of therapeutic antibodies and nanobodies. |
Core Experimental Protocols
Protocol 1: IterativeDe NovoEnzyme Design Using AlphaFold 3
This protocol outlines the cycle for designing a novel enzyme for a target reaction.
Materials & Workflow:
- Define Active Site Geometry: Using the transition state analog (TSA) of the desired reaction, define spatial constraints for catalytic residues (e.g., a catalytic triad, metal coordination site).
- Generate Scaffold Seeds: Use a de novo protein backbone generator (e.g., RFdiffusion, ProteinMPNN) to create thousands of backbone scaffolds that can spatially accommodate the constrained TSA and catalytic residues.
- Sequence Design: Apply a protein language model (e.g., ProteinMPNN, ESM-2) to generate stable amino acid sequences for each scaffold.
- AlphaFold 3 Validation: For each designed sequence, run an AlphaFold 3 prediction with the TSA explicitly specified in the input. This predicts the structure of the designed protein in complex with the TSA.
- Filter & Rank: Filter designs based on:
- High predicted confidence (pLDDT) at the active site.
- Low Predicted Aligned Error (PAE) between the TSA and catalytic residues.
- Correct geometry of the catalytic machinery around the TSA.
- Experimental Expression & Testing: Clone, express, and purify top-ranked designs. Assay for the desired catalytic activity.
- Iterative Re-design: Use negative design data and structural insights from AF3 predictions to guide further design cycles.
Diagram Title: Workflow for De Novo Enzyme Design with AF3 Validation
Protocol 2: Design of a Therapeutic Protein Binder Against a Cell Surface Target
This protocol details steps for designing a novel mini-protein binder against a defined epitope.
Materials & Workflow:
- Target Selection: Obtain an AlphaFold 3-predicted or experimental structure of the target protein (e.g., a GPCR, cytokine).
- Epitope Specification: Define the target epitope (specific residues or region) for binding.
- Binder Scaffold Docking & Design: Use a diffusion model (e.g., RFdiffusion) conditioned on the target epitope to generate plausible binder backbone scaffolds in complex with the target.
- Interface Sequence Design: Use a sequence design tool optimized for interfaces to generate complementary sequences for the binder scaffold.
- AlphaFold 3 in silico Affinity Screening: Run AlphaFold 3 predictions for each designed binder sequence in complex with the full target protein. Rank binders by:
- High interface confidence.
- Low interface PAE (stable interaction).
- Structural complementarity and lack of clashes.
- Specificity Check: Run AF3 predictions of top binders against known paralogs or homologs to assess potential cross-reactivity.
- In Vitro Validation: Express binders, measure binding affinity (SPR, BLI), and assess functional inhibition.
Diagram Title: Therapeutic Binder Design and Specificity Screening
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools for AF3-Guided Protein Design
| Item | Function in the Workflow | Key Provider/Example |
|---|---|---|
| AlphaFold 3 Server/API | Core prediction engine for biomolecular complexes. Provides pLDDT and PAE confidence metrics. | Google DeepMind, Isomorphic Labs |
| ProteinMPNN | Fast, robust neural network for de novo sequence design on provided backbones. Critical for step 3 in Protocol 1 & 2. | University of Washington (Baker Lab) |
| RFdiffusion | Generative model for creating novel protein backbones, can be conditioned on motifs or target surfaces. Used in Protocol 1 & 2. | University of Washington (Baker Lab) |
| ESM-2/ESMFold | Protein language model for sequence design and/or structure prediction. Can be used for inpainting and variant scoring. | Meta AI |
| Transition State Analog (TSA) Libraries | Small molecule structures mimicking reaction transition states. Essential input for enzyme design (Protocol 1). | Commercial chemical vendors (e.g., MolPort, Enamine) |
| Structural Biology Analysis Suite | For analyzing AF3 outputs (pLDDT, PAE, distances, clashes). | PyMOL, ChimeraX, Biopython |
| High-Throughput Cloning & Expression System | For rapid experimental testing of dozens of designs (e.g., yeast surface display, cell-free expression, E. coli vectors). | NEB HiFi Assembly, Twist Bioscience, 96-well expression kits |
| Biophysical Validation Platforms | To confirm binding/activity of designed proteins (e.g., Surface Plasmon Resonance, Bio-Layer Interferometry, Thermal Shift Assays). | Cytiva (Biacore), Sartorius (Octet), Roche (NanoTemper) |
| Tantalum | Tantalum Metal|High-Purity Reagent Grade|RUO | High-purity Tantalum for research applications in electronics, biomedicine, and corrosion studies. For Research Use Only. Not for human use. |
| (S,S)-(-)-Hydrobenzoin | (S,S)-(-)-Hydrobenzoin, CAS:2325-10-2, MF:C14H14O2, MW:214.26 g/mol | Chemical Reagent |
AlphaFold 3 Limitations and Best Practices for Accurate Predictions
Within the broader thesis on AlphaFold 3 biomolecular complex structure prediction research, understanding and mitigating common pitfalls is critical for producing reliable models for drug discovery. This application note details protocols for identifying and addressing low-confidence regions, intrinsically disordered loops, and symmetry-related errors in predicted multimetric complexes.
Table 1: Common AlphaFold 3 Performance Metrics and Pitfall Indicators
| Metric / Region Type | Typical pLDDT / ipTM Score Range | Implication for Model Reliability | Common in Molecule Type |
|---|---|---|---|
| Very High Confidence | pLDDT > 90 | Backbone prediction highly reliable. | Core secondary structures. |
| High Confidence | pLDDT 70-90 | Prediction reliable, side chains may vary. | Stable domains. |
| Low Confidence | pLDDT 50-70 | Caution required, potential errors. | Flexible linkers, surface loops. |
| Very Low Confidence | pLDDT < 50 | Prediction unreliable. Likely disordered. | N/C-terminal tails, disordered regions. |
| Interface Confidence (ipTM) | ipTM > 0.8 | High-confidence oligomeric interface. | Stable complexes. |
| Interface Low Confidence | ipTM < 0.5 | Unreliable quaternary structure. | Weak/transient interactions. |
Table 2: Impact of Symmetry Handling on Complex Prediction Accuracy
| Symmetry Type | Common Issue in Prediction | Typical Result without Constraint | Recommended AlphaFold 3 Protocol Adjustment |
|---|---|---|---|
| Cyclic (C2, C3, etc.) | Asymmetric distortions in symmetric units. | Incorrect interface geometry. | Use symmetry constraints during model generation. |
| Dihedral (D2, D3, etc.) | Loss of perpendicular symmetry axes. | Subunit packing errors. | Template guidance with symmetric templates. |
| Helical | Incorrect rise and twist parameters. | Non-physical filament models. | Multi-sequence alignment (MSA) subsampling for homogeneity. |
Experimental Protocols
Protocol 1: Identifying and Validating Low Confidence Regions
Objective: To flag and biochemically validate regions of a predicted structure with low pLDDT scores. Materials: AlphaFold 3 prediction output (PDB and JSON files), protein expression system, cysteine mutants, fluorescent maleimide probes.
- In silico Analysis: Parse the
predicted_aligned_error.jsonandscores.jsonfiles. Extract residues with pLDDT < 60 and/or high Predicted Aligned Error (PAE) with the rest of the structure. - Mapping: Visualize low-confidence residues (pLDDT < 60) as a red ribbon on the structure using PyMOL or ChimeraX.
- Cysteine Accessibility Assay:
- Design constructs introducing single cysteine residues in low-confidence loop regions (and a high-confidence control region).
- Express and purify proteins under native conditions.
- React with a fluorophore-conjugated maleimide (e.g., Alexa Fluor 488 C5 Maleimide).
- Measure labeling kinetics via stopped-flow fluorescence. Fast labeling indicates solvent exposure and disorder, consistent with low-confidence prediction.
- Protease Sensitivity Assay: Incubate purified protein with a broad-spectrum protease (e.g., Proteinase K). Sample over time and analyze by SDS-PAGE. Low-confidence/disordered regions will be cleaved preferentially.
Protocol 2: Addressing Disordered Loops for Crystallography
Objective: To improve the crystallizability of a protein target by redesigning or truncating predicted disordered termini/loops. Materials: AlphaFold 3 models, PCR cloning equipment, crystallization screens.
- Prediction Analysis: Identify long (>10 residue) loops or termini with consistently low pLDDT (<50) across multiple AF3 runs.
- Design Constructs:
- Truncation: Design primers to remove disordered N/C-terminal tails.
- Loop Replacement: Design primers to replace a predicted disordered loop (residues i to j) with a shorter, stable loop from a homologous protein structure.
- Cloning & Expression: Generate 3-5 construct variants per target. Express and purify using standard protocols.
- Crystallization Trial: Subject all constructs to high-throughput sparse-matrix crystallization screening. Compare hit rates between full-length and engineered constructs.
Protocol 3: Imposing Symmetry in Oligomeric Complex Prediction
Objective: To predict accurate quaternary structures for symmetric complexes by guiding AlphaFold 3. Materials: Multiple sequence alignments (MSAs) for individual subunits, known symmetric templates (optional).
- Symmetry Definition: Determine expected symmetry (e.g., C2, C3) from literature or homologous complexes.
- Input Preparation:
- Method A (Sequence Duplication): For a homodimer, input the same protein sequence twice in the fasta file separated by a colon (e.g.,
>complex\nSequenceA:SequenceA). This explicitly defines the stoichiometry. - Method B (Template Guidance): If a low-resolution symmetric template exists (e.g., from cryo-EM), provide it in the template input field.
- Method A (Sequence Duplication): For a homodimer, input the same protein sequence twice in the fasta file separated by a colon (e.g.,
- AlphaFold 3 Run: Execute prediction with
is_prokaryoteflag set appropriately andnum_multimer_predictions_per_modelincreased to 10-20. - Symmetry Analysis: Use
phenix.ensemble_validationorUSCF Chimera"Matchmaker" to calculate RMSD between symmetry-related subunits post-prediction. Filter models for those with low subunit asymmetry.
Visualization
Diagram 1: AlphaFold 3 Workflow with Pitfall Checkpoints
Diagram 2: Disordered Loop Validation Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Validating AlphaFold 3 Predictions
| Item | Function in Protocol | Example Product/Catalog |
|---|---|---|
| Fluorescent Maleimide | Covalently labels solvent-accessible cysteine residues to probe disorder/accessibility. | Alexa Fluor 488 C5 Maleimide (Thermo Fisher, A10254) |
| Broad-Spectrum Protease | Cleaves unstructured protein regions; used in limited proteolysis to map disordered loops. | Proteinase K (NEB, P8107S) |
| Crystallization Screen Kits | High-throughput screening of conditions for protein crystal growth of redesigned constructs. | MORPHEUS HT-96 (Molecular Dimensions, MD1-46) |
| Site-Directed Mutagenesis Kit | Rapid generation of cysteine mutants or loop truncations for biochemical validation. | Q5 Site-Directed Mutagenesis Kit (NEB, E0554S) |
| Gel Filtration Standards | Assess oligomeric state and monodispersity of complexes post-prediction. | Gel Filtration Markers Kit (Sigma, MWGF1000) |
| Analysis Software | Calculate symmetry (RMSD) and validate ensemble models from multiple AF3 predictions. | PHENIX Suite (phenix.ensemble_validation), ChimeraX |
| UV-123 | UV-123 Light Stabilizer (HALS) for Research | UV-123 is a low-basicity HALS for coatings and polymer research. It prevents UV degradation in acid systems. For Research Use Only. Not for human use. |
| YM-08 | YM-08, MF:C19H17N3OS2, MW:367.5 g/mol | Chemical Reagent |
Challenges with Novel Scaffolds and Unseen Molecular Combinations
Application Notes
The advent of AlphaFold 3 (AF3) represents a paradigm shift in predicting the structure of biomolecular complexes, from proteins and nucleic acids to ligands and post-translational modifications. However, its application to novel chemical scaffolds and unseen molecular combinations—a core task in de novo drug design—presents distinct challenges. This document details these limitations and provides protocols for experimental validation, framed within a thesis on advancing AF3 for early-stage discovery.
Key Quantitative Challenges: While AF3 demonstrates high accuracy on known biomolecule types, its performance degrades on non-canonical inputs. Current benchmarks highlight specific gaps.
Table 1: Performance Metrics of AF3 on Novel/Unseen Combinations
| Prediction Target | Reported Confidence Metric (pLDDT/ipTM) | RMSD vs. Experimental (Ã…) | Key Limitation |
|---|---|---|---|
| Protein + Novel Synthetic Macrocycle | 45-65 (Low) | >5.0 | Poor geometric sampling of constrained ring systems. |
| Protein + Unseen PROTAC-like Binder | 50-70 (Medium) | 4.0 - 8.0 | Inaccurate orientation of linker, poor ternary complex modeling. |
| Antibody + Novel Hapten | 60-75 (Medium) | 3.5 - 6.0 | Limited epitope specificity for small molecule conformers. |
| RNA + Unseen Small Molecule | 40-60 (Low) | >6.0 | High false-positive binding site prediction. |
| Known Protein + Known Ligand (Control) | 70-90 (High) | <2.0 | Baseline for established interactions. |
Data synthesized from recent preprints and benchmark analyses post-AF3 release.
The primary challenges are: 1) Training Data Bias: AF3's training set lacks broad coverage of synthetic chemistry space. 2) Energy Function Limitations: The implicit scoring lacks terms for specific forces crucial for drug-like molecule binding (e.g., halogen bonding, strained ring energetics). 3) Conformational Sampling: The diffusion process may not adequately explore the conformational landscape of novel scaffolds.
Experimental Protocols for Validation
Protocol 1: Orthogonal Validation of AF3-Predicted Novel Ligand Poses
Objective: To experimentally test the geometry and affinity of a novel scaffold bound to a target protein, as predicted by AF3.
Materials: Recombinant target protein. Novel chemical synthesis of the scaffold. Crystallization screens or Cryo-EM grid preparation kits. Surface Plasmon Resonance (SPR) biosensor chips.
Methodology:
- Prediction & Selection: Generate 25 models for the target protein complexed with the novel scaffold using AF3. Cluster models by ligand RMSD and select the top 5 centroid models for experimental testing.
- Biophysical Affinity Screening: Use SPR to measure binding kinetics (ka, kd, KD) of the scaffold to immobilized protein. A sub-micromolar KD supports the possibility of a specific pose.
- Structure Determination Attempt: a. Co-crystallization: Set up 576-condition sparse matrix screens with protein:ligand at 1:5 molar ratio. b. Cryo-EM Single Particle Analysis: For complexes >100 kDa, prepare grids, collect >3,000 movies, and process data. Use the AF3 model for initial docking into the density map.
- Discrepancy Analysis: If an experimental structure is obtained, calculate RMSD of the ligand pose against AF3 predictions. Analyze discrepancies (e.g., rotamer flips, scaffold inversion) to inform model retraining or refinement.
Protocol 2: Assessing Ternary Complex Prediction for Unseen Bifunctional Molecules (e.g., PROTACs)
Objective: To validate AF3's prediction of a ternary complex formed by an E3 ligase, a target protein, and a novel PROTAC molecule.
Materials: Purified E3 ligase (e.g., VHL, CRBN) and target protein. Novel PROTAC compound. Size-Exclusion Chromatography (SEC) columns. Native Mass Spectrometry setup. Cellular lysates for degradation assays.
Methodology:
- In silico Modeling: Input sequences for the E3 ligase and target protein, and the SMILES string for the PROTAC linker connecting two known warheads. Run AF3 in complex mode to generate 20 putative ternary models.
- In vitro Complex Formation: Incubate E3, target, and PROTAC (1:1:5 ratio) for 1 hour at 4°C. Analyze by SEC-MALS (Multi-Angle Light Scattering) to confirm complex formation and stoichiometry.
- Native MS Verification: Directly inject the SEC peak fraction into a native MS instrument to measure the mass of the intact ternary complex.
- Functional Cellular Assay: Treat engineered cell lines expressing tagged target protein with the PROTAC. Monitor target degradation via immunoblotting over 24 hours. Correlation between predicted cooperative binding interface stability and degradation efficacy validates the model's functional relevance.
Visualizations
Title: Validation Workflow for Novel Scaffold Predictions
Title: Root Causes of AF3 Challenges with Novelty
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Validating AF3 Predictions on Novel Combinations
| Reagent/Material | Function & Relevance |
|---|---|
| Biolayer Interferometry (BLI) Biosensors | Label-free, real-time kinetic measurement of novel ligand binding to immobilized target proteins. Crucial for verifying predicted interactions before structural efforts. |
| CrystalDirect Harvesting Plates | Automates crystal harvesting for fragile co-crystals of novel complexes, maximizing success rate from sparse crystallization trials. |
| Ultra-stable Cryo-EM Grids (e.g., UltrAuFoil) | Provides a cleaner, more stable background for imaging low-molecular-weight or heterogeneous complexes involving novel molecules. |
| Native Mass Spectrometry Standards | Pre-calibrated protein complexes enable accurate mass determination of novel ternary complexes (e.g., PROTAC-mediated). |
| DNA-Encoded Library (DEL) Screening Kits | Complements AF3 by providing experimental binding data for millions of diverse, often novel, scaffolds against a target. |
| Alchemical Free Energy Perturbation (FEP+) Software | Molecular dynamics-based method to calculate relative binding affinities for congeneric series, refining AF3's pose rankings for novel scaffolds. |
Within the broader thesis on AlphaFold 3 (AF3) biomolecular complex structure prediction research, the generation and integration of input data remains a cornerstone for model accuracy. While AF3 reduces explicit reliance on deep Multiple Sequence Alignments (MSAs) and external templates compared to its predecessors, their role in conditioning the model, particularly for novel or orphan targets, is critically redefined. This application note details contemporary protocols for MSA construction and template retrieval, framing them as essential, complementary information streams that optimize AF3's internal representations and final prediction quality.
Core Data: Quantitative Impact on Prediction Accuracy
The following table summarizes key performance metrics highlighting the contribution of different input data types to AF3 predictions, as analyzed in recent benchmark studies.
Table 1: Impact of Input Data on AlphaFold 3 Prediction Accuracy (Benchmark Averages)
| Input Data Configuration | Protein-Protein Docking (pLDDT) | Protein-Nucleic Acid (pLDDT) | Protein-Small Molecule (pLDDT) | Interface RMSD (Ã…) Improvement vs. No MSA |
|---|---|---|---|---|
| AF3 (Full Input: MSA + Templates + Ligand Info) | 89.2 | 85.7 | 81.4 | Baseline |
| AF3 (No Evolutionary MSA) | 78.5 | 76.1 | 72.3 | +2.8 Ã… |
| AF3 (No Structural Templates) | 87.1 | 84.0 | 80.1 | +0.5 Ã… |
| AF3 (MSA from Truncated Database) | 82.4 | 79.8 | 75.9 | +1.7 Ã… |
Data synthesized from recent preprints and benchmark analyses on AF3 performance. pLDDT: predicted Local Distance Difference Test (higher is better). RMSD: Root Mean Square Deviation (lower is better).
Application Notes and Detailed Protocols
Protocol 3.1: Generating Optimized MSAs for AF3 Conditioning
Objective: Create deep, diverse, and clean MSAs to provide evolutionary constraints, even for targets with few homologs.
Query Sequence Preparation:
- Input the target protein sequence(s) in FASTA format.
- For complexes, generate separate FASTA files for each chain and a combined file for joint alignment.
Homology Search with MMseqs2:
- Use the ColabFold MMseqs2 server (or local installation) for rapid, sensitive searches against large sequence databases (UniRef30, BFD, MGnify).
- Command (Local):
MSA Curation and Filtering:
- Cluster sequences at >90% identity to reduce redundancy.
- Filter sequences with unusual lengths (<50% or >150% of query length).
- Manually inspect and remove obvious non-homologous sequences or fragments.
Pairing for Complexes (Protein-Protein):
- Use the pair_msa protocol from ColabFold, which identifies paired sequences from the same species across individual MSAs, providing co-evolutionary signals critical for interface prediction.
Protocol 3.2: Retrieving and Selecting Structural Templates
Objective: Identify high-quality structural templates to guide the folding of individual domains and, where available, inter-complex orientations.
Template Search with Foldseek:
- Search the query against the PDB100 database using Foldseek for ultra-fast structural comparisons.
- Command:
Template Evaluation Metrics:
- Prioritize templates by: 1) Lowest E-value, 2) Highest sequence identity (>30% ideal), 3) Coverage of the query sequence (>70%), 4) Resolution of template structure (<3.0 Ã…).
Template Processing for AF3:
- Extract the relevant chains and regions from the template PDB files.
- Align the template sequence to the query sequence using standard pairwise alignment tools (e.g., Clustal Omega).
- Format the template features (atoms, secondary structure) according to AF3's expected input schema.
Visualizing the Integrated AF3 Input Pipeline
AF3 Input Data Conditioning Pipeline
The Scientist's Toolkit: Key Research Reagents & Materials
Table 2: Essential Solutions for MSA and Template-Based Workflows
| Item / Resource | Function / Purpose | Key Consideration for AF3 |
|---|---|---|
| MMseqs2 Software Suite | Rapid, sensitive protein sequence searching and clustering. | Enables generation of large, diverse MSAs from massive databases in minutes. Critical for capturing weak homology. |
| Foldseek | Fast structural alignment for searching the PDB. | Drastically faster than DALI or TM-align for template identification, enabling high-throughput workflows. |
| ColabFold (Server/API) | Integrated pipeline combining MMseqs2, MSAs, template search, and AlphaFold/AlphaFold Multimer. | Simplifies the entire preprocessing pipeline; the pair_msa function is vital for complex prediction. |
| UniRef90/30 Databases | Clustered sets of protein sequences at 90% or 50% identity to reduce redundancy. | Primary sequence databases for MSA construction. UniRef30 provides a broader evolutionary view. |
| PDB100 Database | A clustered subset of the Protein Data Bank, removing highly similar structures. | Standard database for efficient template searches without redundancy. |
| CIF (mmCIF) Format Files | Standard format for representing macromolecular structure data. | AF3 uses mmCIF-formatted template files. Ensure templates are correctly converted and parsed. |
| High-Performance Computing (HPC) Cluster or Cloud GPU | Computational resources for running AF3 inference. | While MSA/template generation can be CPU-based, full AF3 inference requires significant GPU memory (e.g., A100, H100). |
| Inidascamine | Inidascamine, CAS:903884-71-9, MF:C12H17N3O2, MW:235.28 g/mol | Chemical Reagent |
| JPC0323 | JPC0323, CAS:5972-45-2, MF:C22H43NO4, MW:385.6 g/mol | Chemical Reagent |
Within the broader thesis on AlphaFold 3 (AF3) biomolecular complex structure prediction research, a primary technical challenge is the computational scaling for large assemblies. While AF3 demonstrates unprecedented accuracy, its memory and runtime requirements grow significantly with the number of residues and input components, potentially limiting the analysis of large complexes like viral capsids, ribosomes, and transcriptional machinery. This application note details considerations and protocols for managing these resources effectively.
Quantitative Performance Scaling of AF3
The computational demand of AF3 is not linear. Key scaling factors include total number of residues, number of distinct polypeptide chains, and the complexity of pairwise interactions. The following table summarizes approximate resource requirements based on published benchmarks and community reports.
Table 1: Estimated AF3 Resource Scaling for Complexes
| Total Residues | Example Complex | Approx. GPU Memory (GB) | Approx. Runtime* | Key Limiting Factor |
|---|---|---|---|---|
| < 1,000 | Dimeric enzymes | 10-15 | 2-5 minutes | Pairwise MSA processing |
| 1,000 - 3,000 | Heterotrimeric G protein | 15-25 | 10-30 minutes | Template search & representation |
| 3,000 - 6,000 | Small viral capsid subunit | 25-40+ | 1-3 hours | Attention matrix computation |
| > 6,000 | Ribosomal subunit | 40+ (may exceed single GPU) | Several hours | Pairformer stack memory |
*Runtime estimated using a single NVIDIA A100 or H100 GPU.
Protocols for Managing Large Assemblies
Protocol 2.1: Subcomplex Prediction and Docking
This protocol breaks down a large target into manageable subcomplexes for individual prediction, followed by computational docking.
- Subcomplex Definition: Using biological knowledge (e.g., from literature or databases like PDB), decompose the large assembly into stable, interacting subcomplexes. Prioritize pairs or groups with high binding affinity.
- Independent AF3 Prediction: Run AF3 separately for each defined subcomplex. Use the
max_recyclesflag (e.g.,max_recycles=3) to control runtime for each job. - Structural Alignment and Docking: Use the known interface residues from the individual predictions to guide docking.
- Load subcomplex structures (e.g., in PyMOL or ChimeraX).
- Identify and align conserved interface regions using sequence/structure alignment tools.
- Perform rigid-body docking with software like HADDOCK or ClusPro, using the aligned interface as a restraint.
- Validation: Assess the final docked model with metrics like interface pDockQ (from AF3 output) and steric clash analysis.
Protocol 2.2: Strategic Input Curation to Reduce Complexity
Optimize input to minimize unnecessary computational overhead.
- MSA Curation:
- For each chain, gather MSAs using tools like MMseqs2.
- Apply diversity filtering (
--max-seq-idflag) to reduce redundancy. For large complexes, a stricter threshold (e.g., 0.8) is beneficial. - Consider limiting the depth of MSA (e.g., to top 1,000 sequences per chain) if memory is a critical constraint, acknowledging potential accuracy trade-offs.
- Template Selection: Limit the number of templates in the input. Manually curate templates to include only the most relevant (highest sequence identity/coverage) structures from the PDB.
- Composition Input: When predicting a homomultimer, specify the same sequence multiple times in the input, but leverage the multimer logic which is more efficient than treating them as wholly independent chains.
Visualizing the Memory-Bottleneck Workflow
The following diagram illustrates the key stages in the AF3 inference pipeline where memory and runtime bottlenecks commonly occur for large complexes.
Diagram Title: AF3 Inference Pipeline with Key Computational Bottlenecks for Large Complexes
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Research Reagent Solutions for AF3 on Large Complexes
| Item | Function in Workflow | Notes for Large Complexes |
|---|---|---|
| AlphaFold 3 Server/API | Web-based interface for easy access. | Limited to smaller complexes (typically < 2,000 residues). Useful for initial subcomplex scoping. |
| Local AF3 Installation (Open Source) | Full control over parameters and hardware. | Essential for large jobs. Requires high-end GPU (e.g., A100 80GB, H100) and CUDA setup. |
| ColabFold (with AF3 backend) | Streamlined, cloud-Jupyter notebook environment. | Can leverage free/paid cloud GPUs. Requires careful session management for long-running, memory-intensive jobs. |
| MMseqs2 Software Suite | Fast, sensitive homology search for MSA generation. | Critical for curating input. Use --max-seq-id and depth filters to control MSA size. |
| HADDOCK / ClusPro Web Servers | Computational docking platforms. | For integrating AF3-predicted subcomplexes into larger assemblies using interface restraints. |
| PyMOL / ChimeraX | Molecular visualization and analysis software. | For visualizing large assemblies, assessing interfaces, and preparing figures. |
| High-Performance Computing (HPC) Cluster | Provides multiple high-memory GPU nodes. | Necessary for complexes >5,000 residues. Enables parallel subcomplex prediction. |
| M1069 | M1069, MF:C25H30N4O8S, MW:546.6 g/mol | Chemical Reagent |
| Nurr1 agonist 2 | Nurr1 agonist 2, MF:C18H14O3S, MW:310.4 g/mol | Chemical Reagent |
Guidelines for Interpreting Low-Confidence Ligand Poses and Binding Affinities
Application Notes and Protocols
Within the broader thesis on AlphaFold 3 (AF3) for biomolecular complex structure prediction, a critical challenge is the accurate interpretation of low-confidence ligand pose predictions. While AF3 generates predictions for protein-ligand, protein-nucleic acid, and other complexes, its confidence metrics—primarily the predicted aligned error (PAE) and the per-residue pLDDT (predicted Local Distance Difference Test)—require careful contextual analysis. This document provides protocols for evaluating these outputs and integrating them into experimental workflows for drug discovery.
1. Quantitative Metrics for Low-Confidence Assessment
The following table summarizes key AF3 output metrics relevant to ligand binding predictions and their interpretation thresholds.
Table 1: Key AlphaFold 3 Output Metrics for Ligand Pose Assessment
| Metric | Description | High Confidence Range | Low Confidence Range | Interpretation for Ligand Binding |
|---|---|---|---|---|
| pLDDT (Ligand Atoms) | Measures local structure confidence. | 90-100 | <70 | Poses with low ligand pLDDT have highly uncertain atom positions. |
| Interface pLDDT | Average pLDDT of protein residues within 5Ã… of ligand. | >80 | <70 | Low confidence suggests an unreliable protein environment for the docked ligand. |
| Predicted Aligned Error (PAE) at Interface | Expected positional error (Ã…) between ligand and protein residues. | <5 Ã… | >10 Ã… | High PAE indicates low confidence in the relative placement of ligand vs. protein. |
| Predicted RMSD | Internal AF3 estimate of expected Cα RMSD if model aligned on a region. | <2 Å | >5 Å | Applicable to the protein backbone surrounding the binding pocket. |
| Composite Score | (Interface pLDDT) / (Mean Ligand-Protein PAE). | >15 | <5 | A simple heuristic; higher scores suggest more reliable poses. |
2. Experimental Protocol: Orthogonal Validation of Low-Confidence Poses
Protocol 2.1: Computational Cross-Validation Using Molecular Dynamics (MD) Purpose: To assess the stability of a low-confidence AF3-predicted ligand pose. Materials:
- AF3 prediction file (PDB format) for the complex.
- MD simulation software (e.g., GROMACS, AMBER).
- Suitable force field (e.g., CHARMM36, GAFF2) and solvation parameters. Procedure:
- System Preparation: Parameterize the ligand using appropriate tools (e.g.,
antechamberfor AMBER). Solvate the complex in a water box (e.g., TIP3P) and add ions to neutralize charge. - Energy Minimization: Perform 5,000 steps of steepest descent minimization to remove steric clashes.
- Equilibration:
- NVT ensemble: Heat system from 0 K to 300 K over 100 ps, using position restraints on protein and ligand heavy atoms.
- NPT ensemble: Achieve 1 bar pressure over 200 ps, maintaining restraints.
- Production Run: Run an unrestrained simulation for 50-100 ns at 300K and 1 bar. Record trajectories every 10 ps.
- Analysis:
- Calculate the root-mean-square deviation (RMSD) of the ligand relative to its initial AF3-predicted position.
- Compute the ligand-protein interaction fraction (hydrogen bonds, hydrophobic contacts) over time.
- A pose that rapidly diffuses away or shows unstable interactions suggests the AF3 prediction is non-physical.
Protocol 2.2: Experimental Validation via Site-Directed Mutagenesis Purpose: To test the functional relevance of predicted ligand-protein contacts, especially in low-confidence regions. Materials:
- Cloned gene of the target protein.
- Site-directed mutagenesis kit.
- Protein expression and purification system.
- Functional assay (e.g., enzymatic activity, binding assay like SPR or ITC). Procedure:
- Residue Selection: Identify key protein residues predicted to form interactions with the ligand in the AF3 model, even if confidence is low.
- Generate Mutants: Create alanine (or conservative) substitution mutants for at least 3-5 of these residues.
- Express and Purify wild-type and mutant proteins.
- Functional Assay: Measure ligand binding affinity (Kd) or inhibitory concentration (IC50) for each variant.
- Interpretation: A significant loss of binding (>10-fold change in Kd/IC50) for a mutant provides experimental evidence supporting the predicted interaction, increasing confidence in that aspect of the pose.
3. Visualization of the Decision Workflow
Title: Decision Workflow for AF3 Ligand Pose Confidence
4. The Scientist's Toolkit: Key Research Reagents & Solutions
Table 2: Essential Toolkit for Validating AF3 Ligand Predictions
| Item / Reagent | Function / Purpose |
|---|---|
| AlphaFold 3 ColabFold Implementation | Provides accessible, GPU-accelerated platform for generating complex predictions with ligands. |
| Molecular Dynamics Software (GROMACS/AMBER) | Enables physics-based stability assessment of predicted poses through simulation. |
| Site-Directed Mutagenesis Kit (e.g., Q5) | Allows rapid generation of point mutants to test predicted protein-ligand contacts. |
| Surface Plasmon Resonance (SPR) Chip (e.g., Series S CM5) | Immobilization surface for label-free, quantitative measurement of binding kinetics (KD, kon, koff). |
| Isothermal Titration Calorimetry (ITC) Cell | Provides direct measurement of binding affinity (Kd) and thermodynamics (ΔH, ΔS). |
| Cryo-EM Grids (e.g., Quantifoil R1.2/1.3) | For high-resolution structural validation of challenging complexes predicted by AF3. |
| Fragment Library (e.g., 1000+ compounds) | Useful for experimental screening to probe low-confidence pockets suggested by AF3 models. |
When to Trust AlphaFold 3 Outputs and When to Seek Experimental Validation
AlphaFold 3 represents a transformative advance in predicting the structure of biomolecular complexes, including proteins, nucleic acids, ligands, and post-translational modifications. Its accuracy, however, is not uniform across all prediction scenarios. This document provides application notes and protocols to guide researchers in assessing prediction reliability and designing appropriate validation experiments within a structured research thesis.
Quantitative Assessment of AlphaFold 3 Confidence Metrics
Table 1: Key AlphaFold 3 Output Metrics and Interpretation Guidelines
| Metric | Range | High Reliability (Trust) Zone | Low Reliability (Validate) Zone | Interpretation |
|---|---|---|---|---|
| Predicted Aligned Error (PAE) [Ã…] | 0 - >30 | < 5 Ã… | > 15 Ã… | Expected position error of residue i if aligned on residue j. Low inter-domain PAE indicates confident relative positioning. |
| pLDDT (per-residue) | 0 - 100 | > 90 | < 70 | Local confidence measure. >90: high backbone accuracy. <70: low confidence, often disordered. |
| pTM (predicted TM-score) | 0 - 1 | > 0.8 | < 0.5 | Global model confidence. >0.8: high overall accuracy. <0.5: likely incorrect fold. |
| ipTM (interface pTM) | 0 - 1 | > 0.8 | < 0.6 | Specific confidence for interface in a complex. Critical for complex trust assessment. |
| Molecular Similarity (to training set) | N/A | Low Similarity | High Similarity (Template unavailable) | Unique complexes without close homologs in PDB are higher risk for "hallucination." |
Table 2: Decision Matrix for Experimental Validation
| Prediction Scenario | pLDDT (avg) | ipTM | PAE (interface) | Recommended Action | Suggested Validation Method(s) |
|---|---|---|---|---|---|
| Single-domain protein | > 90 | N/A | N/A | Trust for most applications. | Limited validation (e.g., circular dichroism for fold confirmation). |
| Multi-domain protein | > 85 | N/A | < 10 Ã… | Trust domain structures; Validate relative orientation if critical. | SAXS, FRET for inter-domain distance. |
| Protein-Protein Complex | > 80 | > 0.75 | < 8 Ã… | Cautious Trust for hypothesis generation. | Mandatory validation (e.g., X-ray crystallography, cross-linking MS). |
| Protein-Small Molecule | Variable | < 0.7 | > 12 Å | Do Not Trust – High-risk prediction. | Mandatory validation (ITC, SPR, crystallography). |
| Protein-Nucleic Acid | > 75 | > 0.7 | < 10 Å | Use as Guide – Requires validation. | EMSA, cryo-EM, mutagenesis. |
| Membrane Proteins | Often < 70 | Variable | Variable | Extreme Caution – High validation need. | Cryo-EM, NMR in mimetics, functional assays. |
Detailed Experimental Validation Protocols
Protocol 3.1: Cross-linking Mass Spectrometry (XL-MS) for Validating Protein Complexes
Purpose: To obtain experimental distance restraints for validating AlphaFold 3-predicted quaternary structures and interfaces.
Materials (Reagent Solutions):
- Cross-linker: DSSO (Disuccinimidyl sulfoxide) or BS3 – amine-reactive, MS-cleavable.
- Quenching Solution: 1M Ammonium bicarbonate.
- Digestion Buffers: Tris-HCl (pH 8.0), Urea/Guanidine HCl for denaturation.
- Enzymes: Trypsin/Lys-C mix for proteolysis.
- LC-MS/MS System: High-resolution tandem mass spectrometer (e.g., Q-Exactive HF).
Methodology:
- Complex Formation: Incubate purified proteins at predicted stoichiometry (from AF3) in PBS, 30 min, 25°C.
- Cross-linking: Add DSSO to 1-2 mM final concentration. React for 30 min at 25°C.
- Quenching: Add ammonium bicarbonate to 50 mM final concentration, incubate 10 min.
- Digestion: Denature, reduce, alkylate, and digest with trypsin/Lys-C overnight.
- LC-MS/MS Analysis: Inject peptides. Use data-dependent acquisition with stepped higher-energy collisional dissociation (HCD) to fragment both peptides and cross-linker.
- Data Analysis: Use software (e.g., XlinkX, pLink2) to identify cross-linked peptides. Map distance restraints (Cα-Cα ~24-30 Å for DSSO) onto AF3 model.
Protocol 3.2: Surface Plasmon Resonance (SPR) for Validating Binding Affinity
Purpose: To experimentally determine binding kinetics (Ka, Kd) and affinity (KD) for a predicted protein-ligand or protein-protein complex.
Materials (Reagent Solutions):
- Running Buffer: HBS-EP+ (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.05% v/v Surfactant P20, pH 7.4).
- Immobilization Reagents: CMS sensor chip, EDC/NHS amine coupling kit.
- Analytes: Serial dilutions of purified ligand/partner protein in running buffer.
- Regeneration Solution: 10 mM Glycine-HCl (pH 2.0-3.0) or other optimized solution.
Methodology:
- Ligand Immobilization: Activate CMS chip surface with EDC/NHS. Inject protein "A" (predicted receptor) in sodium acetate buffer (pH 4.5-5.5) to achieve ~50-100 RU target density. Deactivate with ethanolamine.
- Analyte Binding: Inject serial dilutions of analyte "B" (predicted partner) at 30 μL/min for 120s association, followed by 300s dissociation in running buffer.
- Regeneration: Inject regeneration solution for 30s to remove bound analyte.
- Data Analysis: Subtract reference cell signal. Fit sensorgrams to a 1:1 binding model to obtain ka, kd, and calculate KD. Compare KD to predicted interface confidence (ipTM).
Protocol 3.3: Mutagenesis and Functional Assay for Interface Validation
Purpose: To test the functional importance of residues predicted by AlphaFold 3 to form a critical binding interface.
Materials (Reagent Solutions):
- Site-Directed Mutagenesis Kit: Q5 or similar high-fidelity polymerase.
- Functional Assay Reagents: Substrate for enzymatic assay, reporter cell line, or co-immunoprecipitation (Co-IP) buffers.
- Lysis/Wash Buffer: 25 mM Tris, 150 mM NaCl, 1% NP-40, pH 7.4, with protease inhibitors.
Methodology:
- Residue Selection: Choose 3-5 residues at the predicted interface with high burial and favorable ΔΔG (from AF3).
- Mutagenesis: Generate alanine (or charge-swap) mutants for selected residues.
- Protein Purification: Express and purify wild-type and mutant proteins.
- Functional/Binding Assay:
- For enzymes: Measure catalytic activity. >10-fold loss in kcat/KM suggests critical interface residue.
- For signaling: Use reporter assay. Loss of signal indicates disrupted complex.
- Direct binding: Perform Co-IP or pull-down. Compare mutant vs. WT binding to partner.
Visualizations
Title: Decision Flowchart: AF3 Trust vs. Validation
Title: Multi-Tier Experimental Validation Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for AlphaFold 3 Validation
| Item | Function in Validation | Example Product/Kit | Critical Notes |
|---|---|---|---|
| Cleavable Cross-linker (DSSO) | Generates MS-identifiable distance restraints for protein complexes. | Thermo Fisher Scientific, DSSO (A33545) | Enables unambiguous identification of cross-linked peptides via MS2 fragmentation. |
| SPR Sensor Chip (CMS) | Gold surface for immobilizing one binding partner to measure real-time binding kinetics. | Cytiva, Series S Sensor Chip CMS | Standard chip for amine coupling of protein ligands. |
| Site-Directed Mutagenesis Kit | Efficiently generates point mutations in plasmids to test predicted interface residues. | NEB, Q5 Site-Directed Mutagenesis Kit (E0554) | High fidelity and efficiency for creating alanine scans. |
| Size-Exclusion Chromatography (SEC) Column | Purifies native complexes and assesses oligomeric state vs. prediction. | Cytiva, HiLoad 16/600 Superdex 200 pg | Critical step before biophysical assays (SPR, XL-MS). |
| Cryo-EM Grids (Quantifoil) | Sample support for high-resolution single-particle cryo-EM validation. | Quantifoil, R1.2/1.3 300 mesh Au grids | Gold grids offer better thermal conductivity. |
| Isothermal Titration Calorimetry (ITC) Cell | Measures binding affinity and thermodynamics in solution without labels. | Malvern Panalytical, VP-ITC Microcell | The "gold standard" for solution-phase KD measurement. |
| Deuterated Solvents & Media | Required for NMR spectroscopy of proteins, especially for backbone assignment. | Cambridge Isotope Laboratories, D2O, ¹âµN/¹³C-labeled growth media | Enables key validation for dynamic/disordered regions. |
| WAY-232897 | WAY-232897, MF:C17H15N3O2S, MW:325.4 g/mol | Chemical Reagent | Bench Chemicals |
| BRD4 Inhibitor-29 | BRD4 Inhibitor-29, MF:C21H28N2O3, MW:356.5 g/mol | Chemical Reagent | Bench Chemicals |
AlphaFold 3 Performance Review: Benchmarks, Comparisons, and Real-World Impact
This application note evaluates the performance of AlphaFold 3 and related deep learning models in the context of the 15th Critical Assessment of Structure Prediction (CASP15) experiment. CASP15, conducted in 2022, represents the most recent blind assessment of protein structure prediction methods, providing an independent benchmark for emerging AI-driven tools like AlphaFold 3. The results are critical for researchers, scientists, and drug development professionals assessing the reliability of computational predictions for biomolecular complex modeling.
The following table summarizes the key quantitative results for top-performing groups and methods in the CASP15 assessment, with a focus on multimeric (complex) targets. AlphaFold 3, while not officially a CASP15 participant, is benchmarked against these results in post-hoc analyses.
Table 1: Summary of Top CASP15 Performance Metrics (Protein Complexes)
| Method / Group | GDT_TS (Global) | GDT_HA (High-Acc) | Interface Contact Score | LDDT (Local) | Rank (Overall) |
|---|---|---|---|---|---|
| AlphaFold-Multimer v2.3 | 87.4 | 76.2 | 0.85 | 0.89 | 1 |
| Baker Group (RoseTTAFold) | 82.1 | 68.5 | 0.79 | 0.85 | 2 |
| Zhang Group (I-TASSER) | 79.8 | 65.2 | 0.75 | 0.83 | 3 |
| Median for all Groups | 65.3 | 45.1 | 0.61 | 0.72 | - |
Data compiled from CASP15 official reports and post-CASP analyses. GDT_TS: Global Distance Test Total Score; GDT_HA: GDT High Accuracy; LDDT: Local Distance Difference Test.
Table 2: AlphaFold 3 Benchmark vs. CASP15 Leaders (Post-hoc Analysis)
| Metric | AlphaFold 3 (Reported) | CASP15 Leader (AF-Multimer) | Performance Delta |
|---|---|---|---|
| Protein-Ligand (RMSD Ã…) | 0.94 | N/A | N/A |
| Protein-Nucleic Acid (TM-score) | 0.92 | 0.81 | +0.11 |
| Antibody-Antigen (Interface Score) | 0.78 | 0.71 | +0.07 |
| Overall Accuracy (Composite) | >90% | 87% | ~3-5% |
Note: Direct comparison is indicative; CASP15 was a blind test, while AF3 benchmarks use curated sets. AF3 shows marked improvement on nucleic acids and small molecules.
Experimental Protocols for Validation
Protocol: CASP15-Style Blind Assessment Workflow
This protocol outlines the standard operating procedure for conducting a blind prediction challenge analogous to CASP, used for internally validating new models like AlphaFold 3.
Objective: To objectively assess the predictive accuracy of a structure prediction method on targets with recently solved, unpublished structures. Materials: Target sequence/structure lists, computational cluster, prediction software, analysis scripts (e.g., LDDT, TM-score, DockQ). Procedure:
- Target Selection & Curation: An independent assessor selects protein complexes and biomolecular assemblies with experimentally determined structures not yet released in the PDB.
- Sequence Release: Only the amino acid/nucleotide sequences of the targets are provided to prediction teams. No structural information is shared.
- Prediction Phase: Teams submit predicted 3D models in a specified format within a defined deadline (e.g., 3 weeks).
- Experimental Structure Release: The experimental reference structures are released by the assessor.
- Quantitative Assessment: The assessor calculates metrics using official scripts:
- For Monomers: GDTTS, GDTHA, LDDT.
- For Complexes: Interface RMSD (iRMSD), Fraction of Native Contacts (FNat), DockQ score.
- For Ligands: Heavy-atom RMSD of the ligand pose.
- Statistical Analysis: Z-scores and global rankings are computed to compare methods.
Protocol: In-house Validation of AlphaFold 3 Predictions
Objective: To benchmark AlphaFold 3 performance against a held-out test set of known biomolecular complexes. Materials: AlphaFold 3 software/license, high-performance GPU cluster, test set (e.g., PDB complex entries post-2022), visualization software (PyMOL, ChimeraX). Procedure:
- Test Set Preparation: Create a non-redundant set of protein-protein, protein-nucleic acid, and protein-ligand complexes with high-resolution experimental structures. Ensure no temporal overlap with the model's training data.
- Model Inference: Run AlphaFold 3 predictions for each target complex using default parameters. For protein-ligand predictions, provide the SMILES string of the ligand.
- Structure Alignment & Metric Calculation:
- Align the predicted model to the experimental structure using the protein backbone.
- Compute standard metrics (RMSD, TM-score, DockQ).
- For ligands, compute the heavy-atom RMSD of the docked pose after aligning the protein receptor.
- Confidence Estimation: Record the predicted aligned error (PAE) and per-residue pLDDT scores. Correlate confidence metrics with observed accuracy.
- Comparative Analysis: Compare results to the performance of AlphaFold-Multimer v2.3 and other baselines on the same test set.
Visualization of Workflows and Relationships
CASP15 Blind Assessment Workflow
AF3 Benchmarking vs CASP15
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools & Resources for Validation
| Item / Reagent | Function / Purpose | Example / Source |
|---|---|---|
| AlphaFold 3 Software | Core prediction engine for biomolecular complexes. Includes models for proteins, nucleic acids, ligands, and post-translational modifications. | Google DeepMind / Isomorphic Labs |
| AlphaFold-Multimer v2.3 | Key baseline comparator; the state-of-the-art method from CASP15 for protein-protein complexes. | GitHub: google-deepmind/alphafold |
| ColabFold | Streamlined, accessible implementation of AlphaFold2/Multimer using MMseqs2 for fast homology search. Useful for rapid prototyping. | GitHub: sokrypton/ColabFold |
| CASP15 Assessment Scripts | Official metric calculation software (LDDT, DockQ, GDT). Critical for ensuring comparable, standardized evaluation. | PredictionCenter.org |
| PDB (Protein Data Bank) | Primary repository of experimental 3D structural data. Source of ground truth and test set curation. | RCSB.org |
| PyMOL / UCSF ChimeraX | Molecular visualization software for inspecting, comparing, and rendering predicted vs. experimental structures. | Schrodinger / RBVI |
| DOCKQ | Specialized quality measure for protein-protein docking predictions. Calculates a continuous score from FNat, iRMSD, and LRMSD. | GitHub: bjornwallner/DockQ |
| pLDDT & PAE Plots | AlphaFold's internal confidence metrics. pLDDT: per-residue confidence (0-100). PAE: predicted error between residue pairs. | Integrated in AF3 output |
| PARP-1-IN-4 | N-(4-Chlorophenyl)-2-(4-(4-chlorophenyl)-1-oxophthalazin-2(1H)-yl)acetamide Supplier | High-purity N-(4-Chlorophenyl)-2-(4-(4-chlorophenyl)-1-oxophthalazin-2(1H)-yl)acetamide for research. This product is For Research Use Only. Not for human or veterinary use. |
| N3-Methyl-5-methyluridine | N3-Methyl-5-methyluridine, MF:C11H16N2O6, MW:272.25 g/mol | Chemical Reagent |
Within the broader thesis on the evolution of biomolecular complex structure prediction, this Application Note details the quantitative performance of AlphaFold 3 (AF3) in predicting the structures of protein-ligand and protein-antibody complexes. These interactions are foundational to drug discovery and therapeutic development. Recent benchmark analyses indicate a paradigm shift in predictive accuracy, moving from the low-confidence regimes of previous tools to high-accuracy predictions for many complexes.
Quantitative Performance Metrics
Benchmarking against experimental structures from the PDB (Protein Data Bank) provides the following key metrics for AF3.
Table 1: AlphaFold 3 Performance on Key Complex Types
| Complex Type | Key Metric (Median) | Benchmark Dataset | Comparison to AlphaFold 2/Previous Tools |
|---|---|---|---|
| Protein-Small Molecule | DockQ score: 0.80 (High Accuracy) | PDB-derived test set | ~50% improvement in ligand RMSD accuracy |
| Protein-Antibody | Interface RMSD (iRMSD): ~1.2 Ã… | Diverse antibody-antigen pairs | Significant improvement in CDR loop and interface prediction |
| Protein-Peptide | lDDT: >85 | Reliably models short peptide interactions | |
| General Protein-Protein | DockQ: 0.81 | Major advance over protein-only docking |
Table 2: Ligand-Specific Pose Accuracy (RMSD in Ångströms)
| Ligand Type | Median RMSD (AF3) | <2.0 Ã… Success Rate |
|---|---|---|
| Drug-like molecules | 1.4 Ã… | 78% |
| Nucleotides | 1.1 Ã… | 89% |
| Ions (e.g., Ca²âº, Zn²âº) | 0.8 Ã… | 95% |
| Cofactors (e.g., NAD) | 2.0 Ã… | 65% |
Experimental Protocol: Benchmarking AF3 on a Protein-Ligand Complex
This protocol outlines the steps to assess AF3's prediction accuracy for a specific target of interest against a known experimental structure.
Materials & Pre-Processing
- Experimental Reference Structure: Obtain the high-resolution crystal or cryo-EM structure (e.g., from the PDB) of the protein-ligand complex.
- Input Sequences:
- Protein sequence(s) in FASTA format.
- Ligand SMILES string or canonical identifier.
- Software/Platform: Access to the AlphaFold Server (https://alphafoldserver.com) or a local installation of the AF3 model.
Procedure
- Input Preparation:
- Separate the protein chain and ligand from the experimental reference structure. Save the protein as a FASTA sequence.
- Note the ligand's 3-letter PDB residue name and map it to its corresponding SMILES string using resources like the PDB Chemical Component Dictionary.
- AlphaFold 3 Submission:
- Navigate to the AlphaFold Server interface.
- Input the protein FASTA sequence(s).
- Specify the ligand by its SMILES string in the appropriate input field for "non-protein molecules."
- Enable "Complex prediction" mode. Do not provide any template structures to assess ab initio performance.
- Submit the job (may require queuing).
- Result Retrieval and Analysis:
- Download the top-ranked predicted model (ranked_0.pdb).
- Align the predicted protein structure to the experimental protein structure using a rigid-body fitting tool (e.g.,
superin PyMOL) based on the protein Cα atoms only. Record the transformation matrix. - Apply the same transformation matrix to the predicted ligand coordinates.
- Calculate the ligand Root-Mean-Square Deviation (RMSD) between the experimental and transformed predicted ligand coordinates using heavy atoms only.
- Calculate the interface RMSD (iRMSD) for all residues/atoms within 5Ã… of the ligand in the experimental structure.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Resources for AF3 Complex Analysis
| Item | Function/Description |
|---|---|
| AlphaFold Server | Primary web interface for running AF3 predictions without local compute. |
| PDB (RCSB Protein Data Bank) | Source of experimental reference structures for benchmarking. |
| PyMOL / ChimeraX | Molecular visualization software for structural alignment, RMSD calculation, and visual inspection. |
| DockQ Tool | Software for calculating DockQ scores, a continuous metric for docking quality. |
| PDB Chemical Component Dictionary | Repository for mapping PDB ligand codes to SMILES strings and standard chemistries. |
| Local ColabFold Implementation | Alternative for batch processing and customized sampling, using the AF3 architecture via MMseqs2. |
| NOC-5 | (1Z)-2-(3-aminopropyl)-1-(hydroxyimino)-2-(propan-2-yl)hydrazin-1-ium-1-olate |
| N-(m-PEG9)-N'-(propargyl-PEG8)-Cy5 | N-(m-PEG9)-N'-(propargyl-PEG8)-Cy5, MF:C63H99ClN2O17, MW:1191.9 g/mol |
Visualizing the AF3 Benchmarking Workflow
Diagram Title: AF3 Protein-Ligand Benchmarking Protocol
Visualizing the Thesis Context of AF3's Impact
Diagram Title: Thesis Context: From Protein Folding to Complex Prediction
Application Notes
Within the broader thesis that AlphaFold 3 represents a paradigm shift from protein-centric to holistic biomolecular interaction modeling, these application notes compare the capabilities of leading structure prediction tools for biomolecular complexes. The central thesis posits that the explicit, integrated treatment of ligands, nucleic acids, and post-translational modifications is critical for accurate in situ biological function prediction.
Core Performance Comparison The following table summarizes key quantitative benchmarking results for protein-protein and protein-ligand complex prediction.
Table 1: Benchmark Performance on Complex Prediction Tasks
| Metric / System | AlphaFold-Multimer v2.3 | RoseTTAFold All-Atom | AlphaFold 3 |
|---|---|---|---|
| Protein-Protein (DockQ ≥ 0.8) | ~60% (on certain benchmarks) | ~50-55% (on certain benchmarks) | Significantly higher (exact % not publicly benchmarked) |
| Protein-Antibody (pLDDT ≥ 80) | Good for epitope, paratope less defined | Moderate | Superior for full paratope-epitope modeling |
| Protein-Small Molecule (RMSD ≤ 2.0Å) | Not Applicable (no ligand capability) | Yes, via explicit all-atom modeling | Yes, with higher accuracy, leveraging diffusion network |
| Protein-DNA/RNA (Interface RMSD) | Limited to protein-only | Good for nucleic acid backbone | State-of-the-Art for full atomic detail |
| Key Architectural Differentiator | Enhanced MSA pairing for proteins | 3-track (sequence, distance, coordinates) all-atom | Joint diffusion, unified IA^3 attention, no templates |
Key Insights:
- AlphaFold-Multimer v2.3 remains a robust, dedicated solution for protein-protein complexes, leveraging deep Multiple Sequence Alignment (MSA) understanding but is fundamentally limited to the protein alphabet.
- RoseTTAFold All-Atom pioneered true all-atom modeling, treating non-protein molecules explicitly in its neural network, providing a crucial proof-of-concept.
- AlphaFold 3 validates the core thesis by integrating all components—proteins, DNA, RNA, ligands, ions—into a single, end-to-end generative architecture (a diffusion model). This eliminates the need for rigid-body docking or multi-stage pipelines, enabling the prediction of conformational changes induced by ligand binding.
Experimental Protocols
Protocol 1: Comparative Prediction of a Protein-Small Molecule Complex Objective: To evaluate the ligand-binding pose prediction accuracy of AlphaFold 3 versus RoseTTAFold All-Atom.
- Input Preparation:
- Obtain the target protein sequence in FASTA format.
- Obtain the ligand SMILES string. For AlphaFold 3, prepare the input as a combined
.pdbfile with the protein (from a homologous structure or predicted monomer) and the ligand placed roughly near the binding site. For RoseTTAFold All-Atom, prepare the ligand.mol2or.sdffile and protein sequence separately.
- Structure Prediction:
- AlphaFold 3 (via Google Cloud Vertex AI): Submit the job using the combined
.pdbfile, specifying the ligand chain ID. Use default settings (numsamples=1, numrecycles=12). - RoseTTAFold All-Atom (Local/Server): Run the
run_roseTTAFold_all_atom.pyscript, providing the protein FASTA, ligand file, and specifying--ligand_mode.
- AlphaFold 3 (via Google Cloud Vertex AI): Submit the job using the combined
- Analysis:
- Align the predicted protein backbone to the experimental reference structure (if available) using PyMOL or UCSF Chimera.
- Calculate the Root-Mean-Square Deviation (RMSD) of the heavy atoms of the predicted ligand pose versus the experimental ligand pose.
Protocol 2: Assessment of Protein-Protein Interface Accuracy Objective: To compare interface precision between AlphaFold-Multimer v2.3 and AlphaFold 3 for a heterodimeric complex.
- Input Preparation: Prepare a paired MSA in A3M format for the two protein sequences (Chain A and B). This is required for AlphaFold-Multimer. For AlphaFold 3, simply provide the two sequences in the input field.
- Structure Prediction:
- AlphaFold-Multimer v2.3 (Local/ColabFold): Use the
colabfold_batchcommand with the--model-type alphafold2_multimer_v3flag and the paired A3M file. - AlphaFold 3: Submit both sequences as a single complex job.
- AlphaFold-Multimer v2.3 (Local/ColabFold): Use the
- Analysis:
- Use the
pdockqtool orDockQscore to evaluate the predicted interface quality against a known structure. - Compute the interface pLDDT (ipLDDT) from the predicted models, focusing on residues within 10Ã… of the partner chain.
- Use the
Visualization
Title: Computational Workflows for Complex Prediction
Title: Logical Thesis Development Path
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Biomolecular Complex Prediction
| Item / Resource | Function / Purpose |
|---|---|
| AlphaFold Server / Google Cloud Vertex AI | Primary platform for running AlphaFold 3 predictions with ligand/nucleic acid support. |
| ColabFold (AF-Multimer v2.3) | Accessible platform for running AlphaFold-Multimer, utilizing MMseqs2 for fast MSA generation. |
| RoseTTAFold All-Atom Server | Web server or local installation for all-atom predictions including small molecules. |
| PDB (Protein Data Bank) | Source of experimental structures for benchmark comparison and input template creation (for AF-Multimer). |
| PubChem | Database to obtain accurate SMILES strings and 3D conformer files for small molecule ligands. |
| Pymol / UCSF Chimera / ChimeraX | Molecular visualization software for analyzing predicted interfaces, aligning structures, and calculating RMSD. |
| DockQ & pdockq | Specialized software tools for quantitatively scoring the quality of predicted protein-protein interfaces. |
| RDKit | Cheminformatics toolkit for processing small molecule files (SMILES, SDF) and generating 3D conformers for input. |
| zr17-2 | (Yl)thio)acetic Acid|RUO|Research Compound |
| Bis-(N,N'-carboxyl-PEG4)-Cy5 | Bis-(N,N'-carboxyl-PEG4)-Cy5, MF:C47H67ClN2O12, MW:887.5 g/mol |
Within the context of AlphaFold 3 research, the prediction of biomolecular complex structures represents a paradigm shift. This application note details the experimental protocols and provides a comparative analysis of traditional structural biology methods—Molecular Docking, Molecular Dynamics (MD) simulations, and Cryo-Electron Microscopy (Cryo-EM)—against the predictive capabilities of AlphaFold 3. This analysis is critical for researchers in drug development to understand the complementary roles of prediction and empirical validation.
Application Notes & Comparative Analysis
The table below summarizes the core characteristics, capabilities, and quantitative performance metrics of each method, based on current literature and benchmark studies.
Table 1: Comparative Analysis of Structural Methods
| Aspect | Molecular Docking | Molecular Dynamics (MD) | Cryo-EM | AlphaFold 3 |
|---|---|---|---|---|
| Primary Purpose | Predict binding pose & affinity of a ligand to a known target. | Simulate physical movements & conformational changes of atoms over time. | Determine high-resolution 3D structures of biomolecules in near-native states. | De novo prediction of protein-ligand, protein-nucleic acid, and multimeric complex structures. |
| Typical System Size | ~10^2 - 10^3 atoms. | ~10^4 - 10^6 atoms (all-atom). | >100 kDa complexes, large assemblies. | Flexible, from small complexes to large assemblies. |
| Temporal Resolution | Static snapshot. | Femtoseconds to milliseconds (enhanced sampling). | Static snapshot, can capture multiple states. | Static ensemble prediction. |
| Key Output Metric | Docking Score (kcal/mol), RMSD of pose. | Root Mean Square Deviation (RMSD), Free Energy (ΔG). | Resolution (Å), Map-to-model FSC. | Predicted Alignment Error (PAE), pLDDT (confidence 0-100). |
| Typical Time per Calculation | Seconds to hours. | Days to months (GPU/CPU clusters). | Weeks to months (sample prep, data collection, processing). | Minutes to hours (per complex). |
| Key Limitation | Relies on a fixed, often rigid receptor structure; scoring function inaccuracies. | Computationally expensive; limited by timescale of biological events. | Sample preparation challenges; requires significant expertise & cost. | Limited explicit dynamics; training data bias; covalent modifications not always modeled. |
| Role in AlphaFold 3 Research | Provides a baseline for ligand pose prediction. | Validates predicted complex stability and refines conformations. | Provides experimental "ground truth" for training and blind testing. | Generates high-accuracy starting models for further investigation. |
Experimental Protocols
Protocol 2.1: Cross-Validation of AlphaFold 3 Prediction using Cryo-EM
Aim: To experimentally validate a protein-protein complex predicted by AlphaFold 3.
- AlphaFold 3 Prediction: Input the sequences of the two interacting partners into the AlphaFold 3 server or local installation. Retrieve the top-ranked model, PAE matrix, and per-residue pLDDT scores.
- Sample Preparation for Cryo-EM:
- Express and purify the individual proteins, then mix at equimolar ratio to form the complex.
- Apply 3 µL of complex (at ~0.5-1 mg/mL) to a glow-discharged Quantifoil grid.
- Blot and plunge-freeze in liquid ethane using a vitrification device (e.g., Vitrobot).
- Data Collection & Processing:
- Collect movie stacks on a 300 keV Cryo-EM microscope with a K3 direct electron detector.
- Use MotionCor2 for motion correction and Gctf for CTF estimation.
- Perform particle picking (e.g., with crYOLO), 2D classification, and multiple rounds of 3D classification in RELION or cryoSPARC to isolate particles of the complex.
- Refine the final 3D reconstruction and calculate the resolution via Fourier Shell Correlation (FSC=0.143).
- Validation & Comparison: Fit the AlphaFold 3 predicted model into the Cryo-EM density map using ChimeraX. Calculate the global RMSD between the predicted atomic coordinates and the refined model from the map.
Protocol 2.2: Refining AlphaFold 3 Models with Molecular Dynamics
Aim: To assess and improve the stability of a predicted protein-ligand complex.
- Initial Model Generation: Use AlphaFold 3 to predict the structure of the protein with a specified small molecule ligand.
- System Preparation:
- Use the
pdbfixerandtleap(AMBER) orCHARMM-GUIto add missing hydrogen atoms, solvate the complex in a TIP3P water box (10 Ã… padding), and add ions to neutralize the system.
- Use the
- Energy Minimization & Equilibration:
- Perform 5,000 steps of steepest descent minimization to remove steric clashes.
- Gradually heat the system from 0 K to 300 K over 100 ps in the NVT ensemble with position restraints on protein heavy atoms.
- Equilibrate for 1 ns in the NPT ensemble (1 atm) with gradual release of restraints.
- Production Simulation: Run an unrestrained MD simulation for 100-500 ns using a GPU-accelerated engine (e.g., AMBER, GROMACS, or OpenMM). Record trajectories every 10 ps.
- Analysis: Calculate the ligand RMSD, protein-ligand interaction fingerprints, and binding free energy (e.g., using MM/PBSA) over the stable simulation period. Compare to the static AlphaFold 3 prediction.
Table 2: Essential Research Reagents & Solutions
| Item | Function / Application |
|---|---|
| HEK293F Cells | Mammalian expression system for producing properly folded, post-translationally modified proteins for Cryo-EM and binding assays. |
| Amylose/SecuritiesResin | For affinity purification of MBP-tagged proteins, a common strategy to stabilize proteins for complex formation. |
| Grid Box (e.g., Quantifoil R1.2/1.3) | Cryo-EM sample support with a regular holey carbon film for vitrification. |
| AMBER/CHARMM Force Fields | Parameter sets defining atomistic interactions for MD simulations (e.g., ff19SB for protein, GAFF2 for small molecules). |
| GPU Cluster (e.g., NVIDIA A100) | High-performance computing resource essential for running AlphaFold 3 predictions and long-timescale MD simulations. |
| RELION / cryoSPARC License | Software suites for high-resolution single-particle Cryo-EM data processing. |
| ChimeraX | Visualization software for analyzing and comparing density maps and atomic models from all methods. |
Visualization of Integrated Workflows
Title: Integrative Structural Biology Workflow with AlphaFold 3
Title: Molecular Dynamics Refinement Protocol
This application note, framed within a broader thesis on AlphaFold 3 (AF3) biomolecular complex structure prediction research, details initial validation studies for the model. Published literature from independent research groups is beginning to assess AF3's accuracy for predicting structures of diverse macromolecular complexes, including proteins, nucleic acids, and small molecule ligands. The following sections present quantitative summaries of these findings, detailed protocols for validation experiments, and essential research tools.
Validation Performance: Comparative Analysis
The following table summarizes key quantitative metrics from published validation studies of AF3, primarily focusing on comparisons to its predecessor, AlphaFold 2 (AF2), and other specialized tools.
Table 1: Summary of Published AlphaFold 3 Validation Metrics
| Complex Type & Study (if available) | Key Metric (vs. AF2) | Benchmark Dataset | Notable Finding |
|---|---|---|---|
| Protein-Ligand | >50% improvement in ligand RMSD (Exact DockQ). Success rate (RMSD < 2.0 Ã…) increased significantly. | PDBbind, PoseBusters | Demonstrates marked improvement in small molecule placement, competitive with docking software. |
| Protein-Nucleic Acid | ~20% improvement in protein-RNA interface prediction (DockQ). Significant gains for protein-DNA complexes. | NPIDR (Nucleic Acid-Protein Interaction Data Resource) | Surpasses AF2 and most specialized tools for nucleic acid partner modeling. |
| Antibody-Antigen | High accuracy for paratope and epitope prediction. Outperforms AF2 and ClusPro in interface RMSD on a benchmark set. | Structural Antibody Database (SAbDab) | Predicts challenging antibody-antigen interfaces without requiring paired sequence alignment. |
| Protein Multimer (General) | Modest improvement over AF2-multimer for many complexes. Superior performance on complexes with conformational changes upon binding. | Benchmark from AF2-multimer paper | Shows robustness across diverse interaction types within a single unified model. |
| Protein-Peptide | Improved modeling of conformational plasticity in bound peptides. Better accuracy for peptides with non-canonical or post-translationally modified residues. | Peptide-protein benchmark sets | Handles the flexibility of short peptide ligands more effectively than rigid docking. |
Note: Comprehensive, large-scale independent benchmarking studies are still in early stages. The above is compiled from initial reports and analyses shared by research groups.
Detailed Experimental Protocol: Validating AF3 Protein-Ligand Predictions
This protocol outlines a standard workflow for computationally and experimentally validating AF3's predictions for a protein-small molecule complex.
Protocol Title:Experimental Validation of an AF3-Predicted Protein-Ligand Structure
Part A: In Silico Prediction and Analysis
Input Preparation:
- Obtain the target protein sequence in FASTA format.
- Define the small molecule ligand of interest using its SMILES string.
- Research Reagent: Ligand SMILES String (A standardized notation representing the 2D molecular structure of the ligand).
Structure Prediction with AF3:
- Access the AF3 server or locally installed version.
- Input the protein sequence and ligand SMILES string. Optionally, provide known structures of individual components as templates (disabled by default for de novo prediction).
- Execute the prediction job. AF3 will generate multiple ranked models (e.g., 5).
Computational Validation Metrics:
- Calculate the Root Mean Square Deviation (RMSD) between the predicted ligand pose and a known experimental reference structure (if available).
- Assess protein-ligand interaction geometry using PoseBusters (a validation suite for ligand structures), which checks for steric clashes, bond length/angle deviations, and protein-ligand atomic clashes.
- Perform molecular dynamics (MD) simulations on the predicted complex to assess stability (e.g., 100 ns simulation).
- Research Reagent: PoseBusters Software (Open-source Python package for validating AI-predicted protein-ligand structures).
Part B: Experimental Structure Determination (X-ray Crystallography)
Protein Expression & Purification:
- Express the recombinant protein in a suitable system (e.g., E. coli, insect cells).
- Purify using affinity and size-exclusion chromatography (SEC).
- Research Reagent: Ni-NTA Agarose (Affinity resin for purifying histidine-tagged recombinant proteins).
Complex Formation & Crystallization:
- Incubate the purified protein with a 2-5 molar excess of the ligand compound.
- Use SEC to isolate the pure complex.
- Set up crystallization trials using commercial screens (e.g., from Hampton Research) via vapor diffusion.
- Research Reagent: Hampton Research Crystal Screen (A sparse-matrix kit of chemical conditions for initial protein crystallization trials).
Data Collection & Structure Determination:
- Flash-cool a crystal in liquid nitrogen.
- Collect X-ray diffraction data at a synchrotron beamline.
- Process data (HKL-3000, XDS). Use the AF3 prediction as a molecular replacement (MR) search model in Phaser (part of the CCP4 or Phenix suite).
- Refine the model with Refmac5 or phenix.refine.
Final Validation & Comparison:
- Calculate the RMSD between the experimentally solved ligand pose and the AF3-predicted pose.
- Statistically compare the protein-ligand interaction networks (hydrogen bonds, hydrophobic contacts) between the prediction and the experimental structure.
Experimental Workflow Visualization
AF3 Validation Workflow: Computation & Experiment
The Scientist's Toolkit: Key Research Reagents & Software
Table 2: Essential Tools for AF3 Validation Studies
| Item Name | Type | Function in Validation |
|---|---|---|
| AlphaFold 3 Server/Code | Software | Core prediction engine for generating biomolecular complex models. |
| PoseBusters | Software | Validates the physical realism and chemical correctness of predicted protein-ligand complexes. |
| PDBbind Database | Database | Provides a curated set of protein-ligand complexes with binding data for benchmarking predictions. |
| HKL-3000 / XDS | Software | Suite for processing raw X-ray diffraction data into usable structure factor amplitudes. |
| CCP4 / Phenix Suite | Software | Comprehensive software packages for crystallographic structure determination, refinement, and analysis. |
| Ni-NTA Agarose | Laboratory Reagent | Affinity chromatography resin for rapid purification of histidine-tagged proteins. |
| Hampton Research Crystal Screens | Laboratory Reagent | Pre-formulated chemical matrices for initial protein crystallization trials. |
| Molecular Dynamics Software (e.g., GROMACS, AMBER) | Software | Simulates the dynamic behavior of the predicted complex to assess stability and conformational flexibility. |
| UNC8153 | UNC8153, MF:C33H37N5O5, MW:583.7 g/mol | Chemical Reagent |
| 12:0 EPC chloride | 12:0 EPC chloride, MF:C34H69ClNO8P, MW:686.3 g/mol | Chemical Reagent |
Quantitative Reception Analysis (2024-Present)
The following tables summarize key quantitative metrics and expert survey data regarding the release of AlphaFold 3 (AF3) by DeepMind/Isomorphic Labs.
Table 1: Benchmark Performance Metrics of AlphaFold 3 vs. Predecessors & Competitors
| Biomolecular Complex Type | AlphaFold 3 Performance (TM-/pTM-score/IQ) | AlphaFold 2/Multimer v2.3 Performance | RoseTTAFold All-Atom Performance | Experimental Accuracy (RMSD Ã…) | Key Benchmark (Reference) |
|---|---|---|---|---|---|
| Protein-Protein | 76.4% (DockQ≥0.8) | 45.7% (DockQ≥0.8) | 51.2% (DockQ≥0.8) | ~1-3 Å | CASP15/Protein Data Bank |
| Protein-Antibody | 81.2% success rate | 62.1% success rate | 58.7% success rate | ~1-4 Ã… | SAbDab benchmark |
| Protein-DNA | 83.1% (ntAF3≥0.8) | 52.9% (ntAF3≥0.8) | 61.4% (ntAF3≥0.8) | ~1.5-4 Å | Nucleic Acid Database |
| Protein-Ligand (Small Molecule) | 64.2% (RMSD≤2.0Å) | Not Applicable | 42.3% (RMSD≤2.0Å) | <2.0 Å | PDBbind v2020 |
| Protein-Post-Translational Modification | Limited quantitative data; qualitative accuracy reported | Not Available | Not Available | N/A | Case studies (e.g., phosphorylated peptides) |
Table 2: Community Adoption & Sentiment Metrics (Post-May 2024 Release)
| Metric | Value/Result | Source/Timeframe |
|---|---|---|
| AlphaFold Server Predictions Run | >1,000,000+ structures | Isomorphic Labs, Oct 2024 |
| Citations of AF3 Nature Paper | ~850 | Google Scholar, Dec 2024 |
| Preprint Downloads/Views | >500,000 | bioRxiv/Publisher Sites |
| Surveyed Researcher Trust in AF3 for Hypothesis Generation | 78% "High/Very High" | Nature Poll (n=1,500), Nov 2024 |
| Critical Blog Posts/Major Criticisms | ~15% of high-impact media coverage | Altmetric analysis |
Detailed Experimental Protocols for Validation Studies
The following protocols are synthesized from key validation studies cited in the reception discourse.
Protocol 2.1: In Silico Benchmarking Against PDB Structures
Objective: To assess the accuracy of AF3 predictions for protein-ligand complexes.
Materials: AlphaFold 3 server/API, local installation of OpenFold or ColabFold (AF3 implementation), benchmark set from PDBbind or PoseBusters, compute cluster (GPU recommended).
Procedure:
1. Curation: Download a non-redundant set of 200 protein-ligand complexes released after April 1, 2023 (to avoid training data contamination) from PDBbind v2024.
2. Input Preparation: For each complex, prepare FASTA sequences for the protein chain(s). For the ligand, generate a SMILES string from the PDB file using RDKit.
3. Prediction: Input protein sequence and ligand SMILES into the AF3 model. Use default settings (num_relax=0 for speed). Run 3 replicates per complex.
4. Analysis:
a. Align the predicted protein structure to the experimental backbone (Cα atoms) using UCSF Chimera matchmaker.
b. Calculate Root-Mean-Square Deviation (RMSD) of the ligand heavy atoms post-alignment.
c. Compute the Interface RMSD (I-RMSD) for all atoms within 5Ã… of the binding partner.
5. Comparison: Repeat steps 3-4 using a state-of-the-art docking tool (e.g., AutoDock-GPU, DiffDock) for the same protein structure.
Protocol 2.2: Experimental Cross-Validation via Cryo-EM
Objective: To experimentally validate a novel AF3-predicted complex structure.
Materials: Cloned genes for target protein and partner, expression system (E. coli/HEK293), purification reagents, AF3 prediction, cryo-EM grid preparation kit, access to 300 keV cryo-EM.
Procedure:
1. Prediction & Cloning: Generate AF3 model of the complex. Design expression constructs based on predicted interacting domains.
2. Expression & Purification: Co-express protein components. Purify the complex via affinity and size-exclusion chromatography (SEC).
3. Sample Vitrification: Apply 3.5 µL of purified complex (0.5-1 mg/mL) to a glow-discharged cryo-EM grid. Blot and plunge-freeze in liquid ethane.
4. Data Collection & Processing: Collect >5,000 micrographs. Process using cryoSPARC: patch motion correction, CTF estimation, blob picker extraction, 2D classification, ab initio reconstruction, and heterogeneous refinement.
5. Model Building & Fitting: Build de novo model using Phenix or Coot. Fit the AF3 prediction into the cryo-EM map using UCSF Chimera fit in map. Calculate map-model correlation (CC) and Q-score.
Visualizations
Title: Scientific Reception Dynamics of AlphaFold 3
Title: AF3 Validation & Feedback Workflow
The Scientist's Toolkit: Key Research Reagents & Solutions
Table 3: Essential Reagents for AlphaFold 3 Validation & Application
| Item | Function in AF3-Related Research | Example Vendor/Resource |
|---|---|---|
| AlphaFold Server | Web-based interface for running AF3 predictions without local compute. | Google DeepMind/Isomorphic Labs |
| ColabFold (AF3 implementation) | Open-source, localizable pipeline integrating MMseqs2 and AF3 logic for batch runs. | GitHub: sokrypton/ColabFold |
| PDBbind Database | Curated set of protein-ligand complexes for benchmarking prediction accuracy. | PDBbind-CN |
| ChimeraX / USCF PyMOL | Molecular visualization software for comparing predicted vs. experimental structures. | RBVI / Schrödinger |
| RDKit | Open-source cheminformatics toolkit for handling ligand SMILES strings and conformers. | RDKit.org |
| Cryo-EM Sample Prep Kit | Glow dischargers, grids (Quantifoil), vitrification robots for experimental validation. | Thermo Fisher Scientific, Gatan |
| SPR/Biacore System | Surface Plasmon Resonance instrument to kinetically validate predicted interactions. | Cytiva |
| Molecular Dynamics Software (e.g., GROMACS) | To refine and assess the dynamic stability of AF3-predicted complexes. | GROMACS.org |
| PKM2 activator 10 | PKM2 activator 10, MF:C19H22F4N4O3S, MW:462.5 g/mol | Chemical Reagent |
| Eicosapentaenoyl serotonin | Eicosapentaenoyl serotonin, MF:C30H40N2O2, MW:460.6 g/mol | Chemical Reagent |
Conclusion
AlphaFold 3 represents a paradigm shift, moving computational structural biology beyond single proteins to the dynamic interactome of life. By delivering unprecedented accuracy in predicting multi-component biomolecular complexes, it provides researchers and drug developers with a powerful, accessible tool for generating testable hypotheses. While not a replacement for experimental methods and with acknowledged limitations in dynamics and novel chemistry, its ability to model protein-ligand, protein-nucleic acid, and decorated protein structures will drastically accelerate early-stage discovery, rational design, and mechanistic studies. The future lies in integrating AlphaFold 3's static snapshots with molecular dynamics for conformational sampling, refining its predictive power for drug affinity, and embedding it into automated, high-throughput discovery pipelines. Its widespread adoption promises to democratize structural insights and catalyze breakthroughs across biomedicine, from next-generation therapeutics to fundamental biological understanding.